

Abstract
NADPH-cytochrome P450 oxidoreductase (CYPOR) and nitric oxide synthase (NOS), two members of the diflavin oxidoreductase family, are multi-domain enzymes containing distinct FAD and FMN domains connected by a flexible hinge. FAD accepts a hydride ion from NADPH, and reduced FAD donates electrons to FMN, which in turn transfers electrons to the heme center of cytochrome P450 or NOS oxygenase domain. Structural analysis of CYPOR, the prototype of this enzyme family, has revealed the exact nature of the domain arrangement and the role of residues involved in cofactor binding. Recent structural and biophysical studies of CYPOR have shown that the two flavin domains undergo large domain movements during catalysis. NOS isoforms contain additional regulatory elements within the reductase domain that control electron transfer through Ca2+-dependent calmodulin (CaM) binding. The recent crystal structure of an iNOS Ca2+/CaM-FMN construct, containing the FMN domain in complex with Ca2+/CaM, provided structural information on the linkage between the reductase and oxgenase domains of NOS, making it possible to model the holo iNOS structure. This review summarizes recent advances in our understanding of the dynamics of domain movements during CYPOR catalysis and the role of the NOS diflavin reductase domain in the regulation of NOS isozyme activities.
Keywords: NADPH-cytochrome P450 oxidoreductase, Nitric oxide synthase, Calmodulin, flavins, heme, diflavin reductase family
Introduction
Enzymes of the diflavin reductase family, for which NADPH-cytochrome P450 oxidoreductase (CYPOR) is the prototype, contain one molecule each of FAD and FMN in a single polypeptide and function in transfer of electrons from NADPH to the ultimate electron acceptor. Notably, these enzymes perform a step-down function from the two-electron donor NADPH to one-electron acceptors including redox cofactors, with the flavins functioning as one-electron carriers (Fig. 1). The microsomal cytochrome P450 (P450) monooxygenase system is composed of CYPOR and P450 [1]. P450 contains an iron protoporphyrin IX (heme) as a cofactor. In humans, there are currently 50 known microsomal P450 monooxygenases, [2] but only one CYPOR [3, 4]. Other physiological electron acceptors of CYPOR include microsomal heme oxygenases [5] and cytochrome b5 (cyt b5) [6]. Each flavin has a unique function: the FAD accepts two reducing equivalents from NADPH and transfers them to FMN one at a time [7, 8]. FMN in turn transfers them to P450s one by one (for reviews see refs. [1, 9–12]. P450 activates molecular oxygen by way of stepwise one-electron reductions during which one atom of the oxygen molecule is inserted into the product (ROH), and the other undergoes two equivalents of reduction to form a water molecule (Eq. 1).
| (Eq 1) |
Figure 1.

Electron transfer components of the microsomal P450 systems and NOS dimer, together with their electron transfer paths.
Other prominent members of the diflavin reductase family include the nitric oxide synthase (NOS) isozymes [13]. NOS catalyzes the NADPH- and oxygen-dependent conversion of l-arginine (l-Arg) to l-citrulline (l-Cit) and nitric oxide (NO) via two consecutive mono-oxygenation reactions (for reviews see refs. [14–16]). All three NOS isoforms are homodimeric flavocytochromes and each monomer consists of two major domains (Fig. 1): an N-terminal oxygenase domain (NOSoxy), which is functionally similar but structurally unrelated to P450; and a C-terminal, flavin-containing, reductase domain (NOSred), which is structurally and functionally similar to CYPOR. The NOSoxy domain contains a P450-like heme and (6R)-5,6,7,8-tetrahydrobiopterin (H4B) and a l-Arg binding site. The two domains are connected by a CaM-binding region. The NOSoxy domain of one monomer accepts electrons from the FMN of the reductase domain (NOSred) of the other monomer (Fig. 1) [17–19].
Three NOS isoforms have been identified in mammals as the products of three distinct genes, and these isoforms share 50–60% sequence identity, but play different physiological roles due to their tissue distribution and regulation. Neuronal NOS (nNOS, NOS I) is located in neurons in the brain and neuromuscular junctions and is involved in neurotransmission. Endothelial NOS (eNOS, NOS III) is located in vascular endothelial cells and is involved in vascular homeostasis, while inducible NOS (iNOS, NOS II), located in macrophages, is expressed only in response to endotoxins or inflammatory cytokines. nNOS and eNOS are constitutively expressed, and their activities are Ca2+/CaM-dependent, whereas iNOS activity is independent of intracellular Ca2+ concentration. The reductase domains in all three NOS isoforms are structurally and functionally related to CYPOR [13] [14–16, 20]. However, NOSred in nNOS and eNOS contains additional regulatory elements, which down regulate the electron transfer (ET) from the reductase domain to the NOSoxy in the absence of CaM, and CaM relieves this suppression. On the other hand, iNOS, which has tightly bound Ca2+/CaM, lacks these regulatory elements except the CaM-binding region. Thus, the NOS activity is regulated by the reductase domain by undergoing dynamic structural rearrangements upon binding to CaM.
In addition to CYPOR and NOSred, which are typical diflavin reductases, other members of this family also include the reductase domain (BMR) of flavocytochrome P450BM3 (P450BM3) from Bacillus megaterium [21] and methionine synthase reductase (MSR) [22–24], both of which have been studied extensively. These enzymes and other members of the growing family, including human cancer-related novel reductase 1 (NR1) [25], flavoprotein subunits of bacterial sulfite reductase (SiR) [26], the less studied pyruvate: NADP+ oxidoreductase from Euglena gracilis [27, 28], and reductase Tah18 protein from yeast [29] share domain structures similar to that of CYPOR, containing both the FNR- and Fld-like folds [30, 31]. Thus, they are likely to have similar functions and mechanism of actions. This review discusses recent advances regarding the structure and function of diflavin reductases. In particular, the structural differences and similarities between CYPOR and NOSred are summarized, and their mechanisms for electron transfer (ET) are discussed.
1. Domain structures in the CYPOR and NOS reductase domain
Both CYPOR and P450 are membrane-bound proteins associated with the cytoplasmic surface of the endoplasmic reticulum (ER) (Fig. 2A). The amino acid sequence [32–35] and crystallographic structures of rat [36] and human [37] CYPOR suggest that the protein has evolved by fusing two ancestral genes that encode proteins related to a FMN-containing flavodoxin (Fld) and a FAD-containing ferredoxin-NADP+ oxidoreductase (FNR) [32, 33] (Figs. 3A and 3B). The Fld sequence corresponds to residues 77 to 228 of rat CYPOR and the FNR sequence lies in residues 267 to 325 and 450 to 678. The first ~55 residues form an anchor, by which the protein is bound to the ER membrane. The amino acids of the connecting domain (CD) are interspersed with the FNR-like domain, (residues 244 to 266 and 326 to 450 in rat CYPOR numbering). The CD, composed mainly of -helices, is tightly bound to the FNR-like domain, and together they form the FAD domain. The FAD and FMN domains are connected by a flexible hinge, which spans 12 residues from Gly-232 to Arg-243 (rat CYPOR numbering) and is located between the F-helix of the FMN domain and strand 6 of the CD [36–38]. Bredt et al. [13] first isolated cDNA of rat nNOS and recognized the sequence similarity between CYPOR and the C-terminal reductase domain of nNOS (Fig. 2B). The reductase domains of all three NOSs bear 30 40% sequence identity with CYPOR. However, the oxygenase domains of NOSs do not reveal any sequence homology with known P450s, although they have a common cysteine residue as the proximal thiolate ligand and exhibit P450-like spectral characteristics [39–41]. In addition, the N-termini of the three NOS isoforms are very different, distinguishing their cellular locations. nNOS contains a PDZ domain of some 200 amino acids, and eNOS contains the myristoylation and palmitoylation motifs (Fig. 2B).
Figure 2.

Domain organization of CYPOR, NOS isoforms, and related enzymes. Dots indicate conserved aromatic amino acid residues and bars above “CD” represent the location of the connecting domains in the primary sequence. H, hinge; C, conseved Cys residue as the thiolate heme ligand; CD, connecting domain. Note that CD consists of two different parts of the linear sequence interspersed with the FNR like domain. (A) Fld, flavodoxin; FNR, ferredoxin-NADP+ oxidoreductase; CYPOR, NADPH-cytochrome P450 oxidoreductase; P450, cytochrome P450; TM, transmembrane domain. (B) CaM, CaM-binding region; H4B, (6R)-5,6,7,8-tetrahydrobiopterin; BF, -finger; AR, autoregulatory region; Myr and Palm, myristoylation and palmitoylation motifs, respectively; Zn, Zinc binding site found at the dimer interface; PDZ, PDZ domain.
Figure 3.

Evolutionary origins of the structures of NADPH-cytochrome P450 reductase (CYPOR) and nNOS reductase domain (nNOS-red) as shown by overlays of the ribbon structures of D. vulgaris flavodoxin (Fld) and spinach ferredoxin-NADP-oxidoreductase (FNR). A, A model of a putative Fld-FNR complex [31]; B, CYPOR structure [36]. The FMN domain, FAD domain, and connecting domain (CD) are marked; C, structure of the nNOS reductase domain (nNOSred) [42] containing regulatory elements, AR, CT and BF shown in red; D, overlay of the structures of Fld, FNR and CYPOR; E, overlay of CYPOR and nNOSred.
To date, only the structure of nNOSred (excluding the CaM-binding region) has been determined, and its overall structure is very similar to that of CYPOR [42] (Fig. 3B and 3C). The reductase domains of all NOS isoforms are structurally and functionally related to CYPOR. However, the NOSred domains include unique regulatory inserts: (i) an approximately 30 amino acid CaM-binding sequence that links the oxygenase- and reductase domains of NOS, (ii) an approximately 45 amino acid autoregulatory region (AR) in the FMN domains of n- and eNOS but not iNOS, (iii) a 21–42 amino acid C-terminal extension (CT) compared to CYPOR, and (iv) a small insertion or -finger (BF) in the connecting domain (CD). Like CYPOR, NOSs also have flexible hinges between the FMN domain and CD, but their lengths are much longer (23–25 residues vs. 12 in CYPOR), and their sequences are not conserved [43].
2. Domain-domain interactions in CYPOR and the NOS reductase domains
Proper protein-protein and/or interdomain interactions are required for rapid and specific ET in multiprotein or multidomain redox enzymes. Thus, the exquisite control of association and dissociation between the domains or proteins is the key factor for rapid and controlled ET [44]. The CYPOR/ P450 and NOS systems are multi-domain enzymes (Figs. 2 and 3), and their functions require specific interprotein/interdomain interactions. In the case of CYPOR, precise and specific interactions between the FMN and FAD/NADPH domains within CYPOR, and between the FMN domain and P450 are required for ET reactions that are necessary for their catalytic actions; i.e., the “FMN domain” must recognize both the FAD domain and P450. The movement of the FMN domain is essential for these sequential electron transfers. The “encounter complex,” consisting of multiple protein conformations, is initially formed by electrostatic interactions leading to the formation of a specific 1:1 complex with single-orientation/conformation (Fig. 4). This complex adopts an optimized geometry between the redox centers for an efficient ET. In both the CYPOR/P450 and NOS systems, the FMN domain shares a common role in the ET process, bearing the conserved patches of acidic amino acid residues involved in the electrostatic interactions with ET partners. The FMN domain has a very strong dipole [45], which is involved in the specific docking with the heme centers. In the CYPOR/ P450 system, which is localized on the ER membrane (Fig. 4), the hydrophobic lipid bilayer presumably serves to concentrate the reactants and hence increase the frequency of their interaction, as well as to maintain their proper orientation for efficient ET [46–48].
Figure 4.

Topology of CYPOR and P450 and the formation of their complex on the ER membrane. CYPOR is in a dynamic equilibrium between the closed and open forms. In the closed form, FMN accepts electrons from FAD and, in its open form, donates electrons to P450. The encounter complex consists of multiple protein orientations leading to the single orientation forming the specific complex. CYPOR is shown in a closed conformation, when the nucleotide (NADPH or NADP+) is bound and opens up in the presence of electron acceptors, e.g., cyt c or P450s (see section 2.1 for detailed discussion).
2.1. Domain movements
In the crystal structures of both rat and human CYPORs [36, 37] and nNOSred [42], the dimethylbenzene edge of the isoalloxazine ring of FMN is covered by the FAD domain, and the distances between FAD and FMN cofactors are 4 Å (C8-C8 atoms) and 5 Å (C7-C7 atoms) (Fig. 3B and 3C). The pyrimidine ring of the FMN isoalloxazine is partially buried by two loops, while the dimethylbezene edge is exposed to solvent. Furthermore, the crystal structure of the ‘closed” form, in which the flavins are close to each other, is difficult to reconcile with the results of mutagenesis studies by Shen and Kasper [49]. Zhao et al [45] proposed that binding of P450 to CYPOR requires a movement of the FMN domain relative to the rest of the CYPOR molecule.
Sevrioukova et al [50] also proposed a similar mechanism in bacterial P450BM3. Hubbard et al [38] reported that the superposition of various CYPOR mutants and wild-type structures revealed the flavin-flavin distance varying from 3.9 Å to 5.8 Å, providing direct evidence for the FMN domain movement. However, the extent of these domain movements is relatively small. In contrast, in the crystal structure of the flavoprotein subunit of E. coli sulfite reductase, the electron density for the entire FMN domain is missing, indicating the flexibility of the FMN domain [51]. Furthermore, the rate of intermolecular ET between the isolated FAD/NADPH and FMN domains of CYPOR results in cytochrome c (cyt c) reductase activity approximately 1.6% that of the intact protein [52], suggesting that the electrostatic interaction between the two domains is relatively weak, consistent with the flexibility between the two domains. The FMN domain of MSR, another member of the diflavin reductase family, is also flexible in solution [53].
Recently, Hamdane and Xia et al [54] demonstrated direct evidence for a large-scale domain movement, revealing the “closed-open” transition. A CYPOR variant with a four amino acid deletion in the hinge region that links the FMN domain to the rest of the protein has been crystallized in three extended conformations (open state), in which the distance between FAD and FMN cofactors ranges 30–60 Å (Fig. 5, left). In this mutant, the rate of ET from FAD to FMN was decreased by three orders of magnitude, and the steady state P450-dependent catalytic activity using CYP2B4 was not detectable. However, when the mutant was reduced by dithionite, i.e., fully reduced FMN was formed, the mutant enzyme catalyzed product formation by CYP2B4 under single turnover conditions at the same rate as wild type. Thus, the authors proposed that the hinge of CYPOR regulates electron transfer from FAD to FMN and from FMN to P450 by adjusting the distance and orientation of the two flavin cofactors. Aigrain et al [55] also provided evidence for a domain movement from the crystal structure of a yeast-human CYPOR chimera. In addition, Xia et al. [56] have used a counter approach that a covalent linkage between the two flavin domains would be expected to interfere with the interaction with P450 (Fig. 5, middle). The mutant CYPOR with an engineered disulfide bond between the FAD/NADPH and FMN domains is indeed essentially incapable of interflavin electron transfer and is unable to support of P450-dependent monoxygenase activity, unless the disulfide bond is reduced, demonstrating that a large conformational change is necessary for the FMN domain to interact with P450. A movie showing possible domain movements of CYPOR on the ER membrane and its docking with P450 2B4 is now included in Supplementary Information.
Figure 5.

