

In various muscles, the two heads of myosin together with part of its α-helical coiled coil make up the minimal unit that is regulated. Reported here, for the first time, is the growth of three-dimensional crystals of such a heavy meromyosin, purified from squid, and their X-ray diffraction to near-atomic resolution (up to ∼5 Å).
Keywords: squid heavy meromyosin, Loligo pealei
Abstract
All muscle-based movement is dependent upon carefully choreographed interactions between the two major muscle components, myosin and actin. Regulation of vertebrate smooth and molluscan muscle contraction is myosin based (both are in the myosin II class), and requires the double-headed form of myosin. Removal of Ca2+ from these muscles promotes a relatively compact conformation of the myosin dimer, which inhibits its interaction with actin. Although atomic structures of single myosin heads are available, the structure of any double-headed portion of myosin, including the ∼375 kDa heavy meromyosin (HMM), has only been visualized at low (∼20 Å) resolution by electron microscopy. Here, the growth of three-dimensional crystals of HMM with near-atomic resolution (up to ∼5 Å) and their X-ray diffraction are reported for the first time. These crystals were grown in off-state conditions, that is in the absence of Ca2+ and the presence of nucleotide analogs, using HMM from the funnel retractor muscle of squid. In addition to the crystallization conditions, the techniques used to isolate and purify this HMM are also described. Efforts at phasing and improving the resolution of the data in order to determine the structure are ongoing.
1. Introduction
Detailed structural information is essential to understand how myosin motors work and how they are regulated. The activated (‘on’-state) myosin motor produces force by cycling between strong and weak actin-binding states. During the past 20 years, many states of the myosin head (referred to as S1) during the contractile cycle have been visualized at atomic resolution. This accomplishment has depended on investigations from a number of laboratories that have taken advantage of specialized features of myosins from different muscle types. Molluscan muscles have been the primary source of myosin used in this laboratory because they have allowed the visualization of an unmodified S1 (molecular mass of ∼130 kDa) containing a full-length lever arm, unlike non-molluscan myosins, which have required truncated or methylated constructs for crystallization (Dominguez et al., 1998 ▶; Rayment et al., 1993 ▶). Using scallop striated muscle, numerous weak actin-binding states of S1 have been visualized (Alamo et al., 2008 ▶; Gourinath et al., 2003 ▶; Himmel et al., 2002 ▶; Houdusse et al., 1999 ▶, 2000 ▶; Risal et al., 2004 ▶; Woodhead et al., 2005 ▶). In contrast, the ability to visualize strong actin-binding rigor-like states of the myosin motor, in the absence of actin, required different isoforms: first, that of nonmuscle myosin V by the Houdusse group (Coureux et al., 2003 ▶) and subsequently squid fast funnel retractor muscle and scallop catch muscle myosin determined in this laboratory (Yang et al., 2007 ▶).
Myosin is generally in a stable resting, or ‘off’, state. In vertebrate striated muscles, the tropomyosin–troponin complex on the thin filament regulates contractile activity. In vertebrate smooth muscle and in molluscan muscles, changes in the conformation of myosin itself control contraction (for a review, see Szent-Györgyi, 2007 ▶; see also Jung et al., 2008 ▶; Jung & Craig, 2008 ▶). In contrast to the on-state, where a single S1 head is the minimal functional unit of the motor, a functional off-state of these regulated myosins requires a larger portion of the molecule. The two heads of the myosin dimer together with (part of) its α-helical coiled-coil tail (molecular mass of ∼375 kDa for HMM; Fig. 1 ▶) is the minimal unit that is regulated (Cremo et al., 1995 ▶; Kalabokis et al., 1996 ▶; Trybus, 1994 ▶). This soluble dimer unit of minimally proteolyzed myosin retains the full sensitivity, activity and cooperativity of myosin without aggregating at low ionic strength (40 mM) owing to the removal of the filament-forming tail region.
Figure 1.
