

Abstract
Glioblastoma, GBM, is the most frequent brain malignancy in adults. Patients with these tumors survive only, approximately, one year after diagnosis and rarely survive beyond two years. This poor prognosis is, in part, due to our insufficient understanding of the complex aggressive nature of these tumors and the lack of effective therapy. In GBM, over-expression of EGFR and/or its constitutively activated variant EGFRvIII is a major characteristic and is associated with tumorigenesis and more aggressive phenotypes, such as, invasiveness and therapeutic resistance. Consequently, both have been major targets for GBM therapy, however, clinical trials of EGFR- and EGFRvIII-targeted therapies have yielded unsatisfactory results and the molecular basis for the poor results is still unclear. Thus, in this review, we will summarize results of recent clinical trials and recent advances made in the understanding of the EGFR/EGFRvIII pathways with a key focus on those associated with intrinsic resistance of GBM to EGFR-targeted therapy. For example, emerging evidence indicates an important role that PTEN plays in predicting GBM response to EGFR-targeted therapy. Aberrant Akt/mTOR pathway has been shown to contribute to the resistant phenotype. Also, several studies have reported that EGFR/EGFRvIII’s cross-talk with the oncogenic transcription factorSTAT3 and receptor tyrosine kinases, (c-Met and PDGFR) potentially lead to GBM resistance to anti-EGFR therapy. Other emerging mechanisms, including one involving HMG-CoA reductase, will also be discussed in this mini-review. These recent findings have provided new insight into the highly complex and interactive nature of the EGFR pathway and generated rationales for novel combinational targeted therapies for these tumors.
Keywords: EGFR/EGFRvIII, glioblastoma, malignant glioma, drug resistance, PTEN, PI3-K/Akt/mTOR, c-Met, STAT3
INTRODUCTION
Gliomas account for approximately 80% of primary brain cancers in adults. Glioblastoma multiforme (GBM) is the most frequent type of gliomas and the most malignant form that is associated with dismal prognosis. Patients with these tumors survive only, approximately, one year after diagnosis and rarely survive beyond two years [1]. This poor prognosis is, in part, due to our insufficient understanding of the complex aggressive nature of these tumors and the lack of effective therapy. As such, vigorous efforts are ongoing to either improve current therapy or to identify and strike new molecular targets for GBM therapy [2]. A number of genes and pathways have emerged as attractive therapeutic targets for GBM, such as, PI3-K/mTOR, PDGFR, VEGF/angiogenesis [3-5], Hedgehog-GLI1 [6] and EGFR/EGFRvIII [5, 7-10].
Given that the genes encoding EGFR and its constitutively activated variant EGFRvIII are frequently amplified and/or over-expressed in GBMs, mono and combinational EGFR-targeted therapies have attracted much attention and are being extensively evaluated pre-clinically and clinically in GBM. Although the pre-clinical studies have shown encouraging results, clinical trials have consistently yielded limited survival benefits [10]. This poor outcome has prompted extensive investigations that aim to shed light on the complex and interactive nature of the EGFR/EGFRvIII signaling pathways and to elucidate molecular mechanisms underlying GBM resistance to EGFR-targeted therapy. This review will thus summarize recent advances made in the fundamental understanding the EGFR- and EGFRvIII-mediated signaling, outcome of clinical trials with EGFR-targeted agents in malignant gliomas, and the potential mechanisms that underlie the resistance of GBM to these therapies.
THE EGFR PATHWAY
Overexpression of EGFR and/or its constitutively activated variant EGFRvIII is frequently found in many human cancers, including GBMs, and is a hallmark for more aggressive tumors that are highly invasive and more resistant to therapy [11, 12]. EGFRvIII is a product of rearrangement with an in-frame deletion of 801 bp of the coding sequence of the extracellular domain, resulting in a deletion of residues 6 through 273 and a glycine insertion as residue 6 [13-16]. EGFR gene amplification is the most frequent genetic alteration in primary GBMs and approximately, half of these tumors carry the rearranged EGFRvIII gene [13-17]. Although EGFRvIII overexpression is mostly concurrent with EGFR gene amplification, it has been reported in a small proportion of primary GBMs without EGFR gene amplification [18]. GBM cells expressing EGFRvIII are more tumorigenic in nude mice compared to those with the wild-type EGFR [19]. Importantly, both EGFR- and EGFRvIII-mediated pathways are of a high degree of biological complexity and consisted of two major signaling modes, namely, the cell-surface and nuclear modes [11, 20, 21].
In the cell-surface signaling mode (Fig. 1a), both EGFR and EGFRvIII function as receptor tyrosine kinases (RTKs) that activate a number of signaling modules, such as, those mediated by PLC-γ, Ras, PI3-K and Janus-activated kinase 2 (JAK2), leading to tumorigenesis and more aggressive tumor behaviors [11, 12]. With regard to signal transducer and activator of transcription 3 (STAT3), cell-surface EGFR and EGFRvIII physically associate with and phosphorylate STAT3 at Y705 and in turn, phosphorylated STAT3 dimerizes and translocates into the cell nucleus to regulate gene expression. The tumor suppressor gene, phosphatase and tensin homolog deleted on chromosome 10 (PTEN), antagonizes the PI3-K pathway and subsequently inhibits the downstream effectors of PI3-K, Akt and mTOR. Interestingly, EGFR also exerts kinase-independent function [22]. In this context, EGFR interacts with and stabilizes sodium/glucose cotransporter 1, thereby maintaining intracellular glucose levels and preventing autophagic cell death [22]. Together, EGFR and EGFRvIII mediate a web of complex signaling networks and impact many important cellular processed.