The closed and open conformations of CYPOR (left and middle panels) and nNOSred (right panel). FAD and FMN are shown with stick models in magenta. In the middle row, ribbon diagrams for CYPOR and nNOS red structures are shown. Distances between FAD and FMN (between arrows indicating C8-C8 carbon atoms pointed by purple lines) are shown in Å in the middle row. The left panel shows relative orientations of FMN domain with respect to the FAD domain observed in WT (gold, PDB code: 1AMO) and three open conformations of the linker-deleted mutant (PDB code: 3FJO), Mol A (red), Mol B (blue) and Mol C (cyan) are shown (see ref [54] for details). In the middle panel, the engineered S-S bond (green) is shown in the closed-form mutant of CYPOR (PDB code: 3OJW, middle panel, see ref. [56] for details); and nNOSred containing the regulatory elements, AR, CT and BF of NOSred (PDB code: 1TLL, see ref. [42] for details) is shown in the right panel. In the bottom row, relative orientations of FMN and FAD are schematically shown by lines representing the isoalloxazine planes. In the wild type structure (left panel), the two planes make ~135 and form a continuous ribbon and aligned, while in the disulfide-engineered mutant the two planes are not aligned (middle panel, bottom). It is not clear whether the difference in the FMN-FAD distances observed in the closed form of wild type CYPOR (4.0 Å) and nNOSred (5.2 Å) and the slight shift and rotation of the two flavin planes observed in the NOSred structure (right panel, bottom). However, it is possible that, in NOS, CaM may unlock the closed conformation to make the distance and orientation between FAD and FMN more favorable for efficient ET between them.
Furthermore, studies of CYPOR by electron-electron double resonance (ELDOR) spectroscopy [57, 58] suggest that the CYPOR molecule adopts multiple “closed” and “open” conformations in solution. Ellis et al [59] demonstrated, using a combination of NMR and small-angle X-ray scattering (SAXS) experiments, that the oxidized human CYPOR exists in solution as a mixture of approximately equal amounts of two conformations, one consistent with the crystal structure (closed form) and one a more extended structure, which presumably is required for interaction with its ET partners. In addition, the relative contributions of each state to the equilibrium are affected by the binding of NADP(H), with the nucleotide bound form favoring the closed form. In contrast, Vincent et al. [60] recently showed, utilizing high resolution NMR measurements together with residue-specific 15N relaxation and 1H-15N residual dipolar coupling data, that the oxidized CYPOR in solution in the absence of bound nucleotide exists in a unique and predominant conformation similar to the closed conformation observed in the crystal structure [36, 37]. It appears to be more reasonable that the closed states that support intramolecular ET are formed by binding of the nucleotide NADP(H), whereas open states that enable ET from FMN to P450 are favored in the absence of bound coenzyme. At present, although the detailed mechanism is unclear, cumulative data suggest that the domain movements are regulated by redox states of the enzyme and NADPH binding and/or NADP+ release. Recently, using a combination of fluorescence resonance energy transfer and stopped flow methods, Pudney et al. [61] have provided evidence that reduction of the flavins in CYPOR induces the opening, while the nucleotide binding induces closing of the CYPOR molecule.
In the constitutive NOS isoforms, NO synthase activity is completely blocked in the absence of CaM (Fig. 5, right), but partial cyt c reductase activity is observed. In the presence of CaM, cyt c reductase activity is increased by 10 20-fold. Craig et al [62] first proposed that a “conformational lock” induced by NADPH binding restricts access of the FMN domain to the oxygenase domain, and that CaM releases this locked conformation. The structure of a CaM-free nNOSred revealed an ionic interaction between Arg1400 in the CT regulatory element and the 2’-phosphate group of the bound NADP+ [63]. Mutations of this residue to Ser or Glu increased cyt c reductase activity, indicating that the interaction between Arg1400 and the 2’-phosphate is responsible for the conformational switch. However, mutation of Arg1400 did not confer the ability to synthesize NO in the CaM-free state, suggesting that additional structural changes induced by the binding of CaM allow an interaction between the FMN domain and the oxygenase heme domain to facilitate efficient ET.
Recently, Xia et al [64] constructed a model for the structure of the entire iNOS molecule, and proposed the movement of the FMN domain in iNOSred in the presence of tightly bound CaM. In the nNOS isoform, Tejero et al [65] have constructed a model for the nNOS-FMN-CaM complex that allows flexibility of the FMN domain. These reports shed some light on the role of regulatory elements that are not present in CYPOR. A more detailed discussion of the iNOS model is presented in section 4.2.
2.2. Domain-domain interactions
In the diflavin reductase family, including CYPOR, NOS isoforms, SiR and MSR, a large-scale conformational rearrangement of the FMN domain is necessary to facilitate specific binding with an electron acceptor for efficient ET. Shimizu et al. [66], Shen and Kasper [49], Bridges et al [67], and Zhao et al [45] carried out mutational analysis and demonstrated the importance of electrostatic interactions involved in the binding of CYPOR to its acceptors. The highly conserved proximal surface of microsomal P450s is relatively electropositive and can interact with the negatively charged surface of the FMN domain of CYPOR (Fig. 6A) [54, 67–69]. A putative model of a P450-CYPOR complex showed the total contact area between the two molecules is ~1500 Å2, of which 870 Å2 is located between the FMN domain and P450 [54]. This electropositive region of P450 (Fig. 6B) also interacts with the negative surfaces of cyt b5, which stimulates catalysis by causing a conformational change in the active site [69]. In addition to these hydrophilic amino acid residues of P450, hydrophobic amino acid residues, Val267 and Leu270 on the proximal site of CYP2B4 also contribute to the CYPOR recognition [70]. The docked model based on the open conformation of the hinge mutant by Hamdane and Xia et al [54] also suggests that the planes of the heme and FMN are almost perpendicular to each other, and the distance between the heme and flavin cofactors is about 12 Å (Fig. 6D). The two residues, Phe429 and Glu439 of P450 lie in between the two cofactors, suggesting that these residues might serve as an ET conduit between the cofactors.
Figure 6.

Docked model of a complex between P450 and CYPOR. (A) A complex of P450 and Mol A of the hinge-deletion mutant of CYPOR; Electrostatic surface of interfaces of P450 (B) and an open form of the mutant CYPOR (C), where blue represents a positively charged surface and red indicates a negatively charged surface. (D) Relative orientation between FMN and Heme and interfaces of docked P450 (E) and CYPOR (F). Five salt-bridge pairs are shown with same letters. Glu242 makes salt bridges with both Arg422 and Arg443 (see ref. [54] for more details).
In addition to the protein-protein interactions, the ER membrane also plays a key role in ET. In the P450 system, the soluble CYPOR, lacking the hydrophobic N-terminal membrane anchor (residues 44 amino acids) is not able to donate electrons to P450 [71], suggesting that the membrane binding facilitates the interactions and proper orientation for efficient ET between the two partners (Fig. 4). Although structural models of the soluble forms of the enzymes have shown flexibility of the position of the FMN domain relative to the FAD/NADPH domain via the connecting domain and hinge, it should be noted that the FMN-binding domain is attached to the ER membrane by its hydrophobic anchor. Therefore, on the ER membrane, the hinge promotes movement of the FAD/NADPH-binding domain relative to the FMN domain and P450, both of which are anchored to the membrane. In the closed conformation, the FAD/NADPH domain moves in closer to the FMN domain to donate electrons to the FMN domain. However, once the FMN is in the reduced state, the FAD/NADPH domain must move away to allow P450 to interact with the FMN domain. Thus, on the ER membrane, it is the movement of FAD/NADPH-binding domain, via the hinge, that is largely responsible for positioning of the FMN domain and facilitating formation of the active complex between the FMN active site and P450 heme (Fig. 4 and refs. [59, 72]). Furthermore, the soluble CYPOR that lacks the N-terminal membrane-binding domain cannot donate electrons to the soluble cyt b5. Thus, effective ET occurs only with the detergent-solubilized intact CYPOR and cyt b5, both of which retain the membrane-binding domains [7, 73], suggesting that the interaction of the CYPOR and P450 (or cyt b5) with the ER membrane is essential for efficient ET (see Fig. 4).
In contrast to the membrane-bound CYPOR/P450 system, in the dimeric and soluble NOS and P450BM3 enzymes, an effective intersubunit ET occurs between monomers. For P450BM3, Kitazume et al. [74] proposed a large rotational and translational motion of the FMN domain, and showed that inter-subunit ET from the FAD domain of one monomer to the FMN domain of the other monomer is obligatory during turnover of the enzyme. On the other hand, the intersubunit ET in NOS occurs between the FMN domain of one monomer and the heme domain of the other monomer of the dimeric enzyme (See Fig. 1). Domain-domain interactions in nNOS are under control of regulatory elements. Arg-1400, discussed above, is located in the CT sequence and conserved in eNOS, but is replaced with Ser in iNOS. Roman et al [75, 76] and Tiso et al [77] reported that a ~ 30 residue extension to the C terminus (CT) is topologically located between the two flavin-binding domains (see Figs. 3 and 5 and ref. [42]), and represses enzyme activities in the absence of CaM. In addition, AR, a ~40 residue insert found in the constitutive NOSs, but not in iNOS, also serves to increase the interfacial area between the FAD- and FMN domains, thereby stabilizing a closed form of the reductase domain [42, 78–80]. The conserved Cys689 and Cys908 in human eNOS are located at the interface between the FAD and FMN domains, and are surrounded by several positively and negatively charged residues. S-Glutathionylation of these amino acid residues disrupts the FAD FMN alignment, interrupting ET between the flavins, thus providing a unique pathway for the redox regulation of cardiovascular function of eNOS [81, 82].
3. Electron transfer mechanism
For CYPOR/P450 and NOS, electron transfer (ET) occurs in at least three steps: i) NADPH to FAD, ii) FAD to FMN, and iii) FMN to the catalytic heme center, P450 or NOSoxy (Figs. 1 and 7). Step i occurs through hydride transfer (NADPH→FAD), and step ii occurs through direct interflavin electron transfer (FAD→FMN), while step iii includes a long range ET through proteins, in which the distance between the two redox cofactors is relatively long. In the structure of the complex between the heme- and FMN-binding domains of P450BM3, the distance between FMN and heme (corresponding to step iii) is ~18 Å [50]. In the modeled structure of the CYPOR and CYP 2B4, this distance is ~12 Å (Fig. 6 and ref. [54]). However, in both cases, the relative orientations between the two cofactors are similar. In iNOS [64] and nNOS [65] the corresponding distance is also ~12–13 Å. P450s contain only heme as prosthetic group, while NOS isoforms include heme and H4B. These prosthetic groups accept electons from FMN in a sequential one-electron transfer, whereby molecular oxygen is activated (see Figs. 10 and 11). ET rates in steps i and ii are much faster than that of step iii, indicating that FMN to heme ET is the rate-limiting step. Although the P450 ET system is not strictly regulated (see Sections 3 4 for details), the NOS system is suppressed by at least three regulatory elements (Figs. 2 and 5), and the suppression is relieved by CaM binding (Fig. 7).
Figure 7.

Electron Transfer Path in CYPOR (A) and NOS (B). The P450 ET system catalyzes the monoxygenation of substrate (RH) (red) (A), while NOS isoforms catalyze the formation of NO from l-Arg (blue) (B), via two- monooxygenation reactions with NG-hydroxy-l-Arginine (NG-hydroxy-l-Arg) as a stable intermediate. In nNOS and eNOS, all three electron-transfer steps are accelerated by CaM binding. The dotted lines from FMN to ferricyanide indicate that, although thermodynamically possible, ET between fully reduced FMN and ferricyanide has not studied in details.
Figure 10.

Redox potentials of the individual redox couples of FAD and FMN of CYPOR and P450 (A), and the catalytic cycle of CYPOR (B). A and B, 1 and 2 indicate the redox pairs shown in A [84, 92]. B, Dots indicate electron equivalents, one-electron (.) and two-electron (..) -reduced states. Numbers under I through VI indicate the total electrons present in the two flavins. Red arrows with dot (.) indicate one-elctron donation/release.
Figure 11.