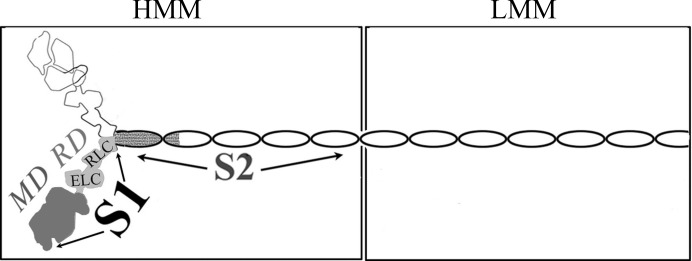
Schematic representation of the domains of myosin. Portions whose atomic structures have been determined are shaded in variations of gray.
Regulatory ions change the conformation of myosin from a generally compact off-state conformation, where the two heads contact each other asymmetrically and fold back towards the tail, to a relatively extended relationship between the tail and independently mobile heads. This change in shape has been determined by sedimentation studies of scallop HMM, where, in the presence of ADP or ATP analogs, calcium yields a slower sedimentation velocity compared with that in EGTA (Stafford et al., 2001 ▶). The compact shape of the off-state of HMM, myosin and/or the thick filament from various species has also been viewed directly by electron microscopy (at a resolution of ∼20 Å) (Brito et al., 2011 ▶; Burgess et al., 2007 ▶; Jung et al., 2008 ▶; Wendt et al., 2001 ▶; Zhao et al., 2009 ▶).
A high-resolution structure of double-headed myosin is nevertheless essential to understand fully how the off-state is achieved in regulated myosins. Crystallizing the far larger and fully sensitive and active HMM has been a long and painfully elusive pursuit. For example, despite many years of effort, we have been able to obtain only needles or fibril microcrystals from the bay scallop (Argopecten irradiens) HMM (unpublished work). It is indeed often difficult to crystallize such a large molecule, especially one that (for the vertebrate smooth muscle isoform) has been shown recently in solution to populate a mixture of disparate conformations, even in the absence of regulatory ions (Kast et al., 2010 ▶; Vileno et al., 2011 ▶). Fortunately, by switching to the funnel retractor muscle of squid (Loligo pealei), we have been able to obtain single crystals of HMM for the first time. We report here the isolation, purification and crystallization of this HMM, as well as the X-ray diffraction results that have been obtained.
2. Materials and methods
2.1. Protein isolation and digestion
Squid (Loligo pealei, common name longfin inshore squid) were obtained from the Marine Biological Laboratory in Woods Hole, Massachusetts, USA. The funnel retractor muscle was harvested and myosin was prepared as described by Stafford et al. (1979 ▶) with slightly longer spin times and higher g-force owing to looser pellets with the squid muscle. All protein work was performed on ice unless otherwise specified. Protein purity was analyzed at all steps by SDS–PAGE gels (Laemmli, 1970 ▶) stained with Coomassie Brilliant Blue R250.
Squid myosin was digested to HMM as described by Stafford et al. (2001 ▶) with the following modifications. The digestion time was 3 min. Saturated ammonium sulfate solution (SAS; pH 6.7) was added to the dialyzed HMM to 48%, then centrifuged. The supernatant was filtered, its volume measured and it was then precipitated with 55% SAS. The pellet was dissolved in a small volume of standard HMM buffer (80 mM NaCl, 20 mM MOPS pH 7, 3 mM NaN3, 2 mM MgCl2, 0.2 mM EGTA, 0.2 mM K-ADP, 0.5 mM DTT, 0.5 µg ml−1 leupeptin) and stored on ice for 3–5 d prior to continuing.