Fig. 1. The EGFR signaling pathway is of critical importance to human cancers and of a high complexity.
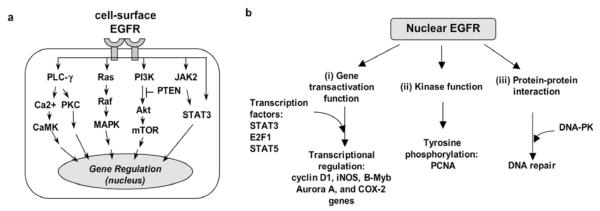
The EGFR signaling pathway exerts its biological effects via two major modes of actions, namely, the cytoplasmic/ traditional (a) and the nuclear (b) signaling modes.
a. The cytoplasmic/traditional EGFR pathway is consisted of five major modules: PLC-γ-CaMK/PKC, Ras-Raf-MAPK, PI-3K-Akt-mTOR, JAK2/STAT3 and STAT3. Activation of these signaling modules often leads to tumorigenesis, tumor proliferation, metastasis, chemoresistance, and radio-resistance.
b. Nuclear EGFR has three key functions: (i) gene transactivation, (ii) tyrosine kinase, and (iii) protein-protein interaction.
Evidence to date indicates that cell-surface EGFR and EGFRvIII differ in their ability to activate their downstream pathways. However, the results are somewhat inconsistent and controversial. For example, Huang et al. [23] conducted a large-scale analysis of phosphotyrosine-mediated signaling pathways using U87MG GBM cells stably expressing EGFRvIII and subsequently found that EGFRvIII preferentially activates PI3-K/Akt over the Ras/MAPK and STAT3 pathways. This observation corroborate the finding reported by Mellinghoff et al. [5] that GBMs with concurrent expression of EGFRvIII and PTEN had a better response to the EGFR kinase inhibitor erlotinib. However, Progent et al. [24] reported that the increased tumorigenic potential of EGFRvIII-expressing GBM, relative to those with EGFR, was associated with Ras/MAPK hyperactivation. Currently, this issue has not been resolved and is likely dependent on cellular context.
In the nuclear signaling mode (Fig. 1b), EGFR has three key functions: (i) gene transactivation [25-28], (ii) tyrosine phosphorylation [29], and (iii) protein-protein interactions [30, 31]. EGFR ligands, oxidative stress and radiation-induced DNA damage stimulate EGFR nuclear transport [11]. Nuclear EGFR is localized on the inner nuclear membrane [32, 33] and in the nucleoplasm [27, 28, 34, 35]. The effect of cetuximab on EGFR nuclear translocalization has been investigated. Liao and Carpenter [36] showed that cetuximab activates EGFR nuclear transport. In contrast, Dittmann et al. [31] reported that cetuximab inhibits radiation-induced EGFR nuclear translocalization. Via its gene transactivation domain, nuclear EGFR activates gene expression [27]. Because of its lack of a DNA-binding domain, nuclear EGFR interacts with DNA-binding transcription factors, STAT3, E2F1 and STAT5, to induce expression of iNOS, B-Myb and aurora A genes, respectively, in breast cancer [25, 26, 28]. Nuclear EGFR retains its tyrosine kinase activity and phosphorylates proliferating cell nuclear antigen (PCNA) to promote cell proliferation [29]. Moreover, nuclear EGFR undergoes protein-protein interactions with DNA-PK to facilitate repair of radiation-induced DNA double-strand breaks in bronchial carcinoma [30, 31].
In GBMs, the nuclear EGFR and nuclear EGFRvIII pathways have been recently investigated. The report by de la Iglesia et al. [37] showed that EGFRvIII is detected in the nucleus of normal astrocytes and primary GBMs. While the consequence of nuclear EGFRvIII was not elucidated, nuclear EGFRvIII appears to interact with STAT3 in normal astrocytes, leading to their malignant transformation [37]. Most recently, our laboratory showed conclusive evidences for the existence of nuclear EGFR and EGFRvIII in GBM cells and its functional interaction with nuclear STAT3 to activate COX-2 gene expression, thus linking EGFR/EGFRvIII to the inflammatory pathway [38]. Nuclear translocalization of both receptors depends on nuclear localization signals located within the juxtamembrane region and when deleted, both receptors fail to enter the cell nucleus. Evidence also suggest a role that nuclear EGFR may play in gliomagenesis [38]. Collectively, the EGFR- and EGFRvIII-mediated pathways are critical for cancer biology and potentially associated with increased proliferation, invasion/metastasis, radio-resistance, and shortened patient survival. These pathways are also highly complex with a profound potential to interact with other important pathways in cancers.