The role of the FAD-FMN pair for activation of molecular oxygen in P450 and the NOS heme center. In P450, introduction of the first electron involves the reduction of ferric P450 (FeIII) and binding O2 (step 1). The addition of the second electron leads to the production of the ferric peroxy form, which is rapidly protonated to the ferric hydroperoxy state (step 2). A further protonation facilitates the ionic O-O bond clevage (step 3), and resulting in a ferryl-oxo form, Compound I, FeIV=O + species [152, 210, 211]. In final steps, 4 and 5, the “oxygen” of the Compound I is transferred to substrate (RH). In NOS, the first electron is also supplied by FMNH−, but the second electron is supplied by the bound H4B (see text for details). H4B functions as a one-electron carrier: H4B ⇔ H4B.+ + e−, and can also function as an electron and proton donor: H4B ⇔ H3B. + e− + H+ (shown in parentheses) [151, 212, 213]. NOS also requires step 6. Figure adapted from [214].
3.1. Properties of flavins
The flavin (Fl) cofactors play a pivotal role in the step-down reaction from two-electron donors to one-electron acceptors (Fig. 1), as they are capable of both one-and two-electron redox chemistry. The flavin cofactors can exist as the oxidized (ox), one-electron reduced semiquinone (sq), and two-electron fully reduced (red) forms (Fig. 8) [83]. The flavins equilibrate between these states with reversible electron transfer occurring across the N(5) and N(1) atoms of the isoalloxazine ring of the flavin. Thus, they show two characteristic one-electron mid-point potentials, Eox/sq for ox-sq couple and Esq/red for sq-red couple. The semiquinone and fully reduced forms can exist in either neutral or anionic forms, with intrinsic pKa values of 8.5 and 6.5, respectively. In CYPOR and NOSred, however, pKa values of the enzyme bound flavins have not yet been determined. Nevertheless, at physiological pH, semiquinones of both enzymes are of blue neutral forms, FADH. and FMNH. [7, 84–88]. In CYPOR, no shift from neutral to red anionic semiquinone is observed within the pH range from 6.5 to 8.5 [86]. The NOS flavin semiquinone is also in the blue neutral state in the pH range from 7 to 9 [88]. On the other hand, the protonation state (FlH2) of the fully reduced forms is not known. In FNR [89] and Fld at physiological pH [90, 91], the relevant reactive forms are the neutral semiquinone (FlH.) and fully reduced anionic (FlH−) forms. In this review, therefore, by analogy to FNR and Fld, we will assume that the fully reduced forms are in the anionic forms, FADH− and FMNH−.
Figure 8.

Isoalloxazine ring of FMN or FAD in its oxidized (Flox), semquinone (one-electron-reduced form, Flsq), or fully reduced (two-electron-reduced form, Flred) state.
In rabbit [84] and human [92] CYPORs and rat NOSred [93], one-electron reduction potentials of both flavins are characterized by a large, positive ΔE (Eox/sq > Esq/red), as reported in Flds (see Fig.10 and ref. [84]). For CYPOR, the values of Eox/sq for FMN (pH 7.0) are 110 mV (for rabbit) and 66 mV (for human); and Esq/red values are 270 mV (for rabbit, [84]) and 269 mV (for human, [92]). The values of FAD Eox/sq at pH 7.0 are 290 mV (for rabbit) and 283 mV (for human); and Esq/red values are 372 mV (for rabbit) and 382 mV (for human). For rat nNOSred [93], the values of Eox/sq and Esq/red for FAD are 283 mV and 310 mV, respectively, while the values of Eox/sq and Esq/red for FMN are 105 mV and 274 mV, respectively [93].
Oxidizd forms of flavins have a sharp absorption peak around 450 nm, and their neutral semiquinone forms have a broad band in the 500 – 700 nm region, with a maximum at 580 to 600 nm. In CYPOR, the neutral semiquinone forms of FAD and FMN can be distinguished by the presence of a 630 nm shoulder in the spectrum of the FMN semiquinone (see Fig. 9A and Fig. 5 of ref. [84]), which is absent in the FAD semiquinone. In NOS isoforms, the neutral semiqunone forms have a broad band in the 500 – 700 nm region, and they share an absorbance peak around 596 nm. In addition to the characteristic absorbance peak at 596 nm, however, the semiquinone state of FAD shows characteristic peak with absorbance at ~520 nm [94–98]. This peak is also observed in neutral semiquinones in FADH of spinach FNR [99], and the Thr66Val mutant of NADH-cyt b5 reductase [100]. This peak is also observed as a ~520 – 530 nm shoulder during the reduction of oxidized nNOSred by NADPH in the presence of CaM (see Fig. 1B (inset) of ref. [101]), and during the reductive titration of oxidized nNOS reductase domain with sodium dithionite (see Fig. 4D, curve d of ref. [102]). Thus, this spectral differece between FAD and FMN semiquinones enables to distinghuish the FAD and FMN semiquinones in the analysis of one-electron transfer reactions between FAD and FMN.
Figure 9.

Reduction of air-stable semiquinone of CYPOR (A) and iNOSred (B). The air-stable semiquinone, 5 µM of CYPOR and iNOSred were reduced by 100 µM NADPH, respectively. Insets, the spectra were recorded at three wavelengths (see text and refs. [97, 112] for details). Figure 9 was adapted from [97].
The origin of the ~520 nm spectral peak for the FAD semiquinone observed in NOSred is interesting. The structures of the FMN domains of CYPOR and NOSred are very similar to that of Fld (Fig. 3). As in D. vulgaris Fld [103], the FMN isoalloxazine rings in CYPOR [36, 37] and NOS [42] are also sandwiched by two aromatic residues, Tyr178 and Tyr140 (rat CYPOR), and Tyr889 and Phe809 (rat nNOS) (Fig. 5). However, unlike the FAD isoalloxazine ring, which has a conformationally mobile aromatic residue, Trp677 (CYPOR) or Phe1395 (rat nNOS) at its re-face [38, 104], the two aromatic residues sandwiching the FMN ring are not known to be mobile during the catalytic cycle. In D. vulgaris Fld [103], the FMN isoalloxazine ring is sandwiched between Tyr98 and Trp60, and its semiquinone spectrum shows characteristic peak with a broad absorbance at ~580 nm, as observed in FMN semiquinone spectra of CYPOR and NOSred. However, the semiquinone form of the Fld Tyr98Ala mutant shows an additional pronounced absorbance transition at 515 nm compared to wild type (see Fig. 2B, dot-dashed line spectrum of ref. [103]). In addition, the C. beijerinckii Fld, whose FMN is staked between Trp90 and the non-aromatic Met56 shares a similar absorbance spectrum to that of Tyr98Ala mutant of D. vulgaris Fld [105]. These data suggest that the spectral peaks observed at 486 nm and 520 nm in CYPOR and NOSred, respectively, for the FAD semiquinone (Fig. 9), but not for the FMN semiquinone [97], is most likely due to the decrease in the π πstacking interactions caused by the mobile aromatic residue (Trp677 of CYPOR and Phe1395 of nNOS) on the re-face of the FAD ring.
The neutral semiquinone form of FMN in both enzymes is likely stabilized by the hydrogen bond between the main chain carbonyl group of the conserved Gly (Gly141 in rat CYPOR, Gly810 in rat nNOS, and Gly61 in D. vulgaris Fld) situated in the FMN-binding loop and N(5)-H of the reduced FMN isoalloxazine ring (see Fig. 8). During the ET reactions, the formation (reduced state, N(5)-H) or breakage (oxidized state, N(5)) of the hydrogen bond between Gly and the N(5) position of FMN isoalloxazine ring results in a significant increase in the reorganizational energy for the ET reactions, leading to the lowering the reactivity of the FMN semiquinone [106]. It is likely that the neutral semiquinone oxidation associated with proton transfer (FMNH. → FMN + e + H+) is kinetically gated by deprotonation of semiquinone [107, 108]. Thus, a low reactivity of FMN semiqunone enables FMN shuttling between the fully reduced and semiquinone oxidation states as a one-electron carrier in these ET systems. However, in P450BM3, this Gly residue, which is conserved in the FMN domains of Fld, CYPOR, and NOS, is absent, resulting in a tighter loop [109, 110], and its FMN semiquinone is an unstable anionic form having a higher reactivity than the neutral semiquinone of Fld, CYPOR, and NOS. Thus, in P450BM3, consistent with this potential reversal, the semiquinone of FMN donates electrons to P450 [50]. Li et al have constructed an nNOS mutant, in which Gly810 was deleted (ΔGly810), thus mimicking P450BM3 [111]. As expected, the FMNox/sq reduction potential is now lower than the sq/red couple as in P450BM3. Interestingly, however, cyt c reductase activity of ΔGly810 in the absence of CaM is at wild type levels and is insensitive to CaM binding, indicating that the ΔGly810 FMN domain is locked in an open position that is no longer sensitive to CaM binding.
The reaction mechanisms of CYPOR and NOSred have been studied by using artificial electron acceptors, such as the relatively large molecule cyt c, and small molecules such as ferricyanide, menadione (MD) and 2,6-dichloroindophenol (DCIP). Although these electron acceptors are not natural physiological partners, they are useful tools for the analysis of reactivity of the FAD/NADPH- and FMN domains ([8, 112, 113] for CYPOR, and [94, 114–116] for NOS). Various studies have established that cyt c accepts electron from the FMN site of the open form of the enzymes, while the ferricyanide can accept electrons mainly from the FAD site of both the open and closed forms of the enzymes [62]. FAD of NOSred of all three NOS isozymes has a high reactivity towards ferricyanide (Eox/red = +425 mV) in the absence of CaM, which increases by 2 3-fold in the presence of CaM. In contrast, the reactivity for MD (Eox/sq = 210 mV) increases by ~10 – 20-fold in the presence of CaM [114–118]. In both the closed and open states of the enzyme, the small molecules, such as ferricyanide and MD, can directly access both the FAD and FMN sites of nNOS. However, MD has a higher affinity for FMN than FAD [116], while ferricyanide has a higher affinity for FAD due presumably to the negative charge repulsion between FMNH− and Fe(CN)6−3. Therefore, differences in the levels of reactivity increases between ferricyanide and MD by CaM suggest that CaM activates ET from FAD to FMN more than from NADPH to FAD. The MD reductase activity increases in the order of iNOS> nNOS> eNOS [119].
3.2. Reduction of FAD by NADPH
In the first step of the reaction of NADPH, the FAD of both CYPOR and NOSred functions as a dehydrogenase flavin (Fig. 1), and accepts a pair of electrons as a hydride ion from NADPH (320 mV). The crystal structure of wild type CYPOR reveals that the indole ring of Trp677 stacks against the re-face of the FAD isoalloxazine ring, thus blocking direct interaction between the flavin ring and the nicotinamide moiety of NADPH [36]. The crystal structure of a mutant in which Trp677 was either deleted or mutated to Gly revealed that the nicotinamide moiety of NADP+ is in van der Waals contact with the isoalloxazine ring with a tilt of ~ 30° between the planes of the two rings [38]. Thus, in the wild type protein, the indole ring of Trp677 presumably moves away from the isoalloxazine ring of FAD, allowing the nicotinamide ring to interact with the flavin allowing the hydride ion transfer to FAD. The local movements of the Gly631 Asn635 loop, located in the FAD-binding cavity, may be coupled with Trp677 movement to allow NADPH/NADP+ binding/release [56]. In spinach FNR, the homologous residue Tyr314, is displaced by the nicotinamide ring [120, 121]. In NOS isoforms, Phe1395 (nNOS numbering in rat) stacks onto the FAD isoalloxazine ring [96, 104, 122] and, in P450BM3, Trp1046 is found at this position [123], strongly suggesting that these residues are homologs of Trp677 of CYPOR and Tyr414 of FNR. In MSR, Trp697 would be the corresponding residue that modulates hydride transfer from NADPH to FAD and intramolecular ET from FAD to FMN [124].
3.3. Intramolecular ET between FAD and FMN
In the second step, the intramolecular ET occurs directly from the FAD to FMN, [36, 42]. The ET rate between FAD and FMN in CYPOR and NOSred can potentially be influenced by several factors [125–128]: (i) distance between and relative orientation of FAD and FMN, (ii) the stabilization of the neutral FMN semiquinone, and (iii) the rate of large-scale confomational movements. Although the crystal structure does not show the dynamic nature of the protein, the structures of CYPOR [36, 37] and nNOSred [42] provide good estimates of the distance and orientation parameters. In rat and human CYPOR [36, 37], the distance between the dimethylbenzene edge of the isoalloxazine rings of FAD and FMN is 4 Å, and the planes of the FAD and FMN molecules are inclined relative to each other at an angle of ~135 (Fig. 5, left panel). This orientation can favor orbital overlap between the extended -orbital systems of both prosthetic groups. The short distance is expected to result in a very fast and efficient ET between the flavin cofactors (up to 1010 s−1 using Dutton’s ruler) [53, 57, 58, 129, 130]. However, an experimentally observed rate of conversion from FADH. - FMNH. to FAD-FMNH− is ~50 s−1 [130], suggesting that the ET between the two flavins is conformationally controlled. While crystal structures of CYPOR have identified discrete closed and open states [36, 54], Hay et al [57], using ELDOR spectroscopy, suggest that in solution there is a continuum of conformational states including multiple closed and open states across the energy landscape. In a mutant CYPOR [56], in which a disulfide bond between the two flavin domains is introduced, the relative orientation of the two flavin rings is twisted about 20° compared to the wild type, and the distance between the two 7-CH3 groups of FAD and FMN is 5.6 Å and that of 8-CH3 methyl groups is 5.2 Å. The increased distance, relative to the corresponding distances of the wild type structure (4.4 Å and 4.0 Å, respectively) (Fig. 5, middle panel), decreases the surface contact area between the two flavin rings. The mutant enzyme, which is in a closed state, has a ferricyanide activity of ~30% of the wild type enzyme, suggesting that the hydride transfer reaction from NADPH to FAD is relatively intact. However, the interflavin ET rate is extremely low, much slower than that expected from application of Dutton’s ruler [127]. Since the protein conformation is fixed in the closed form with the disulfide linkage [56], the slow ET rate between the two flavins is most likely due to (i) decrease in the degree of electronic overlap between the FAD and FMN semiquinones in the mutant CYPOR, and (ii) inability for the enzyme to undergo small conformational changes necessary for optimal interflavin ET that are different from the large scale domain motion, which is necessary for ET between FMN and P450.
The crystal structure of nNOSred [42], which does not include the CaM-binding region, also adopts the closed conformation with the distance between FAD and FMN being ~ 5 Å (Fig. 5, right panel). This closed state is stabilized by the regulatory elements and charged amino acid residues, including Arg1400 (rat nNOS numbering), such that a large conformational change is restricted. As in CYPOR, this short distance (< 5 Å) is expected to allow an extremely rapid interflavin ET rate (FAD to FMN). However, the rate of ET between the two flavins is much slower than that of reduction of the FAD by NADPH [131]. In nNOSred, the relative orientation of FAD and FMN is slightly different from that of CYPOR, resulting in a reduced overlap (see Fig. 5, right panel) [132]. Whether the difference in the observed FMN-FAD distances in the closed form of wild type CYPOR (4A) and nNOSred (5.2 Å) and the slight shift and rotation of the two flavin planes observed in the NOSred structure (Fig. 5, right panel, bottom) are significant is not clear at present. However, the fact that CaM binding activates ET between the two flavins [133, 134] suggests that the intramolecular ET also depends on the degree of electronic orbital overlap between the FAD and FMN; and that CaM binding releases the “locked, closed” conformation altering the relative orientation between the two flavins for more efficient intramolecular one-electron transfer. Moreover, Welland et al [101] have proposed that CaM may also act to increase the rate of hydride transfer from NADPH to FAD due to preferential binding of NADPH to the open form of the enzyme.
The intramolecular ET includes two distinct intramolecular ET reactions (FADH− to FMN and FADH. to FMNH.). ET from FADH− to FMN is energetically more favorable than that from FADH. to FMNH., as judged from the redox potentials (see Section 3-1 and refs. [84, 92, 93]). However, the reduction kinetics of the fully oxidized enzyme, FAD-FMN, by NADPH are quite complex and not completely understood [54, 84, 101, 118, 129, 130, 135]. In contrast, the reduction of the air stable semiquinone, FAD-FMNH. by NADPH is relatively simple [97, 101]. In both CYPOR and the NOSred domains, the FADH.-FMNH− is formed by a single electron transfer from FADH− to FMNH. of FAD−-FMNH. (Scheme 1).
Scheme 1.