2.2. Protein purification
The 55% SAS HMM pellet was dialyzed against 100-fold volume of standard HMM buffer for 4 h, then transferred to a 40-fold to 50-fold volume of start buffer (500 mM KCl, 20 mM potassium phosphate, 3 mM NaN3, 0.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 0.2 mM K-ADP, 0.5 µg ml−1 leupeptin) pH 6.8, and dialyzed overnight. The dialyzed HMM was applied onto a hydroxylapatite (HA) column (Calbiochem, Fast Flow; 1 g HA per 6.7 mg HMM) equilibrated in start buffer and rinsed with 2–3 column volumes. The column was washed with potassium phosphate buffers of increasing molarity and pH (with all other buffer components remaining constant). HMM of varied purity eluted with each buffer. All HMMs were precipitated with SAS as described previously (48% followed by 60%) and dialyzed versus standard HMM buffer (several changes) to remove ammonium sulfate. Dialyzed samples were clarified with a 30 min spin at 100 000g. The 60% pellet samples were used hereafter. Sample sensitivity and Ca2+-activated activity were determined by the coupled assay method (Kalabokis & Szent-Györgyi, 1997 ▶).
The HA-purified HMM (both 50 and 75 mM potassium phosphate) was run over a Superdex 200 column (Pharmacia GE 1×30 cm) at room temperature in 40 mM sodium malonate pH 7, 20 mM 4-(N-morpholino)butanesulfonic acid (MOBS) pH 7.5, 3 mM NaN3, 2.5 mM MgCl2, 0.2 mM EGTA, 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 0.5 µg ml−1 leupeptin, 50 µM adenosine 5′-(β,γ-imido)triphosphate tetralithium salt hydrate (AMPPNP). Purified fractions were combined, AMPPNP was increased to 0.2 mM and the samples were concentrated (Centricon/Microcon spin units). AMPPNP was then increased to 2 mM and the final samples were clarified with a 30 min spin at 80 000g. Protein concentration was determined by the Bradford method (specifically, Bio-Rad Protein Assay), and ATPase sensitivity and activity were determined. Concentrations varied from 3 to 4 mg ml−1; sensitivity was 90+%; activity varied from 0.3 to 0.45 mol−1 s−1.
2.3. Crystallization experiments
Squid HMM-AMPPNP crystals were grown by the sitting-drop vapor-diffusion method at 277 K (using Hampton Research microbridges). Sitting drops were prepared by mixing 2.5 µl precipitant solution [10%(w/v) polyethylene glycol 20000 (PEG 20K), 4% ethylene glycol (EG), 20 mM 2-aminoethane sulfonic acid (taurine), 20 mM MOBS pH 8.0, 0.5 mM TCEP, 0.2 mM EGTA, 2 mM NaN3] with 7.5 µl protein solution. Some drops were set up with a final molarity of 0.14 mM chromium(III) acetylacetonate [Cr(acac)3] (Acros Organics) in the protein and/or precipitant solution. The drops were equilibrated against 1 ml reservoir solution (5% PEG 20 K, 2% EG, 40 mM MOBS pH 8.0, 60 mM sodium malonate, 10 mM taurine, 2 mM NaN3) for 3–4 weeks. Crystals appeared after 2 weeks and grew to dimensions of up to ∼0.5 × 0.1 × 0.05 mm (Fig. 2 ▶).
Figure 2.
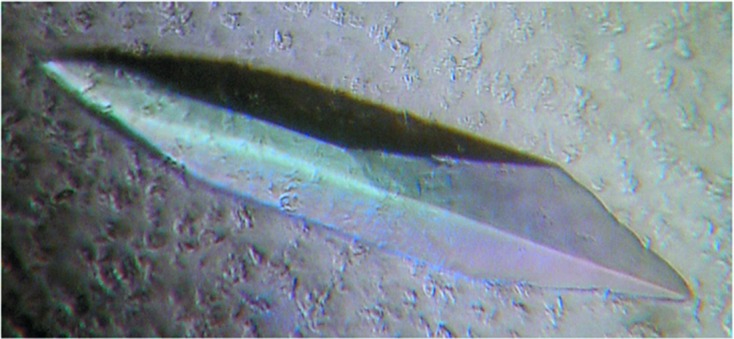
Crystal of squid heavy meromyosin (500 µm in length).