PROGNOSTIC VALUE OF EGFR AND EGFRVIII IN MALIGNANT GLIOMAS
It remains inconclusive regarding the prognostic value of EGFR and EGFRvIII in malignant gliomas. Shinojima et al. [18] evaluated 87 newly diagnosed GBM patients and found EGFR amplification to be an independent, unfavorable predictor for overall survival. In this cohort, EGFRvIII overexpression in the presence of EGFR amplification is the strongest indicator of a poor survival prognosis. In contrast, a number of other studies [39-41] did not observe an association of EGFR amplification with survival in GBM patients. Similarly, Heimberger et al. [42] concluded that overexpression of EGFR and EGFRvIII are not independent predictors of overall survival in a cohort of 54 GBM patients who did not have extensive tumor resection. Analysis of 44 GBM patients by Aldape et al. [43] indicated that EGFRvIII was not predictive of patient survival. It is still unknown whether nuclear EGFRvIII is a prognostic factor for GBM. In other cancer types, high levels of nuclear EGFR predict poor overall survival of patients with breast carcinomas [35], oropharyngeal squamous cell carcinomas [35, 44] and ovarian cancer [45]. Taken together, future investigations are needed to clarify the role of EGFR and EGFRvIII in prognostic prediction of patient survival in malignant gliomas. The predictive value of EGFR and EGFRvIII in EGFR-targeted therapy is discussed in the next section.
EGFR-TARGETED THERAPY IN MALIGNANT GLIOMAS
Five anti-EGFR agents have been approved by the FDA for treating cancer patients, including, three small molecule inhibitors and two antibodies. Chemical structures of the three small molecular weight EGFR inhibitors are listed in Fig. (2). (1) Gefitinib (ZD1839; Iressa) is a small molecular weight EGFR kinase inhibitor that has been approved for locally advanced and metastatic non-small cell lung cancer, NSCLC. (2) Erlotinib (OSI-774; Tarceva), a small molecule EGFR kinase inhibitor, was approved to treat metastatic NSCLC. It has been also approved to be used in combination with gemcitabine for pancreatic cancer that cannot be removed by surgery or has metastasized. (3) Lapatinib (GW572016; Tykerb/Tyverb) is an EGFR/Her-2-dual targeting small molecule inhibitor approved to be combined with other drugs to treat advanced or metastatic breast cancer [46]. It is used in patients whose cancer is Her-2 positive and has failed to respond to other drugs. (4) Cetuximab (C225; Erbitux) is a humanized monoclonal antibody that recognizes the extracellular domain of both EGFR [46] and EGFRvIII [47]. It has been approved for squamous cell carcinoma of the head and neck that has metastasized or recurred after other chemotherapy. It is also used with radiation therapy, as a first-time treatment for advanced squamous cell carcinoma of the head and neck. Cetuximab is also approved for treating metastatic colorectal cancer that has metastasized, after other chemotherapy has failed and for combined used with irinotecan for metastatic colorectal cancer patients who have not responded to irinotecan alone. (5) Panitumumab (ABX-EGF; Vectibix) is a human monoclonal antibody raised against the extracellular domain of EGFR [48]. It has been approved to treat colorectal cancer that has failed other therapies and has metastasized. All these agents, except panitumumab, have been evaluated in phases I & II clinical trials in patients with malignant gliomas. The clinical experiences with these EGFR-targeted therapies are summarized below and in Table 1.
Fig. 2.

Chemical structures of three small molecule EGFR inhibitors.
Table 1.
Overview of Outcome of Clinical Trials Using EGFR-Targeted Therapy in Malignant Gliomas
| Agent | Targets | Phase | Study Design | Outcomes | References |
|---|---|---|---|---|---|
|
Small Molecules Gefitinib (Iressa) |
EGFR | II | single agent 53 recurrent GBM |
6-month EFS: 13% median OS: 39.4 wks median EFS: 8.1 wks No correlation between EGFR and OS or EFS |
Rich 2004 [49] |
| II | single agent 28 GBM, AO & AA |
6-month PFS: 14.3% median OS: 24.6 wks No correlation between EGFR/p-Akt and response |
Franceschi 2007 [50] | ||
| I | gefitinib+sirolimus 34 recurrent GBM & AA |
6-month PFS: 23.5% median PFS: 27.4 wks PR: 14%, SD: 38% |
Reardon 2006 [51] | ||
| I/II | gefitinib+everolimus 22 GBM |
6-month PFS, 4.5% median PFS: 2.6 months median OS: 5.8 months PR: 14%, SD: 38% No correlation between EGFR/PTEN and response |
Kriesl 2009 [52] | ||
| I | gefitinib+TMZ 26 GBM |
Recommendations for phase-2 doses | Prados 2008 [53] | ||
| I | radiosurgery 15 recurrent GBM & AA |
