

In this study, the l,d-transpeptidase LdtMt1 was cloned, expressed and crystallized. Multiwavelength anomalous dispersion experiments have been carried out to obtain experimental phases using data at 2.9 Å resolution from a selenomethionine derivative.
Keywords: peptidoglycan, tuberculosis, Mycobacterium tuberculosis, LdtMt1
Abstract
Mycobacterium tuberculosis is capable of adapting to prolonged periods of dormancy, a state which is resistant to killing by antimycobacterial agents. The l,d-transpeptidation reaction catalysed by the l,d-transpeptidase LdtMt1 is likely to play an essential role in the adaptation of M. tuberculosis to its dormant state. LdtMt1 has been successfully crystallized using vapour-diffusion methods. The crystals of this protein belonged to space group P6522, with unit-cell parameters a = 57.25, b = 57.25, c = 257.96 Å, α = 90, β = 90, γ = 120°. Diffraction data have also been collected from a selenomethionine derivative to 2.9 Å resolution. Model building using the phases derived from the multiwavelength anomalous dispersion experiment is in progress.
1. Introduction
Tuberculosis (TB), an infectious airborne disease, is a major global health problem. Each year, there are about nine million new cases of TB and close to two million deaths (Kaufmann, 2008 ▶; Kaufmann & McMichael, 2005 ▶). The aetiological agent of TB, Mycobacterium tuberculosis, can act both intrapulmonarily and extrapulmonarily (Kaufmann, 2008 ▶; Esposito et al., 2008 ▶, 2010 ▶, 2012 ▶) and is able to survive in humans without producing apparent symptoms (Kaufmann, 2008 ▶). However, this apparent dormancy can develop into active disease even decades after initial infection, when the immune response weakens (Kaufmann, 2008 ▶; Keep et al., 2006 ▶). Since about one-third of the world’s population is infected with dormant M. tuberculosis, the risk of disease reactivation is troublesome. For this reason, the Global Plan to Stop TB 2011–2015 of the World Health Organization recognizes that effective new treatments for latent TB infection are necessary to reach full TB elimination. Over the years, it has become clear that reactivation from dormancy, growth and division of bacteria require modelling of the cell-wall peptidoglycan (PG; Mukamolova, Yanopolskaya et al., 1998 ▶; Mukamolova, Kaprelyants et al., 1998 ▶; Ruggiero et al., 2009 ▶, 2010 ▶; Ruggiero, Marchant et al., 2012 ▶), in a complex mechanism which also involves STPK kinases (Squeglia et al., 2011 ▶; Ruggiero et al., 2011 ▶; Dworkin & Shah, 2010 ▶; Ruggiero, De Simone et al., 2012 ▶). However, the precise nature of M. tuberculosis cells associated with latent tuberculosis is not yet clear.
l,d-Transpeptidases are responsible for an alternate transpeptidation pathway which leads to the formation of 3,3 cross-links instead of the typical 4,3 cross-links catalysed by penicillin-binding proteins (Fig. 1 ▶; Dubée et al., 2012 ▶; Gupta et al., 2010 ▶). The genome of M. tuberculosis contains five putative l,d-transpeptidases (LdtMt1, LdtMt2, MT0202, MT0501 and MT1477, with sequence identities ranging from 35 to 45%), of which only LdtMt1 and LdtMt2 have actually been shown to be endowed with l,d-transpeptidase activity (Gupta et al., 2010 ▶).
Figure 1.

Schematic representation of peptidoglycan transpeptidation. l,d-Transpeptidases cleave the mDap3—d-Ala4 bond and link mDap3 of the donor to the acceptor stems (3,3 cross-link).
Cross-links generated by l,d-transpeptidation (3,3 cross-links; Fig. 1 ▶) are predominant (80%) in the peptidoglycan of M. tuberculosis in the stationary phase (Lavollay et al., 2008 ▶). Consistently, microarray analyses have shown that the gene encoding LdtMt1 is upregulated 17-fold under nutrient starvation (Betts et al., 2002 ▶). Also, analysis of the peptidoglycan structure in a stationary phase culture of a mutant lacking LdtMt2 showed that 3,3 cross-links were synthesized by LdtMt1 (Gupta et al., 2010 ▶). These findings suggested that the l,d-transpeptidation reaction catalysed by LdtMt1 is likely to play an essential role in the adaptation of M. tuberculosis to the stationary phase (Lavollay et al., 2008 ▶; Gupta et al., 2010 ▶) and identified LdtMt1 as an attractive target for the development of efficient drugs.