During the catalytic cycle, the FADH.-FMNH− can donate electrons to electron acceptors, e.g., P450s. In CYPOR, the equilibrium constant Keq lies to the right (Scheme 1) as judged from the absorbance change at 630 nm (Fig. 9A and refs. [84, 97]). In NOS, however, judged from the absorbance changes at ~520 nm (Fig. 9B), Keq is differentially controlled in different NOS isoforms; in iNOS, it lies to the right, but in nNOS to the left, and in eNOS lies far to the left [95, 97, 98]. The three NOS isoforms differ significantly in their rates of NO synthesis (see Section 3 4 for details). In addition, the ET rate between the FMNH− and heme center in eNOS is also slower than other NOS isoforms [136]. To identify contributions of the individual domains of the NOS isoforms, Nishida and de Montellano constructed chimeric proteins, and suggested that the rate-limiting step in NO synthesis involves electron transfer within the reductase domain rather than dioxygen bond breakage, substrate monooxygenation, substrate binding, or product release [137]. Thus, NO formation could depend on the rate of the formation of the active intermediate, FADH.-FMNH−. In eNOS, the shift in Keq results in a much slower second phase of NADPH reduction relative to iNOS and nNOS either with or without CaM [98, 138], indicating that intramolecular ET between FAD and FMN is slower in eNOS. However, the hinge connecting the FAD/NADPH- and FMN domains influences the rate of conversion between the closed and open forms [43]. Thus, the lower activity of eNOS could be attributed to both the rate and extent of the formation of the active intermediate, FADH.-FMNH−; thus both Keq and rates of conversion between the open and closed forms determines the rate-limiting step in each NOS isoform. Taken together, the activity of the reductase domain in NOS isoforms is differently regulated by all of the above factors, and the rate of formation of the active intermediate can be linked to the rate of conversion between the closed and open states. However, the detailed mechanism remains to be explored.
3.4. Intermolecular electron transfer from FMN to catalytic heme centers
The P450 and NOS heme prosthetic groups possess a common cysteinyl thiolate anion as the proximal ligand of the heme iron, and its basicity is crucial for activation of molecular oxygen [139]. The low spin state of heme iron in P450 is stabilized by binding of water at sixth distal position, and has a relatively low redox potential, −200–300 mV. Addition of “type I” substrates displaces the water molecule, yielding a five-coordinate, substrate-bound high-spin ferric complex. Using a native-like membrane bilayer mimic (nanodisc), Das et al showed that substrate binding to CYP3A4 induced a >90% spin conversion to the high spin and shifted the redox potential by +10 to 80 mV depending on the substrates [140]. This increase of the redox potential of P450 increases the rate of the first electron transfer from fully reduced FMN to ferric P450 [141, 142]. The reaction of reduced P450 (FeII) with O2 produces an oxyferrous species with a significantly more positive redox potential (+6 mV) [143] (Fig. 10A). In CYPOR, the second electron is also derived from the active form, FMNH−. For some substrates, however, the addition of cyt b5 (Eox/red = ~ 0 mV) into the purified reconstituted P450 enzyme system affects the catalytic activity, by efficiently donating a second electron to P450 (FeII O2)-RH complex [69, 144]. Cyt b5 can also act as an activator for some P450s, which interact with cyt b5 in a substrate dependent manner [145]. In contrast to the mammalian P450 enzyme system, the fungal cyt b5 reductase/cyt b5 system is an alternative pathway which can donate both first and second electron to fungal P450s [146, 147].
In the absence of substrate, l-Arg, iNOS and nNOS isoforms exist predominantly in the low-spin state [148], but eNOS exists in a mixture of high and low spin states [149]. Binding of l-Arg to eNOS causes the conversion to a pure high spin heme. Binding of l-Arg to iNOS, but not nNOS, induces a significant increase in redox potentials, [148]. The one-electron midpoint potentials of oxygenase domains of NOS isoforms are around −270 mV in the presence of saturating l-Arg, respectively [88]. The Esq/red values for FMN are around −250 mV [88], suggesting that the difference in the activities observed in NOS isoforms cannot be assigned to the thermodynamic parameters. In the NOS system, oxidation of l-Arg occurs as two separate mono-oxygenation reactions (Fig. 11). In both reactions, the first electron (Fig. 11, step 1) is also derived from the active form, FMNH−, but the second electron (Fig. 11, step 2) is derived from the enzyme bound H4B [14, 150, 151]. In the first oxidation reaction, the resulting H4B + radical accepts electrons from the FMN site (Fig. 11, step 6) [14]. In both P450 and NOS systems, Compound I is the common reactive heme species [152, 153]. In NOS, however, the second oxidation reaction may be performed directly by the peroxo-heme species (FeIII OO− (H+)) that reacts by a nucleophilic attack on the guanidinium group of NG hydroxy l arginine (NHA) [150,151]. In step 6, the oxidation of the ferrous (FeII) NO complex by the H4B + radical allows the final release of NO. Thus, the H4B cofactor acts as an electron donor and acceptor during the catalytic cycle. The free form of H4B has a relatively high one-electron redox potential, +270 mV [153]. Although the redox potential of the protein-bound form is unknown, it is unable to reduce the ferric heme [154, 155].
3.5. Catalytic cycle and the rate-limiting step
The FAD-FMN pair of CYPOR and NOSred donates electrons to the catalytic heme centers, including P450 and the NOSoxy. A catalytic cycle may start from the oxidized FAD FMN pair, but as described above, the one-electron reduced form, FAD-FMNH. is stable [5, 7, 132], and its reactivity is extremely low. Furthermore, it could be formed from the oxidized enzyme during the priming reaction (Scheme 2) [9, 156].
Scheme 2.
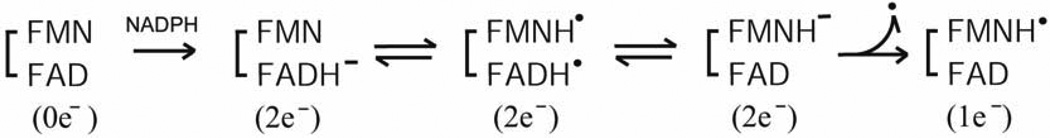
Thus, we can describe the catalytic cycle as starting from the air-stable semiquinone species, FAD-FMNH.. After reduction of FAD-FMNH. by NADPH, interflavin electron transfer occurs from FADH− - FMNH. to produce FADH. - FMNH−. In both P450 and NOS systems, the fully reduced FMN (FMNH−) is the species that donates electrons to the heme catalytic center. As shown in Fig. 10B (lower arrow), the FAD-FMN pair cycles in 1e/3e cycle: 1e(I) 3e(II/III) 2e(IV/V) 1e(I) electron-transfer of flavin cofactors, in which the semiquinone/fully reduced couple (FADH./FMNH−) functions as the one-electron carrier [84, 92, 142]. At high concentrations of NADPH, the intermediate FAD-FMN−, is reduced to a four-electron reduced form, FADH− - FMN− by NADPH (Fig. 10B, upper arrow); thus 2e−/4e− cycle: 2e−(IV/V)-4e−(VI)-3e−(II/III)-2e−(IV/V) electron-transfer is also possible [112, 157]. Since the air stable semiquinone, FAD-FMNH. is stable in liver microsomes [7], the 1e−/3e− cycle is likely the major mechanism in vivo, but the 2e−/4e− cycle is also possible depending on the NADPH/NADP+ ratio [112, 158]. In contrast, FMN of P450BM3 cycles between the oxidized (FMN) and the anionic semiquinone form (FMN −), and donates electrons to P450BM3 [50, 159, 160], utilizing the 0 2 1 0 electron-transfer cycle (Scheme 3) of flavin cofactors.
Scheme 3.
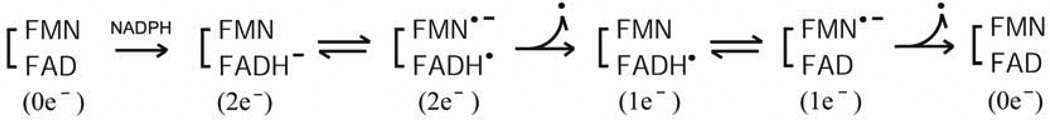
At the enzyme level, the rate-limiting step in both CYPOR [141] and NOS [137] is at or after the introduction of the second electron from the reductase to the heme iron. In both systems, the introduction of the first electron to the heme iron is relatively fast, as compared with turnover numbers [138, 141]. In general, the redox potential difference between the electron donor (FADH.-FMNH−) and acceptor (heme iron FeII/FeIII) is the driving force for ET (Fig. 10A). Therefore, the change of spin state and the associated increase in redox potential that are induced by substrate binding modulate the rate of ET between FMN and heme, and regulate the mono-oxygenase activity. However, its effect is unclear in NOS systems [88]. Large domain movements have been proposed to facilitate relative domain-domain reorientation during the ET process, in which the enzyme (CYPOR) shuttles between open and closed conformations during the catalytic cycle. The fluidity, speed, and frequency of this movement are unclear, but if cyt c reductase activity can be explained by a confomationally gated mechanism, then the rate of conformational changes between the open and closed forms can be expected to be in the order of ~50 – 80 s−1. The catalytic turnover, ~0.2–2 s−1 for microsomal P450s is much slower than cyt c reduction activity of the reductase, indicating that the conformational changes between the FAD/NADPH- and FMN domains cannot be the only factor involved in the rate-limiting step. It is likely that the rate-limiting step for substrate turnover is complex and also influenced by which P450 isoform is involved as well as which substrate is bound to the P450.
In contrast, the overall rate of NO production by all three NOS isozymes primarily depends on the interflavin ET rate of the reductase domains rather than the redox potential difference between the FMNH./FMNH− couple and heme iron FeIII/FeII couple in each enzyme [88, 134]. The ferricyanide activities of all three NOS isoforms are unaffected by the presence or absence of CaM, but, in the presence of CaM, the cyt c reductase activities are significantly different; ~70 s−1 for iNOS, ~30 40 s−1 for nNOS, ~7 s−1 for eNOS. Stopped flow [138] and laser flash photolysis [136] studies have directly measured the rate constants for flavin and heme reductions, and the data clearly demonstrate that the rate of flavin reduction correlates with the rate of heme reduction. However, the rate of NO formation is an order of magnitude slower than that of heme reduction. The turnover numbers for NO synthesis catalyzed by the isoforms have been reported as: ~3.3 s−1 for iNOS, ~1.7 s−1 for nNOS, and ~0.3 s−1 for eNOS [161]. The rate of NO synthesis decreases in the order of iNOS> nNOS> eNOS, consistent with the order of heme reduction, although the catalytic turnover of NO synthesis correlates with NO release from ferric heme NO binding [14, 155]. As shown in Fig. 11, the lower value for the NO formation may reflect the highly complicated ET process invoving the two monooxygenase reactions. In particular, the lower activity of the eNOS isoform is in part caused by the decreased ability of the reductase domain to transfer electrons to the catalytic heme center [98, 138, 161]. Taken together, the rate of conformational change is a major controlling factor for NOS activity, and CaM regulates different NOS isoforms differently [43].
4. Functional insights from models of the full-length NOS
The NOS isoforms are multidomain enzymes, bearing multiple conformations in solution. This flexibility is likely a major cause why crystallization of an intact NOS isozyme has not been successful. Although no full-length structure of either CaM-free or CaM-bound enzyme has been resolved, the 3-D structures of the isolated nNOS NADPH/FAD - FMN module [104], the nNOS NADPH/FAD - FMN module [42], the complex of CaM with the eNOS CaM-binding peptide [162], and the heme domains [163–165] have been solved. In addtion, several schematic models to illustrate the flexibility and relative movement of the FMN module [42, 93, 166–169] have been proposed. However, it was difficult to construct the structure of an entire NOS isoform from these partial structures, especially when no information is available regarding how the two halves of the molecule (i.e., the oxygenase domain and the reductase domain) are connected. The recent crystal structure of the iNOS FMN domain in complex with Ca-CaM revealed the mechanism by which CaM regulates the conformational rearrangements between the reductase and oxygenase domains. Furthermore the structure made it possible to contruct a model for the entire molecule of iNOS (see Fig. 13 and ref. [64]). A model structure of a nNOSoxy-FMN-CaM complex has also been proposed based on the mutational studies on the putative interface between the oxygenase and FMN domains [170, 171]. In the absence of CaM, intra- and inter-molecular/subunit interactions between individual domains are supressed by regulatory factors, as described earlier. Isolated heme domains, as well as the holo enzymes of all three NOSs form dimers indicating that the interactions between the heme domains are relatively strong and specific in each isoform [163–165]. CaM binding alters the interactions between the FAD/NADPH- and FMN domains, and between the FMN and heme-bnding domains to ensure the dimerization of the heme domain and flexibility of the FMN domain in each isoform. In addition, CaM may interact with the FAD/NADPH-, FMN- and heme-binding domains, allowing them to form different conformations, each best-suited for a given step of the entire ET process from NADPH to heme center [167]. A major remaining question is how CaM modulates the interaction between the reductase and oxygenase.
Figure 13.