6-month PFS: 63% median PFS: 7 months median OS: 29 months (all pt’s) median OS: 21 months (GBM) |
Schwer 2008 [54] | ||
| Erlotinib (Tarceva) |
EGFR | II | single agent 67 recurrent GBM & AA |
median PFS: 12 wks (GBM) median PFS: 8.6 wks (AA) limited activity as single agent |
Raizer 20041 |
| II | single agent 58 recurrent GBM |
6-month PFS: 17% median OS: 10 months No correlation between EGFR and response |
Cloughesy 20052 | ||
| II | erlotinib+sirolimus 32 recurrent GBM |
6-month PFS: 3.1% negligible activity p-AKT, but not EGFR/EGFRvIII/PTEN correlates with response. |
Reardon 2009 [55] | ||
| I | Arm 1: erlotinib alone Arm 2: erlotinib+TMZ 83 GBM |
Recommendations for phase-2 doses | Prados 2006 [57] | ||
| I/II | erlotinib+TMZ+RT 97 newly diagnosed GBM |
median OS: 15.3 months biomarkers: pt’s not sensitive to er lotinib No correlation between EGFR/EGFRvIII/ PTEN and response |
Brown 2008 [58] | ||
| II | Arm 1: erlotinib alone Arm 2: TMZ or BCNU 110 recurrent GBM |
6-month PFS: 11.4% (Arm 1) 6-month PFS: 24% (Arm 2) Limited activity of erlotinib No correlation between EGFR/EGFRvIII PTEN/p-Akt and response to erlotinib |
Van den Bent 2009 [59] | ||
| II | erlotinib+carboplatin 43 recurrent GBM |
6-month PSF: 14% median PSF: 9 wks median PS: 30 wks No correlation between EGFR/PTEN/ Akt and PFS or OS |
de Groot 2008 [60] | ||
| I | erlotiniib+RT 19 GBM |
median OS: 55 wks | Krishnan 2006 [61] | ||
| II | erlotinib+ bevacizumab 56 recurrent GBM & AA |
6-month PFS: 25% (GBM) 6-month PFS: 50% (AA) Full results to be reported |
Sathornsumetee 20093 | ||
| Lapatinib (Tykerb/Tyverb) |
EGFR/HER2 | I/II | single agent 7 recurrent GBM (I) 17 recurrent GBM (II) |
No significant lapatinib activity No correlation between EG FRvIII/PTEN and response |
Thiessen 2009 [63] |
|
Antibodies Cetuximab (Erbitux) |
EGFR | I/II | cetuximab+RT+TMZ 17 GBM |
6-month PFS: 81% 12-month PFS: 37% 12-month OS: 87% |
Combs 20094 |
| II | single agent Arm 1: 28 GBM with EGFR amplification Arm 2: 27 GBM with no EGFR amplification |
No significant cetuximab activity No correlation between EGFR and response |
Neyns 2009 [64] | ||
| II | cetuximab+ bevacizumab+ irinotecan 32 recurrent GBM |
Response rates similar to bevacizu mab+irinotecan |
Lassen 20085 |
EFS: event-free survival, OS: overall survival, PFS: progression-free survival, PR: partial response, SD: stable disease.
AA: anaplastic astrocytoma, AO: anaplastic oligodendrogliomas, RT: radiation therapy.
Gefitinib (ZD1839; Iressa)
A phase II clinical trial led by Rich et al. [49] examined the effects of gefitinib as single agent in 57 patients with recurrent GBM and found 6-month EFS to be 13%. The median EFS (event-free survival) time was 8.1 week and the median OS (overall survival) was 39.4 weeks. In this unselected cohort, EGFR expression did not correlate with EFS or OS. Another single agent phase II trial conducted by Franceschi et al. [50] reported that 6-month PFS (progression-free survival) was 14.3%, similar to what was reported by Rich et al. [49] and that there was no correlation between EGFR/p-Akt expression and PFS/OS. Gefitinib-based combination therapy has also been tested clinically in patients with malignant gliomas. Reardon et al. [51] conducted a phase I trial consisted of 34 patients with recurrent GBMs and AAs to examine the efficacy of combined uses of gefitinib and sirolimus (mTOR inhibitor). This study reported encouraging results with two patients (6%) achieving a partial radiographic response and 13 patients (38%) having stable disease. Gefitinib has also been combined with another mTOR inhibitor, everolimus, in a recent phase I/II trial led by Kriesl et al. [52]. This trial enrolled 22 GBM patients and reported disappointing results with only one patient with PSF beyond 6 months. Corroborating results from previous trials, this study observed no correlation between EGFR status and patient response. The study did not find a predictive value for PTEN status in the patient cohort.
A phase I study by Prados et al. [53] compared the effects of Iressa alone and Iressa plus TMZ on 26 GBM patients and found the combinations to be generally safe and recommended phase-2 dose of gefitinib when used in combination with TMZ. Furthermore, gefitinib was evaluated in combination with fractionated stereotactic radiosurgery in a phase I clinical trial of 15 patients with recurrent GBMs and AAs [54]. Schwer et al. reported promising results in which 6-month PFS and 1-year OS were 63% and 40%, respectively. The median OS for the 11 GBM patients was 21 months (range, 9-33 months).