In this study, we have successfully expressed, purified and crystallized LdtMt1 using vapour-diffusion methods. Analysis of the PFAM database (Finn et al., 2006 ▶) shows that LdtMt1 contains two domains: an N-terminal domain, the structure of which cannot unambiguously be predicted, and a C-terminal l,d-transpeptidase catalytic domain (residues 125–249, Ykud family, PF03734). The catalytic domain of LdtMt1 shares 29% sequence identity with that of the l,d-transpeptidase from Enterococcus faecium (Biarrotte-Sorin et al., 2006 ▶). The results obtained here will allow us to gather clues on M. tuberculosis adaptation phenomena and on the molecular basis for the design of enzyme inhibitors with therapeutic interest.
2. Experimental methods
2.1. Cloning, expression and purification
LdtMt1 is thought to contain a signal peptide at its N-terminus (residues 1–28) as predicted by SignalP 3.0 (Bendtsen et al., 2004 ▶). We cloned, expressed and purified LdtMt1 deprived of its signal peptide starting at residue 32. The primers Rv0116cF, 5′-TTCCATGGCGCCACTCCAACCGATCCCA-3′, and Rv0116cR, 5′-CCCAAGCTTACTAGCCGACCACCTCAATGGGA-3′, containing NcoI and HindIII restriction sites were used to amplify the LdtMt1 coding sequence starting at residue Pro32 from the H37Rv strain of M. tuberculosis. The PCR product (660 bp) was cloned into the pETM-11 expression vector (Novagen), giving a protein with a TEV-cleavable N-terminal poly-His tag. Escherichia coli BL21 (DE3) strain was co-transformed with the resulting recombinant plasmid and the pREP4 GroESL plasmid. The overnight culture was used to inoculate 1 l LB medium containing 50 µg l−1 kanamycin; protein induction was performed by the addition of 0.5 mM IPTG at 289 K when an OD600 value of 0.7 was reached. After approximately 16 h, the cells were harvested and the protein was isolated by sonicating cell pellets resuspended in 30 ml lysis buffer {50 mM Tris–HCl, 150 mM NaCl, 5%(v/v) glycerol, 0.01% 3-[(3-cholamidopropyl)dimethylamino]-1-propanesulfonate (CHAPS) pH 7.5} in the presence of a protease-inhibitor cocktail (Roche Diagnostics). The crude cell extract was cleared by centrifugation at 39 000g and the supernatant was loaded onto a 5 ml Ni–NTA column connected to an ÄKTA FPLC system (GE Healthcare) equilibrated with binding buffer [50 mM Tris–HCl, 300 mM NaCl, 5%(v/v) glycerol, 0.01% CHAPS, 10 mM imidazole pH 7.5]. A high NaCl concentration was used to reduce nonspecific binding by impurities during nickel-affinity chromatography (Kim et al., 2008 ▶). After washing with ten volumes of binding buffer, a step gradient of imidazole (40–250–500 mM) was applied to elute the protein. The fractions containing LdtMt1 were pooled and dialysed against 2 l of 50 mM Tris–HCl, 200 mM NaCl, 5%(v/v) glycerol, 0.01% CHAPS pH 7.5 with one exchange at 277 K. After removal of the His tag using TEV protease, the cleaved protein was purified by a second Ni–NTA affinity chromatography step. A final purification step was carried out using a Superdex 200 10/30 (GE Healthcare) column [50 mM Tris–HCl, 200 mM NaCl, 5%(v/v) glycerol, 0.01% CHAPS pH 7.5]. All purification steps were carried out at 277 K. The homogeneity of the protein was tested by SDS–PAGE. Freshly concentrated protein was used for crystallization experiments.
A selenomethionine derivative of LdtMt1 (SeMetLdtMt1) was prepared by growing E. coli BL21 (DE3) cells expressing the recombinant protein in 1 l minimal medium (M9) containing 0.4%(w/v) glucose, 1 mM MgSO4, 0.1 mM CaCl2, 50 µg l−1 kanamycin and 1 mM thiamine at 310 K. After reaching an OD600 of 0.7, an amino-acid mixture (50 mg l−1 Ile, Leu and Val and 100 mg l−1 Phe, Thr and Lys) was added to the culture, which was then shifted to 289 K. After equilibration, 60 mg l−1 seleno-l-methionine was added and induction was performed. The labelled protein was purified as described above.
2.2. Crystallization experiments
Crystallization was performed at 293 K by hanging-drop vapour-diffusion methods. Preliminary crystallization trials were carried out using a crystallization workstation (Hamilton Robotics). 192 high-throughput reagents (Hampton Research) were tested. Optimization of the crystallization conditions was performed by fine-tuning the protein and precipitant concentrations using a drop consisting of 1 µl protein solution and 1 µl precipitant solution and a reservoir volume of 400 µl.