Strategy for modeling of the entire holo iNOS structure. (A) Strategy for the construction of a model structure of iNOS [see ref. [64] and text for details]. (B) Two orthogonal view of the modeled dimer structure of iNOS. The relative orientations of the two cofactors are shown in B, and Trp372 is located between the two cofactors.
4.1. Structure of the iNOS FMN-CaM complex
The iNOS isoform has a much higher affinity for CaM than the two other isoforms. On the basis of the structure of iNOS FMN-CaM complex, the structure of nNOS FMN-CaM complex was also modeled [65]. The structure of the iNOS FMN-CaM complex clearly reveals how CaM strongly binds to this isoform (Fig. 12) [64]. The CaM-binding region of the iNOS polypeptide (residues Leu515-Ser535) forms a tight α-helix, which adopts a relatively random structure in the absence of CaM [64, 172]. Sequence alignment indicates that all three NOS isoforms contain a regulatory β-finger (BF) in the connecting domain (CD), but that of iNOS is six residues shorter than those of nNOS and eNOS. This structure suggests that the bound CaM in nNOS interferes with the regulatory BF located in the CD domain [64], suggesting that BF at least in nNOS would change its conformation upon binding of CaM. Arg530 is conserved in all known iNOS sequences, making salt bridges with Asp80, Glu84 and Glu87 of the CaM C-terminal-lobe (Fig. 12). The corresponding residue in both nNOS and eNOS is Gly, which lacks charge interactions with CaM. The FMN domain and the CaM-binding region are connected by a very short but flexible linker (Ala534-Val537), of which Arg536 is conserved in all NOS isoforms. Although the hinge movement is relatively small, it is the means through which the bound CaM regulates the relative orientation/distance between the FMN and oxygenase domains. Mutation of the corresponding residue in nNOS, Arg752, to Glu or Gln decreases the rate of heme reduction and NO synthase activity, strongly suggesting that this hinge/bridging mechanism controls the CaM dependent NO synthesis. The structure of the FMN domain is essentially identical to the structures of the corresponding domains of rat and human CYPOR [36, 45, 56] rat nNOS [42], and bacterial Flds [173]. The binding of CaM to CaM binding peptides of three NOS isoforms is very fast, indicating that this is not involved in the rate-limiting step in NOS catalysis [174]. In constitutive NOSs, dissociation of bound CaM is fast, but is extremely slow in iNOS, as expected from the tight interactions between the CaM-binding peptide and CaM in iNOS [64].
Figure 12.

The structure of the iNOS CaM·FMN complex and interactions between the FMN domain and the bound CaM. The FMN domain is shown in gold, CaM in green, and Ca2+ with grey balls. The CaM binding region is shown with a gold helix wrapped around by green CaM. FMN is shown with blue sticks. Arg536 located within the hinge between the FMN domain and the CaM binding region makes salt bridges and a hydrogen bond with Glu47 and Asn42 of CaM, respectively. Arg536 also makes several hydrogen bonds with the main chain carbonyl oxygens of Ser562-Cys563-Ala564-Phe565 of the FMN domain. Thus, the Ca2+-CaM binding regulates the hinge motion pivoting at Arg563, which in turn controls ET from FAD to FMN and from FMN to heme.
4.2. Mechanistic implications from the modeled structure of the entire intact molecule
4.2.1. Modeling
In modeling the entire iNOS molecule (Fig. 13), the first key point is that the structures of the FMN domains in iNOS and nNOS are very similar to each other. First, the structure of the chimera FAD-FMN-CaM complex is constructed by overlaying the FMN-binding domain of the iNOS FMN-CaM complex [64] onto the FMN domain of the nNOS FAD FMN structure [42]. The open forms of iNOS reductase domain are constructed by the analogy to CYPOR (see Fig. 5, left for the open forms of CYPOR). As discussed earlier, the structure of the iNOSred is more similar to the CYPOR structure than to the nNOSred (see Fig. 3). Finally, the entire holo iNOS dimer was constructed by docking the oxygenase domain dimer onto the open conformation of the reductase domain (Fig. 5, left), in which the FMN-binding domain of one monomer interacts with the oxygenase domain of the other monomer. The intersubunit electron transfer arrangements shown in the resulting model (Fig. 13) is consistent with previously observed biochemical studies revealing the role of CaM in electron transfer between the two subunits in the dimeric NOS molecule [17–19, 163–165]. The proposed open state model of iNOS [64] will provide insight into how different modules interact with each other during catalysis, including the working models for nNOS and eNOS.
4.2.2. Mechanism
Favorable electrostatic interactions between the FMN-binding domain and P450/or NOSoxy are required for complete redox reactions, including (i) formation of an active donor-acceptor complex, (ii) alignment/positioning of the electron conduit within the donor-acceptor complex, and (iii) dissociation of the oxidized and reduced products [44]. CaM binding causes the destabilization of the FAD-FMN domain interface in the molecule and enables/facilitates the interaction between FMN and heme. Activation of constitutive NOS isoforms by CaM binding is triggered in at least two steps [13–16, 20, 65, 167, 174]. In the first step, an intermediate state is formed that can interact with artificial electron acceptors, such as cyt c, but cannot promote significant heme reduction and NO synthase activity. In the second step, the FMN-binding domain interacts with the oxygenase heme domain. This is the rate-limiting step in NO synthesis. In this model, interactions between the two domains are largely electrostatic, with the oxygenase heme domain being positive and the FMN domain negative [64, 175]. In the iNOS model, the closest distance between FMN and heme (the C8 methyl group of FMN and the vinyl group of the heme B ring) is ~12 Å, through which electron transfer from FMN to heme center would occur. By using a pulsed EPR approach, Astashkin et al [176] have determined the distance to be 13.1 Å between the FMN and heme, which is the shortest distance between heme and FMN. In addition, the FMN fluorescence approach also indicates a heme edge to FMN distance of 12–15 Å [133]. These values are in excellent agreement with those expected from the modeling of iNOS and nNOS [64, 175]. As in CYPOR, the neutral semiquinone, FMNH. state does not donate an electron to the heme center, without the additional reducing equivalents [142, 177]. Therefore, ET occurs only from the FMNH− state to heme centers (Figure 14). Trp372 in iNOS [64] and Trp587 in nNOS [175] are positioned between the heme and FMN cofactors and these aromatic residues may act as an electron conduit between FMN and the heme center.
Figure 14.