Erlotinib (OSI-774; Tarceva)
A 2004 phase II trial led by Raizer et al.1 (meeting abstract) have examined the effects of erlotinib as single agent in 67 patients with recurrent GBMs. This study found erlotinib to have limited activity as a single agent. Another erlotinib single agent phase II trial2 (meeting abstract) enrolled 58 patients with recurrent GBM and reported 6-month PSF of 17%. No correlation between EGFR status and response rates was found in this cohort. Gefitinib has also been used in combination with sirolimus in a recent Phase II trial led by Reardon et al. [55]. This study included 32 patients with recurrent GBM in which 47% of the patients had stable disease. The 6-month PFS was only 3.1% for all patients, but was better for patients not on EIAEDs. The observation with the negative impact of EIAEDs on gefitinib efficacy is consistent with the notions that erlotinib is a substrate of CYP3A [56] and EIAEDs induce CYP3A. These investigators also found no correlation between EGFR/PTEN and patient response, except for p-Akt that was of borderline significance.
Two clinical trials have examined the effects of combination of erlotinib with TMZ in GBM patients. A phase I trial by Prados et al. [57] included 83 GBM patients and reported 6-month PFS to be 10.5%. Co-administration of EIAEDs was found to have reduced exposure to erlotinib, compared to erlotinib alone (33%-71% reduction). Furthermore, Brown et al. [58] conducted a phase I/II trials with 97 newly diagnosed GBM patients and found the cohort to be sensitive to erlotinib, in contrast to the disappointing outcome from patients with recurrent GBMs. Furthermore, a phase II trial by van den Bent et al. [59] was recently completed in 110 patients with recurrent GBMs. This study has compared the effects of erlotinib alone and TMZ (or BCNU). Unfortunately, erlotinib appeared to have insufficient single-agent activity in unselected GBM as indicated by the 6-month PFS of 11.4% in the erlotinib arm, compared to 24% in the other arm with TMZ or BCNU. No clear biomarker was identified to associate with improved outcome to erlotinib. Furthermore, erlotinib was also used in combination with carboplatin to treat 43 recurrent GBM patients in a phase II study led by de Groot et al. [60]. Although this combination was well tolerated but only yielded modest activity in unselected patients with 6-month PFS of 14%. Similar to other reports, no correlation was observed between EGFR/Akt/PTEN expression and PFS/OS.
Krishnan et al. [61] examined the effects of combined use of radiotherapy and erlotinib in 19 GBM patients in a phase I trial. Median survival was 55 weeks. Most recently, Sathornsumetee et al.3 (meeting abstract) completed a single-arm phase II trial to evaluate the efficacy of erlotinib plus bevacizumab (Avastin, anti-VEGF antibody) in 56 patients with recurrent GBMs and AAs. They have reported encouraging outcomes with 6-month PFS of 25% for GBMs and 50% for AAs. It is worth noting that in May 2009, bevacizumab was approved by FDA to treat GBM that have progressed. This approval was based on the promising results of two clinical trials, NCT00345163 conducted by Friedman et al. with 167 patients [62] and a NCI study 06-C-0064E (56 patients). Overall, responses were observed in 20-26% of patients and the median duration of response was approximately 4 months.
Lapatinib (GW572016; Tykerb/Tyverb)
Lapatinib targets both EGFR and Her-2 [46]. The phase I/II trial by Thiessen et al. [63] enrolled a total of 24 patients with recurrent GBM. Accrual was ceased because of the lack of phase II efficacy. Overall, lapatinib did not show significant activity in unselected GBM patients. Lapatinib plasma clearance was increased by approximately ten-fold when given with EIAEDs. Similar to other reports, EGFRvIII and PTEN co-expression did not predict a favorable response. Another phase II trial (NCT00103129) using lapatinib was recently completed in recurrent GBMs and gliosarcomas, and the results are being prepared.
Cetuximab (C225; Erbitux)
Cetuximab is a humanized monoclonal antibody that recognizes the extracellular domain of both EGFR [46] and EGFRvIII [47]. A single-arm phase I/II trial by Combs et al.4 (meeting abstract) evaluated the efficacy of cetuximab+radiation therapy+TMZ combination in 17 GBM patients. The results are encouraging with 6-month PFS of 81%, 12-month PFS of 37% and 12-month OS of 87%. A stratified phase II trial based on EGFR copy number was recently completed by Neyns et al. [64]. In this study with cetuximab as single agent, a total of 55 GBM patients were evaluated in which 28 and 27 patients were with and without an increased EGFR copy number, respectively. However, no significant correlation was found between response, survival and EGFR copy number. Another recently completed phase II trial evaluated the efficacy of cetuximab+bevacizumab+irinotecan combination in 32 patients with recurrent GBMs5 (meeting abstract). In this study, Lassen et al. found the combination to be well tolerated but have similar benefits compared to bevacizumab+irinotecan combination. Further evaluation of this regimen is not planned by the investigators.