2.3. Data collection and processing
Preliminary diffraction data at 2.9 Å were collected in-house at 100 K using a Rigaku MicroMax-007 HF generator producing Cu Kα radiation and equipped with a Saturn 944 CCD detector. Higher diffraction data for both native LdtMt1 and SeMetLdtMt1 were collected on synchrotron beamline BM14 at the ESRF, Grenoble, France at 100 K. Cryoprotection of the crystals was achieved by rapid soaking (1–2 s) in a solution of 14%(v/v) glycerol. An oscillation range of 1° and an X-ray dose corresponding to about 4 s exposure were adopted for all experiments. The data sets were scaled and merged using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). Multiwavelength anomalous diffraction (MAD) was carried out using three different wavelengths determined from the selenium absorption spectrum and the native data set (Table 1 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the highest-resolution shell.
| SeMet derivative | Native | |||
|---|---|---|---|---|
| Peak | Inflection point | Remote | ||
| Beamline | BM14 | BM14 | BM14 | BM14 |
| Space group | P6522 | P6522 | P6522 | P6522 |
| Unit-cell parameters (Å) | ||||
| a | 56.80 | 56.85 | 56.84 | 57.114 |
| b | 56.80 | 56.85 | 56.84 | 57.114 |
| c | 256.76 | 257.08 | 257.05 | 256.997 |
| Resolution range (Å) | 50.00–2.95 (3.01–2.95) | 50.00–2.95 (3.01–2.95) | 50.00–2.90 (2.95–2.90) | 50.00–1.90 (1.93–1.90) |
| Wavelength (Å) | 0.9784 | 0.9786 | 0.9724 | 0.9786 |
| Mosaicity (°) | 0.9 | 0.8 | 0.8 | 0.5 |
| Average multiplicity | 9.0 (6.2) | 6.1 (5.6) | 6.7 (6.9) | 9.6 (5.6) |
| Unique reflections | 5590 | 5742 | 6076 | 20581 |
| Completeness (%) | 97.8 (98.5) | 97.4 (98.8) | 99.5 (99.5) | 98.9 (96.0) |
| R merge † (%) | 7.4 (44.5) | 7.0 (45.1) | 4.7 (34.8) | 7.6 (41.3) |
| Average I/σ(I) | 18.1 (2.1) | 23.2 (3.0) | 25.6 (4.1) | 29.9 (2.6) |
R
merge = 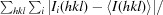
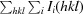 , where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of the intensity of reflection hkl.
, where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of the intensity of reflection hkl.
2.4. Structure determination
The C-terminal part of LdtMt1 (residues 123–249) shares 29% sequence identity with the l,d-transpeptidase from E. faecium (Biarrotte-Sorin et al., 2006 ▶; PDB entry 1zat). Using this structure, we attempted molecular replacement (MR) using the program Phaser (McCoy et al., 2007 ▶). However, all trials were unsuccessful. The structure of the enzyme was solved by MAD using data sets collected at wavelengths optimized for SeMet (Table 1 ▶) and a data set from a native crystal. Phases were derived using SHELXD (Sheldrick, 2008 ▶) implemented in the Auto-Rickshaw pipeline (Panjikar et al., 2005 ▶). These phases were improved by solvent-flattening and phase-extension routines using the program RESOLVE (Terwilliger, 2003b ▶).
3. Results and discussion
The gene of LdtMt1 consists of 660 bp encoding the protein sequence (residues 32–251) deprived of its N-terminal signal peptide. This construct has been successfully cloned, expressed, purified and crystallized using vapour-diffusion methods. The purified LdtMt1 showed a single band of approximately 24 kDa on SDS–PAGE, which is in good agreement with the molecular mass derived by mass spectrometry (23 935 Da). The initial screening using commercially available solutions revealed some initial indications of crystallization in four conditions containing polyethylene glycol (PEG) 3350. Fine-tuning of crystallization conditions using the hanging-drop method produced crystals suitable for X-ray diffraction studies (Fig. 2 ▶). The best crystals of LdtMt1 with dimensions of 0.05 × 0.05 × 0.2 mm were obtained by mixing protein solution at 5 mg ml−1 with 0.16 M ammonium citrate tribasic pH 7.0, 16%(w/v) PEG 3350 (Fig. 2 ▶).
Figure 2.