To prepare crystals for freezing, the concentration of cryoprotectant PEG 400 in the drop was increased slowly up to 5, 10, 15 and then 20%, with corresponding supplements of PEG 20 K up to 10–12% and EG at not more than 3%. Each crystal was picked up in a loop and frozen by immersion in liquid nitrogen, where it was stored until diffraction experiments.
2.4. X-ray data collection and analysis
Well over 5000 crystals were screened, primarily at the Brookhaven National Laboratory (BNL) and Cornell High Electron Synchrotron Source (CHESS) synchrotrons, with the best data set obtained at 100 K at CHESS using beamline A1 (λ = 0.939 Å). Diffraction data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). X-ray data were collected from a squid HMM-AMPPNP chromium crystal. Data-processing statistics shown in Table 1 ▶ are from a single crystal which diffracted to 5.07 Å resolution, with a complete data set to 7 Å resolution (Fig. 3 ▶ and Table 1 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the highest-resolution shell.
| No. of crystals | 1 |
| Beamline | A1, Cornell High Energy Synchrotron Source |
| Wavelength | 0.977 |
| Detector | Quantum 210 CCD detector |
| Crystal–detector distance (mm) | 480 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 360 |
| Exposure time per image (s) | 60 |
| Resolution range (A) | 50–5.07 (5.16–5.07) |
| Space group† | P212121 |
| Unit-cell parameters (A,°) | a = 143.9, b = 167.6, c = 531.0, α = β = γ = 90 |
| Mosaicity (°) | 0.300 |
| Total no. of reflections | 259264 |
| Unique reflections | 44222 |
| Multiplicity | 3.8 (3.7) |
| 〈I/σ(I)〉 | 14.9 (1.7) |
| Completeness (%) | 83.7 (46.3) |
| R merge ‡ (%) | 0.202 (0.412) |
| Overall B factor from Wilson plot (Å2) | 247.7 |
| Optical resolution | 3.89 |
Space-group assignment is based on the low metric tensor distortion index for primitive orthorhombic determined by HKL-2000 and the presence of h = odd, k = odd and l = odd systematic absences in the h00, 0k0 and 00l directions, respectively. We do not yet know the exact number of molecules in the asymmetric unit. Values of 1, 2, 3 and 4 would yield solvent contents of 86.4, 72.8, 59.3 and 45.7%, respectively.
R
merge = 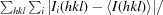
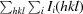 .
.
Figure 3.

X-ray diffraction image of squid HMM crystal. The five resolution rings in the right image are at 21.54, 10.79, 7.22, 5.45 and 4.39 Å; the latter two pass through the boxed region, which is magnified in the left image. Note that the diffraction is anisotropic, i.e. diffraction is observed past 4.4 Å resolution in some directions but to only ∼7 Å resolution in other directions, and a second minor lattice is seen at low resolution. (These factors may account in part for the relatively high R merge given in Table 1 ▶.)
3. Results and discussion
Regulated myosins are in the off-state in the absence of calcium and the presence of ATP. This off-state is not specific to ATP, since numerous ATP analogs have yielded a similar post-rigor structure in S1 (Himmel et al., 2002 ▶). Certain analogs, however, may be more representative than others of ATP bound in a physiological environment. Sedimentation rates of scallop HMM with AMPPNP are the most similar to those observed with ATP with or without Ca2+ (Stafford et al., 2001 ▶). It made sense to utilize this slowly hydrolyzing ATP analog for our crystallization experiments, since HMM remains fully functional (normal sensitivity and activity) in the presence of non-cross-linked AMPPNP, enabling a mimicking of an in vivo ATP-HMM. To maintain the sample in its off-state, no Ca2+ was ever introduced and at least 0.2 mM EGTA was present.