THE EFFICIENCY OF EGFR-TARGETED AGENTS IN PENETRATING BLOOD-BRAIN BARRIER (BBB)
Brain tumor chemotherapy often encounters a major hurdle, BBB, which serves as a physical barrier against systemically administered anti-cancer agents. The information with regard to the efficiency of EGFR-targeted agents in penetrating BBB and concentrating in the GBM tumors are limited and somewhat, controversial. This can be due to the following reasons. Although pre-clinical pharmacokinetic studies provided some information using non-tumor bearing animal models, these results do not always corroborate the clinical data. In addition, only a small proportion of the clinical trials in malignant gliomas concurrently conducted pharmacokinetic studies and most of these studies collected blood samples to determine plasma clearance of the agents rather than analyzing drug concentrations in the intracerebral fluids and/or glioma tissues. Therefore, the degrees of anti-EGFR agents in penetrating BBB and accumulating in GBM tumors are often estimated from indirect evidence, such as, the extent of inhibition of EGFR phosphorylation in the tumors, disease progression and patient survival.
Gefitinib
It is a widely accepted concept that compounds with certain biochemical properties, including higher lipophilicity, are expected to penetrate BBB [65]. Since gefitinib is highly water-soluble, it was initially predicted to have a low capacity to penetrate BBB. Consistent with this notion, AstraZeneca initially reported that only very limited amount of radiolabeled [14C]-gefitinib was detected in the CNS in non-tumor-bearing rats. In contrast to these predictions and results, a 2002 study by Heimberger et al. [66] showed that intracranial EGFR-expressing GBM xenografts responded to orally administrated gefitinib. In 2003, another study led by Cappuzzo et al. [67] showed in a total of four NSCLC patients that their brain metastases completely or partially responded to gefitinib treatments. The observed penetration of gefitinib through BBB may likely be the consequences of the altered BBB integrity as a result of intracranial tumors and/or prior exposures to chemotherapy. This speculation is supported by a study conducted by Hofer et al. [68] showing a 10-13-fold higher gefitinib concentrations in the GBM tissues than in plasma from two patients receiving gefitinib treatments and prior first-line chemotherapy. The reasonable extent of gefitinib accumulation in GBM tissues may also be due to the fact that CYP3A, the key cytochrome P450 enzyme that metabolizes gefitinib [56], is expressed at a low level in GBM tissues [68]. Conversely, enzyme-inducing antiepileptic drugs (EIAEDs) induce CYP3A and Reardon et al. [51] reported that GBM exposure to gefitinib was significantly lowered by the concurrent use of EIAEDs. Therefore, EIAEDs-induced inactivation of gefitinib may contribute to the observed lack of clinical efficacy of gefitinib in inhibiting EGFR phosphorylation in GBM tumors [69].
In addition to CYP3A, gefitinib appears to be a substrate of the drug efflux protein p-glycoprotein that is highly expression in BBB6 (meeting report) [70]. Interestingly, it has been reported that gefitinib inhibits the activity of another drug efflux protein, breast cancer resistance protein (BCRP, ABCG2), and reverses the resistance of non-glioma cancer cells to a series of anti-cancer agents, 7-ethyl-10-hydroxycamptothecin (SN-38), topotecan, and mitoxantrone [71]. Together, these findings indicate that gefitinib can penetrate BBB of GBM-carrying animals and GBM patients; however, its overall efficiency in BBB penetration and accumulation in GBM may be compromised by the concurrent use of EIAEDs that induce CYP3A and by drug efflux protein p-glycoprotein that is expressed in BBB.
Erlotinib
There is limited information with regard to the efficiency of erlotinib in crossing BBB. Sakaria et al. [72] reported that erlotinib sensitized intracranial GBM tumors to radiation therapy in nude mice. In the clinical setting, however, Lassman et al. [69] reported that erlotinib showed a very low efficiency in penetrating BBB and in inhibiting tumoric EGFR phosphorylation. This is consistent with the facts that BBB expresses high levels of drug efflux proteins, such as, p-glycoprotein, BCRP and multidrug resistance protein 2 (MRP2; ABCC2) [70, 73]6 (meeting report) and that erlotinib is a substrate of these proteins [70]. Furthermore, erlotinib is also a substrate of CYP3A [56] and this can potentially render erlotinib sensitive to EIAEDs-mediated inactivation. Corroborating this observation, Prado et al. [57] showed that co-administration of EIAEDs reduced the exposure of GBM to erlotinib. Another clinical trial led by van den Bent et al. [59] also demonstrated that the use of EIAEDs significantly increased erlotinib clearance. A recent phase I trial with erlotinib and sirolimus by Reardon et al. [55] reported that PFS was better for GBM patients not on EIAEDs; however, median OS did not differ significantly by EIAED status. Another GBM trial with bevacizumab plus erlotinib reported no survival difference between EIAED and non-EIAED groups3 (meeting abstract).
Lapatinib, Cetuximab, and Panitumumab
Very little is known about the extent to which these three agents penetrate BBB. The phase I/II trial by Thiessen et al. [63] concluded that lapatinib did not show significant activity in GBM patients and that lapatinib clearance was significantly increased by the concurrent use of EIAEDs. Corroborating this observation, lapatinib has been shown to be a substrate of drug efflux proteins within BBB, including, p-glycoprotein and BCRP [74, 75]. For cetuximab, Eller et al. [76] showed that intracranially grown GBM xenografts responded to systemically administered cetuximab treatments. In this animal study, radiation therapy was observed to augment the efficacy of cetuximab. Thus far, no information is available on the ability of panitumumab to cross BBB, despite a study [77] reported that panitumumab, in combination with AMG 102 (a HGF neutralizing antibody), was effective in targeting subcutaneous GBM xenografts.