Proposed model for CaM-mediated regulation of NOS. (A) The conformational rearrangements of functional domains in the NOS dimer. H1 indicates the hinge between the FMN- and FAD domains, and H2 represents the hinge between the FMN- and heme domains. Upon binding to CaM, H2 is divided into H2a and H2b. (B) Mechanism of redox-linked conformational selection. Positive (plus) and negative (minus) surface charges of the FAD-, FMN-, and heme domains are indicated in red. The blue dots indicate hydrated water molecules; the FMN semiquinone state has a tighter water-network. Upon futher reduction of the semiquinone to fully reduced, FMNH− state, the water-network becomes loose and more flexible [179]. In the protein-protein interface between heme and FMN domains of nNOS, the following residues are involved in these electrostatic interactions: Lys423, Lys620, Lys660 (heme domain) and Glu762, Asp813, Glu816, and Glu819 (FMN domain) [175].
The FMN-binding domain has a strong dipole moment (677 Debye) with the positive and negative poles located toward the two terminal helices and near the area of the FMN binding site, respectively [45, 49, 178]. A redox-linked conformational change is proposed in the catalytic cycle of both the P450 and NOS systems [72], and a selective dynamic transition between “opening” and “closing” may be modulated by NADPH binding and NADP+ release [58]. In this regard, we propose that in the neutral (blue) FMN semiquinone state, the FMN domain has a high affinity for the FAD/NADPH binding domain, while the anionic fully reduced FMN has a high affinity for the heme domain (see Fig. 14), and that the protein conformational changes are important for orientation of domains for complex formation while electrostatic interactions are important factors for the association and dissociation of the active electron transfer complex. The conserved negatively charged region of the FMN binding site surrounding the solvent-exposed dimethyl benzene ring of FMN is involved in the interaction with both the FAD and heme domains. Recently, Chang et al [179] have shown that, in D. vulgaris Fld, solvation dynamics and protein conformation near the functional site (i.e., in the vicinity of the FMN isoalloxazine ring) are closely linked to the flavin redox states, which in turn can be correlated to the intermolecular ET properties. Analogous to Fld, we propose that, in the more ordered semiquinone conformation, the FMN domain has high affinity for the FAD/NADPH domain in order to accept electrons from FAD, whereas the less ordered anionic fully reduced form can easily adopt its conformation to interact with the oxygenase domain. It is interesting to note that the FMN CaM complex of NOS functions as a one-electron carrier that can recognize different domains during the redox-linked conformational rearrangements (Fig. 14). Similar mechanisms may also operate in the microsomal P450 system, in which FMN semiquinone has a higher affinity for the FAD/NADPH domain, while its reduced state has a higher affinity for P450s. An exquisite balance of associaton and dissociation between the ET partners (i.e., between the FAD/NADPH and FMN domains) is required for rapid and productive ET reactions in the P450 and NOS systems. It is likely that a similar strategy adopting this “redox-linked conformational changes” is utilized in many other multidomain redox enzymes.
5. Functional significance of missense mutations in the diflavin reductase family
Studies of naturally-ocuring mutations in diflavin reductase genes and resulting phenotypes have contributed to the elucidation of structure-function of this enzyme family. The human CYPOR gene is located on chromosome 7q11.2 [35] and contains 16 exons. The human gene encodes 680 amino acids and shares 94% amino acid sequence identity with the rat enzyme. As expected, the overall 3D structure of human CYPOR is very similar to that of rat CYPOR [36, 37]. The CYPOR/P450 system is a major microsomal oxidative enzyme system. Although there are numerous functional P450 genes in mammals (e.g., 74 in rat, 92 in mouse, 59 in human), there is only one CYPOR gene in each species [3, 4]. The human genome project has identified 59 functional P450 genes: seven encode mitochondrial enzymes, all of which play key roles in the biosynthesis of sterols, and 50 encode microsomal enzymes. The remaining two encode prostacyclin synthase (CYP8A1) and thromboxane-A synthase (CYP5A1). Of the 50 microsomal enzymes, 20 participate in the biosynthesis of endobiotic [2] substrates, such as steroids and eicosanoids; 17 principally metabolize xenobiotic compounds; and 13 are orphan enzymes [180]. Neary all microsomal P450s accept electrons from CYPOR, presumably reacting via conserved patches in the FMN domain of CYPOR (Fig. 6). Site-specific mutational analyses have been used to determine amino acid residues involved in catalysis [49, 181]. CYPOR transfers electrons to three steroidogenic microsomal P450s, including 17 α-hydroxylase/17, 20 lyase (CYP17A1), 21-hydroxylase (CYP21A2), and P450 aromatase (CYP19A1), as well as to CYP51 and the non-P450 protein squalene monooxygenase in the cholesterol biosynthesis pathway. Thus, missense mutations of the human CYPOR gene (CYPOR/POR) cause multiple defects associated with disruption of cholesterol biosynthesis and steroidogenesis, such as ambiguous genitalia, polycystic ovary, and/or Antley-Bixler syndrome, a skeletal malformation featuring craniosynostosis. Since the first report in 2004 [182], over 40 mutation sites have been identified in the human CYPOR/POR gene [183–190] (summarized by Fluck and Pandey [191]).
Several known CYPOR missense mutations interfere with: (1) cofactor binding (NADP(H), FAD, or FMN), (2) interaction between the FAD and FMN domains, and (3) interaction of CYPOR with P450s. In addition, many of these mutations also result in unstable proteins due to lack of cofactor binding and/or improper polypeptide folding. Tyr181Asp, Arg457His, Tyr459His and Val492Glu mutations result in low cyt c and 17α hydroxylase/17, 20 lyase activities, in which Tyr181Asp cause decreased affinity for FMN-binding [185, 187, 192], while Arg457His and Val492Glu cause decreased FAD-binding affinity [37, 182, 193]. The results of Tyr181Asp and Tyr459His mutations are entirely consistent with the hypothesis that the aromatic residues are required for binding of FAD and FMN (see Fig. 2). The overall 3D structures of Arg457His and Val492Glu variants are similar to wild type, with local disruption of hydrogen bonding and salt bridging involving the FAD pyrophosphate moiety, leading to weaker FAD binding, resulting in unstable protein, and loss of catalytic activity, all of which can be rescued by cofactor addition [37]. Addition of exogenous FAD to either variant protects the CYPOR protein from limited trypsin digestion, supporting the role of the cofactor in conferring stability to its structure. In addition to electron transfer steps, the level of expression of CYPOR may also be affected by polymorphisms of the promoter regions [194, 195]. In addition to endobiotic substrates, the mutations also affect biotransformatin of xenobiotic compounds [196–198] and, since CYPOR transfers electrons to heme oxygenases, heme oxygenase activities are also decreased [199, 200].
Methionine synthase reductase (MSR), another diflavin reductase, catalyzes the NADPH-dependent reductive methylation of methionine synthase (MS), which is a cobalamin-dependent enzyme [124]. The MSR (MSR) gene is located on chromosome 5q15.2-q15.3. The reaction catalyzed by MS involves a methyl group transfer from methylenetetrahydrofolate to homocysteine, via a Cob(I)-alamin cofactor, to form methionine, tetrahydrofolate, and the intermediate methylcob-(III)-alamin. MSR transfers electrons to the oxidized, inactive form (Cob(II)-alamin) of MS to regenerate the active form (Cob(I)-alamin). Like CYPOR/P450, the electron transfer system involves two components, MSR and MS, and electron transfer sequence is: NADPH→MSR (FAD→FMN)→MS(CoII). MSR gene deficiency in human is a recessive genetic disorder affecting folate and methionine metabolism and is characterized by elevated levels of plasma homocysteine, because MS becomes inactivated and is not regenerated without MSR. A number of mutations have been described in MSR in homocystinuric patients [201]. Ala129 is located adjacent to a cluster of conserved acidic residues found in the FMN binding site, and the Ala129Thr mutation destabilizes the FMN binding, resulting in an FMN depleted protein [202] and a decrease in the rate of electron flow from MSR to MS. The mutant transfers electrons to ferricyanide as efficiently as wild type, but the rates of cyt c and MD reduction are significantly decreased, indicating that, like CYPOR, cyt c and MD accept electrons mainly from the FMN-binding site. In addition, reconstitution of the mutant with FMN partially restores its ability to reduce cyt c and to reactivate MS, suggesting that patients harboring the Ala129Thr mutation may be responsive to riboflavin therapy.
Nitric oxide (NO) has important functions in the cardiovascular, immune, and neuronal systems [1]. The three human NOS isoforms are encoded by separate genes, localized on 12q24.2 for nNOS (NOS1), 17q11.2 for iNOS (NOS2) and 7q36 for eNOS (NOS3) [203]. Human point mutations and knockout mouse models for each of the three NOS isoforms have served to elucidate the physiological functions of each isoforms. In mice, the knockout of eNOS gene (NOS III−/−) shows an incomplete return of spontaneous circulation (ROSC) rate and worsens post-ROSC left-ventricular function [204]. Global ablation of the single CYPOR gene results in embryonic lethality [3, 4]. However, mice carry multiple NOS genes. When any one of the NOS isoforms is knocked out, the mice are still viable [205], and compensatory interactions among NOS isoforms have been reported [206]. Mice with all three NOS genes knocked-out (NOS−/−) are also viable, but their survival and fertility are markedly reduced as compared to wild-type mice [207, 208]. The relatively mild phenotypes associated with individual NOS knockouts highlight the importance of compensatory interactions between the NOS isoforms and also suggest the presence of additional genes which compensate for NOS functions. For example, an iNOS sequence derived from human hepatocytes has been found to contain three different amino acid changes within the iNOSred domain (Ala805Asp, Phe831Ser, and Leu832Pro). The phenotype of single Ala805Asp mutation is comparable to wild type, while a significant (2–4 fold) reduction of iNOS activity was observed in both the Phe831Ser and Leu832Pro point mutants [209]. The modeling of the reductase domain suggests altered hydrogen bonding, van der Waals, and hydrophobic interactions of these mutants, supporting the importance of the hydrophobic interactions in the protein stability and catalytic activity. Functional variations in the three NOS mutations have yet to be carefully characterized on the basis of the structure as well as thorough biochemical analysis. The structural model of iNOS (see Fig. 14 and ref. [64]) is an important step forward in the analysis of mutations in these multi-domain enzymes.
Supplementary Material
This review covers structure and mechanism of NADPH-cytochrome P450 oxidoreductase.
-
►
The enzyme catalyzes electron transfer from NADPH to FAD to FMN to cytochrome p450.
-
►
The enzyme contains distinct FAD and FMN domains that are connected by a flexible hinge.
-
►
Two flavin domains undergo large domain movements during catalysis.
Acknowledgements
We thank Dr. Anna L. Shen, McArdle Laboratory, University of Wisconsin-Madison, for helpful discussions and suggestions. Parts of the work performed in the Kim laboratory were supported by National Institutes of Health Grant GM52682 (JJPK). The authors acknowledge many collaborators including Drs. BS Masters, CB Kasper, AL Shen, and L Waskell. Major contributors in the Kim laboratory include Ming Wang, Dave Roberts, Paul Hubbard, and Ila Misra; and in the Iyanagi laboratory include Sigenobu Kimura, Zhi-Wen Guan, Hiroyuki Matsuda, Keita Yamamoto and Yoshitaka Nishino.
Abbreviations used
- CYPOR
NADPH-cytochrome P450 oxidoreductase
- CYPOR/POR
CYPOR gene
- P450
cytochrome P450
- cyt c
cytochrome c
- cyt b5
cytochrome b5
- NOS
nitric oxide synthase
- iNOS
inducible NOS
- nNOS
neuronal NOS
- eNOS
endotherial NOS
- NOSred
the reductase domain of NOS
- NOSoxy
the oxygenase domain of NOS
- CaM
Calmodulin
- FNR
ferredoxin-NADP+ oxidoreductase
- Fld
flavodoxin
- FMN domain
FMN containing flavodoxin-like domain
- FAD domain
FAD-containing FNR-like domain plus the connecting domain
- CD
connecting domain
- BF
β-finger
- NR
human cancer-related novel reductase
- MS
methionine synthase
- MSR
methionine synthase reductase
- P450BM3
Bacillus megaterium flavocytochrome P450BM3
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- heme
iron protoporpyrin IX
- ET
electron transfer
- MD
menadione
- ER
endoplasmic reticulum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roman LJ, Masters BS. In: Textbook of Biochemistry with Clinical Correlations. Devlin TM, editor. 2010. pp. 425–456. [Google Scholar]
- 2.Guengerich FP. Chemical Res. Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 3.Shen AL, O'Leary KA, Kasper CB. J. Biol. Chem. 2002;277:6536–6541. doi: 10.1074/jbc.M111408200. [DOI] [PubMed] [Google Scholar]
- 4.Otto DM, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, Adams RH, Tickle C, Wolf CR. Mol. Cell. Biol. 2003;23:6103–6116. doi: 10.1128/MCB.23.17.6103-6116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schacter BA, Nelson EB, Marver HS, Masters BS. J. Biol. Chem. 1972;247:3601–3607. [PubMed] [Google Scholar]
- 6.Guengerich FP. Arch. Biochem. Biophys. 2005;440:204–215. doi: 10.1016/j.abb.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Iyanagi T, Mason HS. Biochemistry. 1973;12:2297–2308. doi: 10.1021/bi00736a018. [DOI] [PubMed] [Google Scholar]
- 8.Vermilion JL, Coon MJ. J. Biol. Chem. 1978;253:8812–8819. [PubMed] [Google Scholar]
- 9.Iyanagi T. Int. Rev. Cytol. 2007;260:35–112. doi: 10.1016/S0074-7696(06)60002-8. [DOI] [PubMed] [Google Scholar]
- 10.Munro AW, Girvan HM, McLean KJ. Nat. Prod. Rep. 2007;24:585–609. doi: 10.1039/b604190f. [DOI] [PubMed] [Google Scholar]