Our digestion of squid myosin produced one major band of HMM (along with a non-removable ∼70 kDa fragment; the C-terminus of the HMM was not determined). This differs from the two HMMs obtained from digestion of scallop myosin (owing to alternative digestion sites in the tail) and on SDS gels the heavy-chain portion runs in between the two heavy chains observed in scallop HMM. This initial crude squid HMM is very stable when stored as the 55% SAS pellet, remaining fully sensitive and active for weeks, with no degradation, thus offering a safe ‘stopping’ point for the protein prior to continuing with the purification. The 50 mM potassium phosphate portion from the HA column contained the most S1 and had the lowest overall yield of HMM, but it also contained the least amount of higher-molecular-weight contamination. Following a sizing-column run, this sample exhibited the greatest ability to crystallize. The sizing-column-purified 75 mM potassium phosphate HMM also crystallized well. The final purified HMM is stable for at least 1 month. Since no sequencing was performed on our trypsin-derived HMM samples, it is unknown whether there are multiple isoforms present (Matulef et al., 1998 ▶; Shaffer & Kier, 2012 ▶) in the initial digestion product, nor whether subsequent purification separates them.
Although crystals were obtained from flash-frozen/thawed HMM, use of fresh, non-frozen material increased the size of the crystals (as well as the reproducibility). This result shows a marked change from results with S1, where previously frozen material produced crystals equal to fresh material. The first HMM-AMPPNP crystals (elongated hexagonal shaped) grew to 0.25 × 0.05 × 0.03 mm and diffracted up to 10 Å resolution. Optimization of crystal conditions resulted in larger crystals (0.5 × 0.1 × 0.05 mm) and consisted of using sitting drops (10 µl), MOBS buffer pH 8, TCEP, adding 10 mM taurine, a naturally produced osmolyte in squid (Cohen, 1958 ▶) that can stabilize protein structure (Bolen, 2004 ▶; Jeruzalmi & Steitz, 1997 ▶), and exchanging NaCl for sodium malonate. Freezing with PEG 400, in combination with these optimized conditions, improved crystal diffraction to 8 Å resolution. With regard to the cryocooling procedure harming the crystals, both non-frozen and frozen (then annealed) crystals were checked for diffraction at room temperature; neither diffracted, so this gives no definitive answer. Given the challenge of freezing the HMM crystals, many dehydrating reagents were tested, but none gave positive results. Numerous reagents, additives and detergents not included in any kits were also explored, such as raffinose, ectoine, aspartame, sucralose, melizitose, carnitine, carnosine, ionic liquids (IOLITECs kit), nanospheres, as well as the compound trifluoperazine (TFP; it has been shown that micromolar concentrations of TFP bind to scallop myosin, locking it in a conformation resembling the off-state; Patel et al., 2000 ▶). There was no improvement, however, in the diffraction quality of the crystals grown in the presence of any of these additives and many crystals did not diffract at all. There was also no success with numerous metals (both gold and SmCl3 yielded only small poor-quality crystals and crystals grown in the presence of iridium diffracted very poorly). Only with the addition of Cr(acac)3 (0.14 mM in the crystallization drop), along with the optimized conditions discussed above, was an improvement in diffraction obtained, as well as increasing the number of crystals giving any diffraction at all. It should be noted that the presence of chromium in the crystal was not verified by fluorescence. The probability of having an HMM crystal that diffracted was extremely low, so several sources were used for screening. A nearly complete data set to 8 Å resolution was achieved at BNL. A crystal screened at CHESS showed diffraction past 4.4 Å resolution in some directions (Fig. 3 ▶). From this crystal, a nearly complete data set was obtained to 7 Å resolution and a partial data set to 5.07 Å resolution (Table 1 ▶).