MOLECULAR MECHANISMS UNDERLYING GLIOMA RESISTANCE TO EGFR-TARGETED THERAPY
Although the gain-of-function mutations within the EGFR kinase domain is commonly found in lung cancer [78, 79] and lead to their hyper-sensitivity to EGFR inhibitors, these somatic mutations are not present in gliomas [80]. Thus, extensive investigations are being directed at identifying other mechanisms that may account for GBM resistance to EGFR-targeted therapy. One of the key directions has been focused on the PTEN-Akt-mTOR signaling axis. The tumor suppressor gene PTEN is frequently mutated and/or deleted in GBMs and consequently, renders the PI3-K downstream effectors (Akt and mTOR) hyperactive in these tumors. Accumulating evidence suggests EGFRvIII and PTEN co-expression to be a predictor for the responsiveness of GBM to EGFR inhibitory agents [5]. However, there are also reports showing a lack of correlation between EGFRvIII-PTEN status and GBM response to the therapy, suggesting other resistance factors may also be involved. For example, several studies have provided evidence supporting the notion that inhibition of a dominant oncogene, such as EGFR/EGFRvIII, by targeted therapy can alter the hierarchy of RTKs and non-receptor TKs resulting in the activation of other TKs, such as, c-mesenchymal-epithelial transition factor (c-Met), platelet-derived growth factor receptor (PDGFR) and JAK2, in order to facilitate tumor survival [77, 81, 82]. Another potential mechanism of resistance can be derived from HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway that produces metabolites to activate EGFR signaling [83, 84]. Our laboratory has recently shown that the JAK2-STAT3 pathway is constitutively activated in the majority of GBMs and that STAT3 undergoes multi-level interactions with EGFR, leading to the resistance of GBM cells to Iressa [81]. Detailed discussions for each of above mentioned mechanisms are provided below.
PTEN-PI3-K-Akt-mTOR Signaling Axis
GBMs commonly contain mutations and deletion of the PTEN tumor suppressor gene [85]. The PTEN gene encodes a lipid phosphatase that metabolizes phosphatidylinositol 3,4,5-trisphosphate, the product of PI3-K, and thereby antagonizes PI3K-mediated signaling pathway. Consistent with this notion, loss of PTEN correlates with increased activity of PI3-K’s downstream effector Akt and the substrate of Akt, mTOR (mammalian target of rapamycin) [86].
Emerging evidence indicates that PTEN expression is a molecular determinant of the response of EGFRvIII-expressing GBMs to EGFR kinase inhibitors. In the study led by Mellinghoff et al. [5] which contained a total of 82 patients from two institutes, recurrent malignant gliomas co-expressing EGFRvIII and PTEN demonstrated significantly better clinical response to erlotinib. This clinical association was confirmed using in vitro experiments with cultured GBM cells [5]. In agreement with this finding, a phase I trial led by Haas-Kogan et al. [87] reported that GBMs with high levels of EGFR and low levels of phosphorylated Akt had a better response to erlotinib. Furthermore, Sarkaria et al. [88] showed in serially passaged GBM xenografts that erlotinib-sensitive tumors commonly express PTEN and amplified EGFR. In contrast to these observations, a phase II trial with GBM patients reported that the expression of PTEN was not associated with patient response to erlotinib while low p-Akt expression was of borderline significance to an improved outcome [59]. Results of another phase I/II trial showed that EGFRvIII and PTEN co-expression did not predict better responsiveness of GBM patients to lapatinib and that lapatinib did not yield significant activity in GBM patients [63].
Wang et al. [89] reported that rapamycin enhanced the sensitivity of PTEN-deficient GBM cells to erlotinib treatment, providing a rationale for the combination of mTOR and EGFR kinase inhibitors in treating GBM with PTEN deficiency. In light of this interesting in vitro observation, the efficacy of this combination has been evaluated clinically in GBM patients. Results of a recent phase II clinical trial of erlotinib plus sirolimus showed that the combination was well tolerated in adult patients with heavily pre-treated, recurrent GBM, but unfortunately, yielded negligible activity among these unselected GBM patients [55]. This study led by Reardon et al. also reported that tumor markers, including, EGFR, EGFRvIII, and PTEN failed to show an association with PFS, except for increased p-Akt expression which only achieved borderline significance. Taken together, these observations clearly indicate that the role of PTEN expression as a determinant of GBM responsiveness to EGFR-targeted therapy is still unclear and that the combination of mTOR- and EGFR-targeted therapies is in need of improvement.
c-Met and PDGFR
The c-Met RTK has been shown to be co-activated in GBM cells with increased levels of EGFR/EGFRvIII [23, 77, 82]. This co-expression/co-activation has been shown to be a result of transcription-independent and -dependent mechanisms. In a transcription-independent fashion, activated EGFR associates with c-Met and the association facilitates c-Met phosphorylation in the absence of its only known ligand, HGF [90]. In support of this finding, c-Met is constitutively phosphorylated in the absence of HGF in human cancer cells. On the other hand, HGF transcriptionally activates the expression of EGFR ligands, TGF-α and heparin binding-EGF, leading to EGFR activation [91].