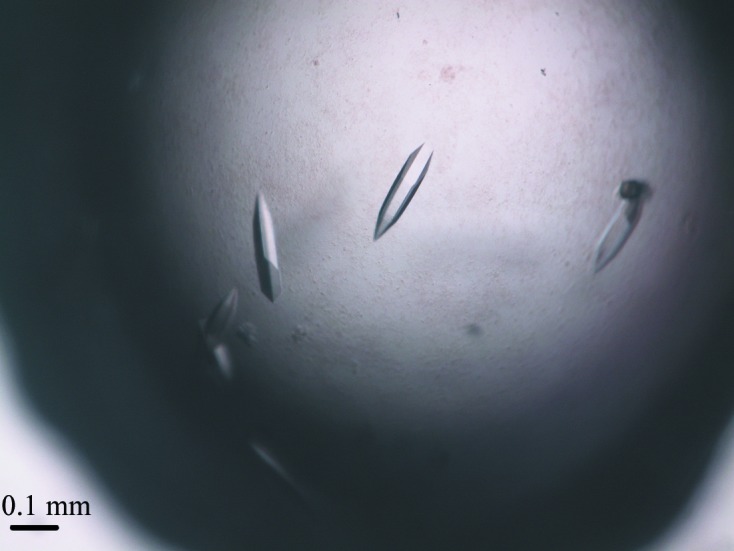
Image of typical LdtMt1 crystals grown by vapour diffusion using 0.16 M ammonium citrate tribasic pH 7.0, 16%(w/v) polyethylene glycol 3350 as precipitant.
The crystals diffracted to 1.90 Å resolution on the BM14 beamline at the ESRF (Fig. 3 ▶) and belonged to space group P6522, with unit-cell parameters a = 57.25, b = 57.25, c = 257.96, α = 90, β = 90, γ = 120.00 (Table 1 ▶). Matthews coefficient calculations suggested the presence of one molecule in the asymmetric unit (V M = 2.5 Å3 Da−1, 52% solvent content; Matthews, 1968 ▶). All attempts to solve the structure by MR using the available structure of the l,d-transpeptidase from E. faecium (29% sequence identity between residues 123–249; PDB entry 1zat) were unsuccessful. This suggested that the available model was not suitable for use in MR. Therefore, a SeMet derivative of the protein was prepared in order to perform MAD experiments. Crystals of SeMetLdtMt1 were obtained using the same procedure as adopted for the native protein. The best crystals of SeMetLdtMt1 grew using protein solution at 3 mg ml−1 mixed with 0.18 M ammonium citrate tribasic pH 7.0, 18%(w/v) PEG 3350 and diffracted to 2.95 Å resolution on the BM14 beamline at the ESRF. For peak and inflection wavelength determination, a fluorescence scan was recorded from a single SeMetLdtMt1 crystal.
Figure 3.

Diffraction pattern of a native LdtMt1 crystal.
The Auto-Rickshaw pipeline was used to combine phases derived from the three wavelengths corresponding to peak, inflection and remote regions of the fluorescence scan (Panjikar et al., 2005 ▶). SHELXD identified four selenium sites in the asymmetric unit of the protein: three in the protein sequence plus one at the N-terminus (Sheldrick, 2008 ▶). The initial set of phases was improved using the solvent-flattening and phase-extension methods implemented in the program RESOLVE (Terwilliger, 2003a ▶,b ▶). Manual model-building sessions aimed at defining the complete structure of LdtMt1 are in progress (Fig. 4 ▶). This work will produce precious information for understanding the structural features associated with LdtMt1 activity and inhibition.
Figure 4.

(2F o − F c) electron-density map contoured at 2.0σ. The density clearly shows a β-sheet region of the N-terminal domain of LdtMt1.
Acknowledgments
This work was funded by the MIUR (PRIN 2009 – prot. 200993WWF9) and by the Mizutani Foundation of Glycoscience (reference No. 120012). The research leading to this publication received funding from the European Community’s Seventh Framework Program (FP7/2007–2013) under grant agreement No. 226716. The pREP4 GroESL plasmid was kindly provided by Hélène Barreteau (CERMAV–CNRS Grenoble) and Stéphane Mesnage (Department of Molecular Biology and Biotechnology, University of Sheffield).