- 11.Laursen T, Jensen K, Moller BL. Biochim. Biophys. Acta. 2011;1814:132–138. doi: 10.1016/j.bbapap.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Mowat CG, Gazur B, Campbell LP, Chapman SK. Arch. Biochem. Biophys. 2010;493:37–52. doi: 10.1016/j.abb.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 14.Stuehr DJ, Tejero J, Haque MM. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daff S. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Feng C. Coordination Chemistry Reviews. 2012;256:393–411. doi: 10.1016/j.ccr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siddhanta U, Prest A, Fan B, Wolan D, Rousseau DL, Stuehr DJ. J. Biol. Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 18.Panda K, Ghosh S, Stuehr DJ. J. Biol. Chem. 2001;276(26):23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 19.Sagami I, Daff S, Shimizu T. J. Biol. Chem. 2001;276:30036–30042. doi: 10.1074/jbc.M104123200. [DOI] [PubMed] [Google Scholar]
- 20.Spratt DE, Duangkham Y, Taiakina V, Guillemette G. The Open Nitric Oxide Journal. 2011;3(Suppl 1-M3):16–24. [Google Scholar]
- 21.Ruettinger RT, Wen LP, Fulco AJ. J. Biol. Chem. 1989;264:10987–10995. [PubMed] [Google Scholar]
- 22.Leclerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng HH, Rommens JM, Scherer SW, Rosenblatt DS, Gravel RA. Proc. Natl. Acad. Sci. USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olteanu H, Benerjee R. J. Biol. Chem. 2001;276:35558–35563. doi: 10.1074/jbc.M103707200. [DOI] [PubMed] [Google Scholar]
- 24.Wolthers KR, Scrutton NS. Biochemistry. 2007;46:6696–6709. doi: 10.1021/bi700339v. [DOI] [PubMed] [Google Scholar]
- 25.Paine MJ, Garner AP, Powell D, Sibbald J, Sales M, Pratt N, Smith T, Tew DG, Wolf CR. J. Biol. Chem. 2000;275:1471–1478. doi: 10.1074/jbc.275.2.1471. [DOI] [PubMed] [Google Scholar]
- 26.Ostrowski J, Barber MJ, Rueger DC, Miller BE, Siegel LM, Kredich NM. J. Biol. Chem. 1989;264:15796–15808. [PubMed] [Google Scholar]
- 27.Inui H, Yamaji R, Saidoh H, Miyatake K, Nakano Y, Kitaoka S. Arch. Biochem. Biophys. 1991;286:270–276. doi: 10.1016/0003-9861(91)90040-p. [DOI] [PubMed] [Google Scholar]
- 28.Nakazawa M, Inui H, Yamaji R, Yamamoto T, Takenaka S, Ueda M, Nakano Y, Miyatake K. FEBS Lett. 2000;479:155–156. doi: 10.1016/s0014-5793(00)01882-2. [DOI] [PubMed] [Google Scholar]
- 29.Netz DJ, Stumpfig M, Dore C, Muhlenhoff U, Pierik AJ, Lill R. Nat Chem Biol. 2010;6:758–765. doi: 10.1038/nchembio.432. [DOI] [PubMed] [Google Scholar]
- 30.Aliverti A, Pandini V, Pennati A, de Rosa M, Zanetti G. Arch. Biochem. Biophys. 2008;474:283–291. doi: 10.1016/j.abb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Medina M. FEBS J. 2009;276:3942–3958. doi: 10.1111/j.1742-4658.2009.07122.x. [DOI] [PubMed] [Google Scholar]
- 32.Porter TD, Kasper CB. Proc. Natl. Acad. Sci. USA. 1985;82:973–977. doi: 10.1073/pnas.82.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter TD, Kasper CB. Biochemistry. 1986;25:1682–1687. doi: 10.1021/bi00355a036. [DOI] [PubMed] [Google Scholar]
- 34.Haniu M, Iyanagi T, Miller P, Lee TD, Shively JE. Biochemistry. 1986;25:7906–7911. doi: 10.1021/bi00372a018. [DOI] [PubMed] [Google Scholar]
- 35.Yamano S, Aoyama T, McBride OW, Hardwick JP, Gelboin HV, Gonzalez FJ. Mol. Pharmacol. 1989;36:83–88. [PubMed] [Google Scholar]
- 36.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Proc. Natl. Acad. Sci. USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia C, Panda SP, Marohnic CC, Martasek P, Masters BS, Kim JJ. Proc. Natl. Acad. Sci. USA. 2011;108:13486–13491. doi: 10.1073/pnas.1106632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubbard PA, Shen AL, Paschke R, Kasper CB, Kim JJ. J. Biol. Chem. 2001;276:29163–29170. doi: 10.1074/jbc.M101731200. [DOI] [PubMed] [Google Scholar]
- 39.Masters BS. Biochem. Biophys. Res. Commun. 2005;338:507–519. doi: 10.1016/j.bbrc.2005.09.165. [DOI] [PubMed] [Google Scholar]
- 40.Omura T. Biochem.Biophys. Res. Commun. 2005;338:404–409. doi: 10.1016/j.bbrc.2005.08.267. [DOI] [PubMed] [Google Scholar]
- 41.Richards MK, Marletta MA. Biochemistry. 1994;33:14723–14732. doi: 10.1021/bi00253a010. [DOI] [PubMed] [Google Scholar]
- 42.Garcin ED, Bruns CM, LIoyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. J. Biol. Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 43.Haque MM, Panda K, Tejero J, Aulak KS, Fadalla MA, Mustovich AT, Stuehr DJ. Proc. Natl. Acad. Sci. USA. 2007;104:9254–9259. doi: 10.1073/pnas.0700332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bashir Q, Scanu S, Ubbink M. FEBS J. 2011;278:1391–1400. doi: 10.1111/j.1742-4658.2011.08062.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Q, Modi S, Smith G, Paine M, McDonagh PD, Wolf CR, Tew D, Lian LY, Roberts GC, Driessen HP. Protein Sci. 1999;8:298–306. doi: 10.1110/ps.8.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taniguchi H, Imai Y, Iyanagi T, Sato R. Biochim. Biophys. Acta. 1979;550:341–356. doi: 10.1016/0005-2736(79)90220-7. [DOI] [PubMed] [Google Scholar]
- 47.Backes WL, Kelley RW. Pharmacol. Ther. 2003;98:221–233. doi: 10.1016/s0163-7258(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 48.Cojocaru V, Balali-Mood K, Sansom MS, Wade RC. PLoS Comput Biol. 2011;7:1–14. doi: 10.1371/journal.pcbi.1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen AL, Kasper CB. J. Biol. Chem. 1995;270:27475–27480. doi: 10.1074/jbc.270.46.27475. [DOI] [PubMed] [Google Scholar]
- 50.Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Proc. Natl. Acad. Sci. USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gruez A, Pignol D, Zeghouf M, Coves J, Fontecave M, Ferrer JL, Fontecilla-Camps JC. J. Mol. Biol. 2000;299:199–212. doi: 10.1006/jmbi.2000.3748. [DOI] [PubMed] [Google Scholar]
- 52.Smith GC, Tew DG, Wolf CR. Proc. Natl. Acad. Sci. USA. 1994;91:8710–8714. doi: 10.1073/pnas.91.18.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leferink NG, Pudney CR, Brenner S, Heyes DJ, Eady RR, Hasnain SS, Hay S, Rigby SE, Scrutton NS. FEBS Lett. 2012;586:578–584. doi: 10.1016/j.febslet.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. J. Biol. Chem. 2009;284:11374–11384. doi: 10.1074/jbc.M807868200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aigrain L, Pompon D, Morera S, Truan G. EMBO Rep. 2009;10:742–747. doi: 10.1038/embor.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia C, Hamdane D, Shen AL, Choi V, Kasper CB, Pearl NM, Zhang H, Im SC, Waskell L, Kim JJ. J. Biol. Chem. 2011;286:16246–16260. doi: 10.1074/jbc.M111.230532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hay S, Brenner S, Khara B, Quinn AM, Rigby SE, Scrutton NS. J. Am. Chem. Soc. 2010;132:9738–9745. doi: 10.1021/ja1016206. [DOI] [PubMed] [Google Scholar]
- 58.Pudney CR, Heyes DJ, Khara B, Hay S, Rigby SE, Scrutton NS. FEBS J. 2012;279:1534–1544. doi: 10.1111/j.1742-4658.2011.08442.x. [DOI] [PubMed] [Google Scholar]
- 59.Ellis J, Gutierrez A, Barsukov IL, Huang WC, Grossmann JG, Roberts GC. J. Biol. Chem. 2009;284:36628–36637. doi: 10.1074/jbc.M109.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vincent B, Morellet N, Fatemi F, Aigrain L, Truan G, Guittet E, Lescop E. J. Mol. Biol. 2012;420:296–309. doi: 10.1016/j.jmb.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 61.Pudney CR, Khara B, Johannissen LO, Scrutton NS. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1001222. e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Craig DH, Chapman SK, Daff S. J. Biol. Chem. 2002;277:33987–33994. doi: 10.1074/jbc.M203118200. [DOI] [PubMed] [Google Scholar]
- 63.Tiso M, Konas DW, Panda K, Garcin ED, Sharma M, Getzoff ED, Stuehr DJ. J. Biol. Chem. 2005;280:39208–39219. doi: 10.1074/jbc.M507775200. [DOI] [PubMed] [Google Scholar]
- 64.Xia C, Misra I, Iyanagi T, Kim JJ. J. Biol. Chem. 2009;284:30708–30717. doi: 10.1074/jbc.M109.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tejero J, Haque1 MM, Durra D, Stuehr DJ. J. Biol. Chem. 2010;285:25941–25949. doi: 10.1074/jbc.M110.126797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shimizu T, Tateishi T, Hatano M, Fujii-Kuriyama Y. J. Biol. Chem. 1991;266:3372–3375. [PubMed] [Google Scholar]
- 67.Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. J. Biol. Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 68.Johnson EF. Drug Metab. Dispos. 2003;31:1532–1540. doi: 10.1124/dmd.31.12.1532. [DOI] [PubMed] [Google Scholar]
- 69.Im SC, Waskell L. Arch. Biochem. Biophys. 2011;507:144–153. doi: 10.1016/j.abb.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kenaan C, Zhang H, Shea EV, Hollenberg PF. Biochemistry. 2011;50:3957–3967. doi: 10.1021/bi1020748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Black SD, Coon MJ. J. Biol. Chem. 1982;257:5929–5938. [PubMed] [Google Scholar]
- 72.Iyanagi T. Biochem. Biophys. Res. Commun. 2005;338:520–528. doi: 10.1016/j.bbrc.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 73.Enoch HG, Strittmatter P. J. Biol. Chem. 1979;254:8976–8981. [PubMed] [Google Scholar]
- 74.Kitazume T, Haines DC, Estabrook RW, Chen B, Peterson JA. Biochemistry. 2007;46:11892–11901. doi: 10.1021/bi701031r. [DOI] [PubMed] [Google Scholar]
- 75.Roman LJ, Miller RT, de La Garza MA, Kim JJ, Masters BSS. J. Biol. Chem. 2000;275:21914–21919. doi: 10.1074/jbc.M002449200. [DOI] [PubMed] [Google Scholar]
- 76.Roman LJ, Martasek P, Miller RT, Harris DE, de La Garza MA, Shea TM, Kim JJ, Masters BS. J. Biol. Chem. 2000;275:29225–29232. doi: 10.1074/jbc.M004766200. [DOI] [PubMed] [Google Scholar]
- 77.Tiso M, Tejero J, Panda K, Aulak KS, Stuehr DJ. Biochemistry. 2007;46:14418–14428. doi: 10.1021/bi701646k. [DOI] [PubMed] [Google Scholar]
- 78.Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM, Martasek P, Roman LJ, Masters BS, Jones CL, Weissman BA, Lane P, Liu Q, Gross SS. J. Biol. Chem. 1997;272:29769–29777. doi: 10.1074/jbc.272.47.29769. [DOI] [PubMed] [Google Scholar]
- 79.Daff S, Sagami I, Shimizu T. J. Biol. Chem. 1999;274:30589–30595. doi: 10.1074/jbc.274.43.30589. [DOI] [PubMed] [Google Scholar]
- 80.Lane P, Gross SS. Acta Physiol Scand. 2000;168:53–63. doi: 10.1046/j.1365-201x.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 81.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen CA, Lin CH, Druhan LJ, Wang TY, Chen YR, Zweier JL. J. Biol. Chem. 2011;286:29098–29107. doi: 10.1074/jbc.M111.240127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kao YT, Saxena C, He TF, Guo L, Wang L, Sancar A, Zhong D. J. Am. Chem. Soc. 2008;130:13132–13139. doi: 10.1021/ja8045469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iyanagi T, Makino N, Mason HS. Biochemistry. 1974;13:1701–1710. doi: 10.1021/bi00705a023. [DOI] [PubMed] [Google Scholar]
- 85.Vermilion JL, Ballou DP, Massey V, Coon MJ. J. Biol. Chem. 1981;256:266–277. [PubMed] [Google Scholar]
- 86.Brenner S, Hay S, Munro AW, Scrutton NS. FEBS J. 2008;275:4540–4557. doi: 10.1111/j.1742-4658.2008.06597.x. [DOI] [PubMed] [Google Scholar]
- 87.Stuehr DJ, Iketa-Saito M. J. Biol. Chem. 1992;267:20547–20550. [PubMed] [Google Scholar]
- 88.Gao YT, Smith SM, Weinberg JB, Montgomery HJ, Newman E, Guillemette JG, Ghosh DK, Roman LJ, Martasek P, Salerno JC. J. Biol. Chem. 2004;279:18759–18766. doi: 10.1074/jbc.M308936200. [DOI] [PubMed] [Google Scholar]
- 89.Corrado ME, Aliverti A, Zanetti G, Mayhew SG. Eur. J. Biochem. 1996;239:662–667. doi: 10.1111/j.1432-1033.1996.0662u.x. [DOI] [PubMed] [Google Scholar]
- 90.Hoover DM, Drennan CL, Metzger AL, Osborne C, Weber CH, Pattridge KA, Ludwig ML. J. Mol. Biol. 1999;294:725–743. doi: 10.1006/jmbi.1999.3152. [DOI] [PubMed] [Google Scholar]
- 91.Ishikita H. J. Biol. Chem. 2007;282:25240–25246. doi: 10.1074/jbc.M702788200. [DOI] [PubMed] [Google Scholar]
- 92.Munro AW, Noble MA, Robledo L, Daff SN, Chapman SK. Biochemistry. 2001;40:1956–1963. doi: 10.1021/bi001718u. [DOI] [PubMed] [Google Scholar]
- 93.Welland A, Garnaud PE, Kitamura M, Miles CS, Daff S. Biochemistry. 2008;47:9771–9780. doi: 10.1021/bi800787m. [DOI] [PubMed] [Google Scholar]
- 94.Guan ZW IT. Arch. Biochem. Biophys. 2003;412:65–76. doi: 10.1016/s0003-9861(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 95.Guan Z-W, Kamatani D, Kimura S, Iyanagi T. J. Biol. Chem. 2003;278:30859–30868. doi: 10.1074/jbc.M301929200. [DOI] [PubMed] [Google Scholar]
- 96.Dunford AJ, Marshall KR, Munro AW, Scrutton NS. Eur. J. Biochem. 2004;271:2548–2560. doi: 10.1111/j.1432-1033.2004.04185.x. [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto K, Kimura S, Shiro Y, Iyanagi T. Arch. Biochem. Biophys. 2005;440:65–78. doi: 10.1016/j.abb.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 98.Nishino Y, Yamamoto K, Kimura S, Kikuchi A, Shiro Y, Iyanagi T. Arch. Biochem. Biophys. 2007;465:254–265. doi: 10.1016/j.abb.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 99.Batie CJ, Kamin H. J. Biol. Chem. 1984;259:11976–11985. [PubMed] [Google Scholar]
- 100.Kimura S, Kawamura M, Iyanagi T. J. Biol. Chem. 2003;278:3580–3589. doi: 10.1074/jbc.M209838200. [DOI] [PubMed] [Google Scholar]
- 101.Welland A, Daff S. FEBS J. 2010;277:3833–3843. doi: 10.1111/j.1742-4658.2010.07787.x. [DOI] [PubMed] [Google Scholar]
- 102.Dunford AJ, Rigby SE, Hay S, Munro AW, Scrutton NS. Biochemistry. 2007;46:5018–5029. doi: 10.1021/bi7001339. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Z, Swenson RP. Biochemistry. 1996;35:15980–15988. doi: 10.1021/bi962124n. [DOI] [PubMed] [Google Scholar]
- 104.Zhang J, Martasek P, Paschke R, Shea T, Masters BS, Kim JJ. J. Biol. Chem. 2001;276:37506–37513. doi: 10.1074/jbc.M105503200. [DOI] [PubMed] [Google Scholar]
- 105.Ludwig ML, Pattridge KA, Metzger AL, Dixon MM, Eren M, Feng Y, Swenson RP. Biochemistry. 1997;36:1259–1280. doi: 10.1021/bi962180o. [DOI] [PubMed] [Google Scholar]
- 106.Marcus RA, Sutin N. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 107.Astuti Y, Topoglidis E, Briscoe PB, Fantuzzi A, Gilardi G, Durrant JR. J. Am. Chem. Soc. 2004;126:8001–8009. doi: 10.1021/ja0496470. [DOI] [PubMed] [Google Scholar]
- 108.Damiani MJ, Nostedt JJ, O'Neil MA. J. Biol. Chem. 2011;286:4382–4391. doi: 10.1074/jbc.M110.194696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen HC, Swemson RP. Biochemistry. 2008;47(52):13788–13799. doi: 10.1021/bi800954h. [DOI] [PubMed] [Google Scholar]
- 110.Kasim M, Chen HC, Swenson RP. Biochemistry. 2009;48:5131–5141. doi: 10.1021/bi900607q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li H, Das A, Sibhatu H, Jamal J, Sligar SG, Poulos TL. J. Biol. Chem. 2008;283:34762–34772. doi: 10.1074/jbc.M806949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Iyanagi T, Makino R, Anan FK. Biochemistry. 1981;20:1722–1730. doi: 10.1021/bi00510a004. [DOI] [PubMed] [Google Scholar]
- 113.Iyanagi T, Yamazaki I. Biochim. Biophys. Acta. 1970;216:282–294. doi: 10.1016/0005-2728(70)90220-3. [DOI] [PubMed] [Google Scholar]
- 114.Adak S, Ghosh S, Abu-Soud HM, Stuehr DJ. J. Biol. Chem. 1999;274:22313–22320. doi: 10.1074/jbc.274.32.22313. [DOI] [PubMed] [Google Scholar]