In addition to continued efforts to improve the quality, resolution and reproducibility of the X-ray diffraction, efforts are also under way to phase the data. Heavy-atom-derivatized crystals that have diffracted include those with 5-amino-2,4,6-triiodoisophthalic acid (I3C; 9–10 Å resolution) and a tantalum bromide cluster (10–12 Å resolution) and we are currently working on obtaining data sets. No molecular-replacement solution using electron-microscopy-derived models (Wendt et al., 2001 ▶; Zhong Huang & Kenneth Taylor, personal communication) has yet been found.
Sequence database references: squid regulatory light chain: P08052 (Todarodes pacificus); squid essential light chain: P05945 (Todarodes pacificus); squid myosin heavy chain: O44934 (Loligo pealei; also referenced as Doryteuthis pealeii). Conflicts with O44934: Glu238 to Lys; Val744 to Ala; Ser864 to Arg; Ala1495 to Glu; His1531 to Arg; Gly1548 to Ala (changes submitted in 2009).
Acknowledgments
We thank Jerry Brown for helpful discussions, his attempts to phase the current data by molecular replacement and for his work on the manuscript. We also thank Karen Allen for suggesting the use of chromium(III) acetylacetonate to improve diffraction, colleagues and the staff of the BNL and CHESS synchrotrons (in particular Marian Szebenyi and Irina Kriksunov) for assistance with and/or the carrying out of data collection and former members of the Cohen laboratory for their help with screening the first HMM crystals obtained. This work is supported by NIH/NIAMS grant No. AR017346 to CC. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAMS or NIH.
References
- Alamo, L., Wriggers, W., Pinto, A., Bártoli, F., Salazar, L., Zhao, F. Q., Craig, R. & Padrón, R. (2008). J. Mol. Biol. 384, 780–797. [DOI] [PMC free article] [PubMed]
- Bolen, D. W. (2004). Methods, 34, 312–322. [DOI] [PubMed]
- Brito, R., Alamo, L., Lundberg, U., Guerrero, J. R., Pinto, A., Sulbarán, G., Gawinowicz, M. A., Craig, R. & Padrón, R. (2011). J. Mol. Biol. 414, 44–61. [DOI] [PMC free article] [PubMed]
- Burgess, S. A., Yu, S., Walker, M. L., Hawkins, R. J., Chalovich, J. M. & Knight, P. J. (2007). J. Mol. Biol. 372, 1165–1178. [DOI] [PubMed]
- Cohen, C. (1958). J. Biophys. Biochem. Cytol. 4, 489–490. [DOI] [PMC free article] [PubMed]
- Coureux, P. D., Wells, A. L., Ménétrey, J., Yengo, C. M., Morris, C. A., Sweeney, H. L. & Houdusse, A. (2003). Nature (London), 425, 419–423. [DOI] [PubMed]
- Cremo, C. R., Sellers, J. R. & Facemyer, K. C. (1995). J. Biol. Chem. 270, 2171–2175. [DOI] [PubMed]
- Dominguez, R., Freyzon, Y., Trybus, K. M. & Cohen, C. (1998). Cell, 94, 559–571. [DOI] [PubMed]
- Gourinath, S., Himmel, D. M., Brown, J. H., Reshetnikova, L., Szent-Györgyi, A. G. & Cohen, C. (2003). Structure, 11, 1621–1627. [DOI] [PubMed]
- Himmel, D. M., Gourinath, S., Reshetnikova, L., Shen, Y., Szent-Györgyi, A. G. & Cohen, C. (2002). Proc. Natl Acad. Sci. USA, 99, 12645–12650. [DOI] [PMC free article] [PubMed]