References
- Bendtsen, J. D., Nielsen, H., von Heijne, G. & Brunak, S. (2004). J. Mol. Biol. 340, 783–795. [DOI] [PubMed]
- Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A. & Duncan, K. (2002). Mol. Microbiol. 43, 717–731. [DOI] [PubMed]
- Biarrotte-Sorin, S., Hugonnet, J. E., Delfosse, V., Mainardi, J. L., Gutmann, L., Arthur, M. & Mayer, C. (2006). J. Mol. Biol. 359, 533–538. [DOI] [PubMed]
- Dubée, V., Triboulet, S., Mainardi, J. L., Ethève-Quelquejeu, M., Gutmann, L., Marie, A., Dubost, L., Hugonnet, J. E. & Arthur, M. (2012). Antimicrob. Agents Chemother. 56, 4189–4195. [DOI] [PMC free article] [PubMed]
- Dworkin, J. & Shah, I. M. (2010). Nature Rev. Microbiol. 8, 890–896. [DOI] [PubMed]
- Esposito, C., Cantisani, M., D’Auria, G., Falcigno, L., Pedone, E., Galdiero, S. & Berisio, R. (2012). FEBS Lett. 586, 659–667. [DOI] [PubMed]
- Esposito, C., Carullo, P., Pedone, E., Graziano, G., Del Vecchio, P. & Berisio, R. (2010). FEBS Lett. 584, 1091–1096. [DOI] [PubMed]
- Esposito, C., Pethoukov, M. V., Svergun, D. I., Ruggiero, A., Pedone, C., Pedone, E. & Berisio, R. (2008). J. Bacteriol. 190, 4749–4753. [DOI] [PMC free article] [PubMed]
- Finn, R. D., Mistry, J., Schuster-Böckler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M., Khanna, A., Durbin, R., Eddy, S. R., Sonnhammer, E. L. & Bateman, A. (2006). Nucleic Acids Res. 34, D247–D251. [DOI] [PMC free article] [PubMed]
- Gupta, R., Lavollay, M., Mainardi, J. L., Arthur, M., Bishai, W. R. & Lamichhane, G. (2010). Nature Med. 16, 466–469. [DOI] [PMC free article] [PubMed]
- Kaufmann, S. H. (2008). Nature (London), 453, 295–296. [DOI] [PubMed]
- Kaufmann, S. H. & McMichael, A. J. (2005). Nature Med. 11, S33–S44. [DOI] [PMC free article] [PubMed]
- Keep, N. H., Ward, J. M., Cohen-Gonsaud, M. & Henderson, B. (2006). Trends Microbiol. 14, 271–276. [DOI] [PubMed]
- Kim, Y. et al. (2008). Adv. Protein Chem. Struct. Biol. 75, 85–105. [DOI] [PMC free article] [PubMed]
- Lavollay, M., Arthur, M., Fourgeaud, M., Dubost, L., Marie, A., Veziris, N., Blanot, D., Gutmann, L. & Mainardi, J. L. (2008). J. Bacteriol. 190, 4360–4366. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mukamolova, G. V., Kaprelyants, A. S., Young, D. I., Young, M. & Kell, D. B. (1998). Proc. Natl Acad. Sci. USA, 95, 8916–8921. [DOI] [PMC free article] [PubMed]
- Mukamolova, G. V., Yanopolskaya, N. D., Kell, D. B. & Kaprelyants, A. S. (1998). Antonie Van Leeuwenhoek, 73, 237–243. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Panjikar, S., Parthasarathy, V., Lamzin, V. S., Weiss, M. S. & Tucker, P. A. (2005). Acta Cryst. D61, 449–457. [DOI] [PubMed]
- Ruggiero, A., De Simone, P., Smaldone, G., Squeglia, F. & Berisio, R. (2012). Curr. Protein Pept. Sci. 13, 756–766. [DOI] [PMC free article] [PubMed]
- Ruggiero, A., Marasco, D., Squeglia, F., Soldini, S., Pedone, E., Pedone, C. & Berisio, R. (2010). Structure, 18, 1184–1190. [DOI] [PubMed]
- Ruggiero, A., Marchant, J., Squeglia, F., Makarov, V., De Simone, A. & Berisio, R. (2012). J. Biomol. Struct. Dyn., doi:10.1080/07391102.2012.698243s. [DOI] [PubMed]
- Ruggiero, A., Squeglia, F., Marasco, D., Marchetti, R., Molinaro, A. & Berisio, R. (2011). Biochem. J. 435, 33–41. [DOI] [PubMed]
- Ruggiero, A., Tizzano, B., Pedone, E., Pedone, C., Wilmanns, M. & Berisio, R. (2009). J. Mol. Biol. 385, 153–162. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Squeglia, F., Marchetti, R., Ruggiero, A., Lanzetta, R., Marasco, D., Dworkin, J., Petoukhov, M., Molinaro, A., Berisio, R. & Silipo, A. (2011). J. Am. Chem. Soc. 133, 20676–20679. [DOI] [PubMed]
- Terwilliger, T. C. (2003a). Acta Cryst. D59, 45–49. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2003b). Methods Enzymol. 374, 22–37. [DOI] [PubMed]