- 115.Matsuda H, Kimura S, Iyanagi T. Biochim. Biophys. Acta. 2000;1459:106–116. doi: 10.1016/s0005-2728(00)00117-1. [DOI] [PubMed] [Google Scholar]
- 116.Fu J, Yamamoto K, Guan ZW, Kimura S, Iyanagi T. Arch. Biochem. Biophys. 2004;427:180–187. doi: 10.1016/j.abb.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 117.Wolthers KR, Schimerlik MI. Biochemistry. 2001;40:4722–4737. doi: 10.1021/bi0023495. [DOI] [PubMed] [Google Scholar]
- 118.Haque MM, Kenney C, Tejero J, Stuehr DJ. FEBS J. 2011;278:4055–4069. doi: 10.1111/j.1742-4658.2011.08310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garner AP, Paine MJ, Rodriguez-Crespo I, Chinje EC, Ortiz De Montellano P, Stratford IJ, Tew DG, Wolf CR. Cancer Res. 1999;59:1929–1934. [PubMed] [Google Scholar]
- 120.Deng Z, Aliverti A, Zanetti G, Arakaki AK, Ottado J, Orellano EG, Calcaterra NB, Ceccarelli EA, Carrillo N, Karplus PA. Nat. Struc. Biol. 1999;6:847–853. doi: 10.1038/12307. [DOI] [PubMed] [Google Scholar]
- 121.Tejero J, Perez-Dorado I, Maya C, Martinez-Julvez M, Sanz-Aparicio J, Gomez-Moreno C, Hermoso JA, Medina M. Biochemistry. 2005;44:13477–13490. doi: 10.1021/bi051278c. [DOI] [PubMed] [Google Scholar]
- 122.Konas DW, Zhu K, Sharma M, Aulak KS, Brudvig GW, Stuehr DJ. J. Biol. Chem. 2004;279:35412–35425. doi: 10.1074/jbc.M400872200. [DOI] [PubMed] [Google Scholar]
- 123.Joyce MG, Ekanem IS, Roitel O, Dunford AJ, Neeli R, Girvan HM, Baker GJ, Curtis RA, Munro AW, Leys D. FEBS J. 2012;279:1694–1706. doi: 10.1111/j.1742-4658.2012.08544.x. [DOI] [PubMed] [Google Scholar]
- 124.Meints CE, Gustafsson FS, Scrutton NS, Wolthers KR. Biochemistry. 2011;50:11131–11142. doi: 10.1021/bi2012228. [DOI] [PubMed] [Google Scholar]
- 125.Leys D, Scrutton NS. Curr. Opin. Struct. Biol. 2004;14:642–647. doi: 10.1016/j.sbi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 126.Davidson VL. Acc. Chem. Res. 2008;41:730–738. doi: 10.1021/ar700252c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 128.Simondsen RP, Weber PC, Salemme FR, Tollin G. Biochemistry. 1982;21:6366–6375. doi: 10.1021/bi00268a008. [DOI] [PubMed] [Google Scholar]
- 129.Gutierrez A, Grunau A, Paine M, Munro AW, Wolf CR, Roberts GC, Scrutton NS. Biochem. Soc. Trans. 2003;31:497–501. doi: 10.1042/bst0310497. [DOI] [PubMed] [Google Scholar]
- 130.Gutierrez A, Paine M, Wolf CR, Scrutton NS, Roberts GC. Biochemistry. 2002;41:4626–4637. doi: 10.1021/bi0159433. [DOI] [PubMed] [Google Scholar]
- 131.Konas DW, Takaya N, Sharma M, Stuehr DJ. Biochemistry. 2006;45:12596–12609. doi: 10.1021/bi061011t. [DOI] [PubMed] [Google Scholar]
- 132.Iyanagi T. Flavins and Flavoproteins. Tokyo, Japan: ARchiTeck Inc; 2005. pp. 453–464. [Google Scholar]
- 133.Ghosh DK, Ray K, Rogers AJ, Nahm NJ, Salerno JC. FEBS J. 2012;279:1306–1317. doi: 10.1111/j.1742-4658.2012.08525.x. [DOI] [PubMed] [Google Scholar]
- 134.Matsuda H, Iyanagi T. Biochim. Biophys. Acta. 1999;1473:345–355. doi: 10.1016/s0304-4165(99)00193-2. [DOI] [PubMed] [Google Scholar]
- 135.Knight K, Scrutton NS. Biochem. J. 2002;367:19–30. doi: 10.1042/BJ20020667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Feng C, Taiakina V, Ghosh DK, Guillemette JG, Tollin G. Biochim. Biophys. Acta. 2011;1814:1997–2002. doi: 10.1016/j.bbapap.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nishida CR, Ortiz de Montellano PR. J. Biol. Chem. 1998;273:5566–5571. doi: 10.1074/jbc.273.10.5566. [DOI] [PubMed] [Google Scholar]
- 138.Miller RT, Martasek P, Omura T, Siler Masters BS. Biochem. Biophys. Res. Commun. 1999;265:184–188. doi: 10.1006/bbrc.1999.1643. [DOI] [PubMed] [Google Scholar]
- 139.Yoshioka S, Takahashi S, Ishimori K, Morishima I. J. Inorg. Biochem. 2000;81:141–151. doi: 10.1016/s0162-0134(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 140.Das A, Grinkova YV, Sligar SG. J. Am. Chem. Soc. 2007;129:13778–13779. doi: 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Imai Y, Sato R, Iyanagi T. J Biochem. 1977;82:1237–1246. doi: 10.1093/oxfordjournals.jbchem.a131811. [DOI] [PubMed] [Google Scholar]
- 142.Oprian DD, Vatsis KP, Coon MJ. J. Biol. Chem. 1979;254:8895–8902. [PubMed] [Google Scholar]
- 143.Koppenol WH. J. Am. Chem. Soc. 2007;129:9686–9690. doi: 10.1021/ja071546p. [DOI] [PubMed] [Google Scholar]
- 144.Noshiro M, Ullrich V, Omura T. Eur. J. Biochem. 1981;116:521–526. doi: 10.1111/j.1432-1033.1981.tb05367.x. [DOI] [PubMed] [Google Scholar]
- 145.Kaspera R, Naraharisetti SB, Evangelista EA, Marciante KD, Psaty BM, Totah RA. Biochem. Pharmacol. 2011;82:681–691. doi: 10.1016/j.bcp.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Syed K, Kattamuri C, Thompson TB, Yadav JS. Arch. Biochem. Biophys. 2011;509:26–32. doi: 10.1016/j.abb.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ichinose H, Wariishi H. Arch. Biochem. Biophys. 2012;518:8–15. doi: 10.1016/j.abb.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 148.Presta A, Weber-Main AM, Stankovich MT, Stuehr DJ. J. Am. Chem. Soc. 1998;120:9460–9465. [Google Scholar]
- 149.Tsai AL, Berka V, Chen PF, Palmer G. J. Biol. Chem. 1996;271:32563–32571. doi: 10.1074/jbc.271.51.32563. [DOI] [PubMed] [Google Scholar]
- 150.Woodward JJ, Nejatyjahromy Y, Britt RD, Marletta MA. J. Am. Chem. Soc. 2010;132:5105–5113. doi: 10.1021/ja909378n. [DOI] [PubMed] [Google Scholar]
- 151.Santolini J. J. Inorg. Biochem. 2011;105:127–141. doi: 10.1016/j.jinorgbio.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 152.Cooper HL, Groves JT. Arch. Biochem. Biophys. 2010;507:111–118. doi: 10.1016/j.abb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gorren AC, Kungl AJ, Schmidt K, Werner ER, Mayer B. Nitric Oxide. 2001;5:176–186. doi: 10.1006/niox.2001.0332. [DOI] [PubMed] [Google Scholar]
- 154.Wei CC, Crane BR, Stuehr DJ. Chem. Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 155.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. J. Biol. Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 156.Massey V. Biochem. Soc. Trans. 2000;28:283–296. [PubMed] [Google Scholar]
- 157.Backes WL, Reker-Backes CE. J. Biol. Chem. 1988;263:247–253. [PubMed] [Google Scholar]
- 158.Iyanagi T, Suzaki T, Kobayashi S. J. Biol. Chem. 1981;256:12933–12939. [PubMed] [Google Scholar]
- 159.Daff SN, Chapman SK, Turner KL, Holt RA, Govindaraj S, Poulos TL, Munro AW. Biochemistry. 1997;36:13816–13823. doi: 10.1021/bi971085s. [DOI] [PubMed] [Google Scholar]
- 160.Murataliev MB, Klein M, Fulco A, Feyereisen R. Biochemistry. 1997;36:8401–8412. doi: 10.1021/bi970026b. [DOI] [PubMed] [Google Scholar]
- 161.Roman LJ, Martasek P, Masters BS. Chem. Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 162.Aoyagi M, Arvai AS, Tainer JA, Getzoff ED. EMBO J. 2003;22:766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 164.Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 165.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Nat. Struct. Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 166.Ghosh DK, Salerno JC. Front. Biosci. 2003;8:193–209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- 167.Roman LJ, Masters BSS. J. Biol. Chem. 2006;281:23111–23118. doi: 10.1074/jbc.M603671200. [DOI] [PubMed] [Google Scholar]
- 168.Panda K, Haque MM, Garcin-Hosfield ED, Durra D, Getzoff ED, Stuehr DJ. J. Biol. Chem. 2006;281:36819–36827. doi: 10.1074/jbc.M606129200. [DOI] [PubMed] [Google Scholar]
- 169.Jones RJ, Gao YT, Simone TM, Salerno JC, Smith SM. Nitric Oxide. 2006;14:228–237. doi: 10.1016/j.niox.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 170.Feng C, Tollin G, Holliday MA, Thomas C, Salerno JC, Enemark JH, Ghosh DK. Biochemistry. 2006;45:6354–6362. doi: 10.1021/bi060223n. [DOI] [PubMed] [Google Scholar]
- 171.Feng C, Tollin G, Hazzard JT, Nahm NJ, Guillemette JG, Salerno JC, Ghosh DK. J. Am. Chem. Soc. 2007;129:5621–5629. doi: 10.1021/ja068685b. [DOI] [PubMed] [Google Scholar]
- 172.Matsubara M, Hayashi N, Titani K, Taniguchi H. J. Biol. Chem. 1997;272:23050–23056. doi: 10.1074/jbc.272.37.23050. [DOI] [PubMed] [Google Scholar]
- 173.Watenpaugh KD, Sieker LC, Jensen LH. Proc. Natl. Acad. Sci. U S A. 1973;70:3857–3860. doi: 10.1073/pnas.70.12.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu G, Berka V, Tsai A-L. J. Inorg. Biochem. 2011;105:1226–1237. doi: 10.1016/j.jinorgbio.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tejero J, Hannibal L, Mustovich A, Stuehr DJ. J. Biol. Chem. 2010;285:27232–27240. doi: 10.1074/jbc.M110.138842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Astashkin AV, Elmore BO, Fan W, Guillemette JG, Feng C. J. Am. Chem. Soc. 2010;132:12059–12067. doi: 10.1021/ja104461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Miller RT, Martasek P, Nishimura JS, Panda S, Harris DE, Roman LJ, Masters BS. Flavins and Flavoproteins 1999. Berlin: Agency for Scientic Publications; 1999. pp. 151–154. [Google Scholar]
- 178.Frago S, Lans I, Navarro JA, Hervás M, Edmondson DE, De la Rosa MA, Gómez-Moreno C, Mayhew SG, Medina M. Biochim. Biophys. Acta. 2010;1797:262–271. doi: 10.1016/j.bbabio.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 179.Chang CW, He TF, Guo L, Stevens JA, Li T, Wang L, Zhong D. J. Am. Chem. Soc. 2010;132:12741–12747. doi: 10.1021/ja1050154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guengerich FP, Cheng Q. Pharmacol. Rev. 2011;63:684–699. doi: 10.1124/pr.110.003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shen AL, Porter TD, Wilson TE, Kasper CB. J. Biol. Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 182.Fluck CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, Jabs EW, Mendonca BB, Fujieda K, Miller WL. Nat. Genet. 2004;36:228–230. doi: 10.1038/ng1300. [DOI] [PubMed] [Google Scholar]
- 183.Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, Stewart PM, Shackleton CH. Lancet. 2004;363:2128–2135. doi: 10.1016/S0140-6736(04)16503-3. [DOI] [PubMed] [Google Scholar]
- 184.Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A. Am. J. Med. Genet. Part A. 2004;128A:333–339. doi: 10.1002/ajmg.a.30169. [DOI] [PubMed] [Google Scholar]
- 185.Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, Jabs EW, Van Vliet G, Sack J, Fluck CE, Miller WL. Am. J. Hum. Genet. 2005;76:729–749. doi: 10.1086/429417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, Matsuo N, Sato S, Homma K, Nishimura G, Hasegawa T, Ogata T. J. Clin. Endocrinol. Metab. 2005;90:414–426. doi: 10.1210/jc.2004-0810. [DOI] [PubMed] [Google Scholar]
- 187.Agrawal V, Huang N, Miller WL. Pharmacogenet. Genomics. 2008;18:569–576. doi: 10.1097/FPC.0b013e32830054ac. [DOI] [PubMed] [Google Scholar]
- 188.Nicolo C, Flück CE, Mullis PE, Pandey AV. Mol. Cell. Endocrinol. 2010;321:245–252. doi: 10.1016/j.mce.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 189.Pandey AV, Kempna P, Hofer G, Mullis PE, Fluck CE. Mol. Endocrinol. 2007;21:2579–2595. doi: 10.1210/me.2007-0245. [DOI] [PubMed] [Google Scholar]
- 190.Tomkova M, Marohnic CC, Baxova A, Martasek P. Cas Lek Cesk. 2008;147:261–265. [PubMed] [Google Scholar]
- 191.Fluck CE, Pandey AV. Endocr. Dev. 2011;20:63–79. doi: 10.1159/000321221. [DOI] [PubMed] [Google Scholar]
- 192.Marohnic CC, Panda SP, McCammon K, Rueff J, Masters BSS, Kranendonk M. Drug Metab. Dispos. 2010;38:332–340. doi: 10.1124/dmd.109.030445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Marohnic CC, Panda SP, Martasek P, Masters BS. J. Biol. Chem. 2006;281:35975–35982. doi: 10.1074/jbc.M607095200. [DOI] [PubMed] [Google Scholar]
- 194.Tee MK, Huang N, Damm I, Miller WL. Mol. Endocrinol. 2011;25:715–731. doi: 10.1210/me.2010-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Soneda S, Yazawa T, Fukami M, Adachi M, Mizota M, Fujieda K, Miyamoto K, Ogata T. J. Clin. Endocrinol. Metab. 2011;96:1881–1887. doi: 10.1210/jc.2011-1337. [DOI] [PubMed] [Google Scholar]
- 196.Hart SN, Wang S, Nakamoto K, Wesselman C, Li Y, Zhong XB. Pharmacogenet. Genomics. 2008;18:11–24. doi: 10.1097/FPC.0b013e3282f2f121. [DOI] [PubMed] [Google Scholar]
- 197.Kranendonk M, Marohnic CC, Panda SP, Duarte MP, Oliveira JS, Masters BS, Rueff J. Arch. Biochem. Biophys. 2008;475:93–99. doi: 10.1016/j.abb.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gomes AM, Winter S, Klein K, Turpeinen M, Schaeffeler E, Schwab M, Zanger UM. Pharmacogenomics. 2009;10:579–599. doi: 10.2217/pgs.09.7. [DOI] [PubMed] [Google Scholar]
- 199.Pandey AV, Fluck CE, Mullis PE. Biochem. Biophys. Res. Commun. 2010;400:374–378. doi: 10.1016/j.bbrc.2010.08.072. [DOI] [PubMed] [Google Scholar]
- 200.Marohnic CC, Huber WJ, Iii, Connick J, Reed JR, McCammona K, Panda SP, Martásek P, Backes WL, Masters BSS. Arch. Biochem. Biophys. 2011;513:42–50. doi: 10.1016/j.abb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wilson A, Leclerc D, Rosenblatt DS, Gravel RA. Hum. Mol. Genet. 1999;8:2009–2016. doi: 10.1093/hmg/8.11.2009. [DOI] [PubMed] [Google Scholar]
- 202.Gherasim CG, Zaman U, Raza A, Banerjee R. Biochemistry. 2008;47:12515–12522. doi: 10.1021/bi8008328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Andreakis N, D'Aniello S, Albalat R, Patti FP, Garcia-Fernandez J, Procaccini G, Sordino P, Palumbo A. Mol. Biol. Evol. 2011;28:163–179. doi: 10.1093/molbev/msq179. [DOI] [PubMed] [Google Scholar]
- 204.Beiser DG, Orbelyan GA, Inouye BT, Costakis JG, Hamann KJ, McNally EM, Vanden Hoek TL. Resuscitation. 2011;82:115–121. doi: 10.1016/j.resuscitation.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huang PL. J. Am. Soc. Nephrol. JASN. 2000;11(Suppl 16):S120–S123. [PubMed] [Google Scholar]
- 206.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 207.Morishita T, Tsutsui M, Shimokawa H, Sabanai K, Tasaki H, Suda O, Nakata S, Tanimoto A, Wang KY, Ueta Y, Sasaguri Y, Nakashima Y, Yanagihara N. Proc. Natl. Acad. Sci. USA. 2005;102:10616–10621. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tsutsui M, Shimokawa H, Otsuji Y, Yanagihara N. Pharmacol. Therapeutics. 2010;128:499–508. doi: 10.1016/j.pharmthera.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 209.Naureckiene S, Kodangattil SR, Kaftan EJ, Jones PG, Kennedy JD, Rogers KE, Chanda PK. Protein J. 2008;27:309–318. doi: 10.1007/s10930-008-9139-y. [DOI] [PubMed] [Google Scholar]
- 210.Watanabe Y, Iyanagi T, Oae S. Tetrahedron Lett. 1982;23:533–536. [Google Scholar]
- 211.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 212.Woodward JJ, Chang MM, Martin NI, Marletta MA. J. Am. Chem. Soc. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- 213.Stoll S, NejatyJahromy Y, Woodward JJ, Ozarowski A, Marletta MA, Britt RD. J. Am. Chem. Soc. 2010;132:11812–11823. doi: 10.1021/ja105372s. [DOI] [PubMed] [Google Scholar]
- 214.Iyanagi T. Seikagaku. 2011;83:273–293. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.