- Houdusse, A., Kalabokis, V. N., Himmel, D., Szent-Györgyi, A. G. & Cohen, C. (1999). Cell, 97, 459–470. [DOI] [PubMed]
- Houdusse, A., Szent-Gyorgyi, A. G. & Cohen, C. (2000). Proc. Natl Acad. Sci. USA, 97, 11238–11243. [DOI] [PMC free article] [PubMed]
- Jeruzalmi, D. & Steitz, T. A. (1997). J. Mol. Biol. 274, 748–756. [DOI] [PubMed]
- Jung, H. S., Burgess, S. A., Billington, N., Colegrave, M., Patel, H., Chalovich, J. M., Chantler, P. D. & Knight, P. J. (2008). Proc. Natl Acad. Sci. USA, 105, 6022–6026. [DOI] [PMC free article] [PubMed]
- Jung, H. S. & Craig, R. (2008). J. Mol. Biol. 383, 512–519. [DOI] [PMC free article] [PubMed]
- Kalabokis, V. N. & Szent-Györgyi, A. G. (1997). Biochemistry, 36, 15834–15840. [DOI] [PubMed]
- Kalabokis, V. N., Vibert, P., York, M. L. & Szent-Györgyi, A. G. (1996). J. Biol. Chem. 271, 26779–26782. [DOI] [PubMed]
- Kast, D., Espinoza-Fonseca, L. M., Yi, C. & Thomas, D. D. (2010). Proc. Natl Acad. Sci. USA, 107, 8207–8212. [DOI] [PMC free article] [PubMed]
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Matulef, K., Sirokmán, K., Perreault-Micale, C. L. & Szent-Györgyi, A. G. (1998). J. Muscle Res. Cell Motil. 19, 705–712. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Patel, H., Margossian, S. S. & Chantler, P. D. (2000). J. Biol. Chem. 275, 4880–4888. [DOI] [PubMed]
- Rayment, I., Rypniewski, W. R., Schmidt-Bäse, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G. & Holden, H. M. (1993). Science, 261, 50–58. [DOI] [PubMed]
- Risal, D., Gourinath, S., Himmel, D. M., Szent-Györgyi, A. G. & Cohen, C. (2004). Proc. Natl Acad. Sci. USA, 101, 8930–8935. [DOI] [PMC free article] [PubMed]
- Shaffer, J. F. & Kier, W. M. (2012). J. Exp. Biol. 215, 239–246. [DOI] [PMC free article] [PubMed]
- Stafford, W. F., Jacobsen, M. P., Woodhead, J., Craig, R., O’Neall-Hennessey, E. & Szent-Györgyi, A. G. (2001). J. Mol. Biol. 307, 137–147. [DOI] [PubMed]
- Stafford, W. F. III, Szentkiralyi, E. M. & Szent-Györgyi, A. G. (1979). Biochemistry, 18, 5273–5280. [DOI] [PubMed]
- Szent-Györgyi, A. G. (2007). Adv. Exp. Med. Biol. 592, 253–264. [DOI] [PubMed]
- Trybus, K. M. (1994). J. Biol. Chem. 269, 20819–20822. [PubMed]
- Vileno, B., Chamoun, J., Liang, H., Brewer, P., Haldeman, B. D., Facemyer, K. C., Salzameda, B., Song, L., Li, H.-C., Cremo, C. R. & Fajer, P. G. (2011). Proc. Natl Acad. Sci. USA, 108, 8218–8223. [DOI] [PMC free article] [PubMed]
- Wendt, T., Taylor, D., Trybus, K. M. & Taylor, K. (2001). Proc. Natl Acad. Sci. USA, 98, 4361–4366. [DOI] [PMC free article] [PubMed]
- Woodhead, J. L., Zhao, F.-Q., Craig, R., Egelman, E. H., Alamo, L. & Padrón, R. (2005). Nature (London), 436, 1195–1199. [DOI] [PubMed]
- Yang, Y., Gourinath, S., Kovács, M., Nyitray, L., Reutzel, R., Himmel, D. M., O’Neall-Hennessey, E., Reshetnikova, L., Szent-Györgyi, A. G., Brown, J. H. & Cohen, C. (2007). Structure, 15, 553–564. [DOI] [PubMed]
- Zhao, F.-Q., Craig, R. & Woodhead, J. L. (2009). J. Mol. Biol. 385, 423–431. [DOI] [PMC free article] [PubMed]