Combination of the c-Met inhibitor (SU11272) and erlotinib has been shown to yield significantly higher anti-proliferative effects than single agents on GBM cells with c-Met/EGFR co-activation [23, 82]. Using xenograft models, Lal et al. [92] showed that a neutralizing anti-HGF monoclonal antibody (L2G7) synergizes with erlotinib to inhibit the growth of the PTEN-null/HGF(+)/c-Met(+)/EGFRvIII(+) U87MG GBM tumors. Similar positive results were reported by Pillary et al. [77] using the humanized HGF-specific antibody AMG 102 (Amgen) in combination with panitumumab. The AMG 102/479-panitumumab combination will be evaluated in a phase I trial in colon cancer, colorectal cancer, gastrointestinal cancer, metastatic colorectal cancer and rectal cancer with wild-type KRAS (clinical trial # NCT00788957; http://ClinicalTrials.gov).
It is worth noting that there are several other anti-HGF and anti-c-Met agents that are under various phases of clinical trials in different cancer types. For example, the c-Met kinase inhibitor foretinib (GSK1363089; XL880; GSK) is being evaluated in papillary renal cell carcinoma, metastatic gastric cancer and hepatocellular carcinoma (NCT00920192). Another c-Met kinase inhibitor, PF-02341066 by Pfizer, will be tested in adults with lymphoma and in young patients with CNS cancers and large cell lymphoma (clinical trial # NCT00939770). AMG 208 (Amgen), a small molecule inhibitor of c-Met, will be evaluated in a phase I trial in solid tumors (clinical trial # NCT00813384). Interestingly, concurrent activation of c-Met and PDGFR appears to be a frequent event in GBM and this has been suspected to be a mechanism for GBM resistance to EGFR kinase inhibitors [23, 82]. Stommel et al. [82] showed that the combination of erlotinib, SU11274 (c-Met inhibitor) and imatinib (Gleevec; abl-PDGFR inhibitor) significantly inhibits the in vitro growth of GBM cells, compared to single agents and dual-drug combinations.
HMG-CoA Reductase
HMG-CoA (3-hydroxy-3-mehylglutaryl CoA) reductase is the rate-limiting step of the mevalonate pathway that catalyzes the conversion HMG-CoA to mevalonate [93]. The mevalonate pathway produces end products, such as, dolichol, cholesterol, geranylgeranyl pyrophosphate and farnesyl pyrophosphate that can directly affect EGFR activity, as well as, indirectly modulate the activity of EGFR-mediated downstream molecules [84]. Dolichol is involved in N-linked glycosylation of several RTKs [94] and the ligand-binding domain of EGFR is glycosylated to allow for ligand-binding, cell-surface localization and proper conformation [95]. Cholesterol modulates EGFR kinase activity [83]. Finally, the EGFR downstream signaling molecule, Ras, is post-translationally modified by geranylgeranyl transferase and farnesyl transferase that use geranylgeranyl pyrophosphate and farnesyl pyrophosphate, respectively. Although the levels of HMG-CoA reductase in tumors have not been shown to be elevated compared to normal tissues, the growth of some cancer cells appears to be more dependent on the metabolites of the mevalonate pathway. This is likely due to the fact that cancer cells, but not normal tissues, frequently express high levels of EGFR and Ras, whose activity is enhanced by these metabolites.
Inhibitors that target HMG-CoA reductase, also known as statins used to reduce cholesterol levels, have been shown to demonstrate anti-cancer activity [96]. For example, a Japanese-conducted randomized control trial of 81 patients with hepatocellular carcinoma showed that the HMG-CoA reductase inhibitor, pravastatin, prolonged the survival of 5-FU-treated patients from 9 months to 18 months [94]. However, no significant benefit was reported in a phase I/II trial led by Larner et al. [97] using lovastatin in patients with malignant gliomas. While the mechanism underlying statins-mediated anti-cancer effects remain unclear, statins are known to inhibit EGFR autophosphorylation and thereby, target EGFR-expressing cancer cells. For example, several studies reported that combined targeting of HMG-CoA reductase and EGFR yielded synergistic killing effects in several cancer types, including GBM [98]. Combination of lovastatin and EGFR kinase inhibitory agents, AG1478 and Iressa, led to growth-inhibitory effects on breast cancer, colon carcinoma and NSCLC [99, 100]. The combination of cetuximab and the HMG-CoA reductase inhibitor (fluvastatin) suppressed the growth of hepatocellular cancer [101]. Lovastatin significantly enhanced the sensitivity of GBM cells to gefitinib [98]. The synergistic effects of Iressa with lovastatin were observed in GBM cells with EGFR or EGFRvIII expression independent of PTEN status, thereby providing a rationale for combining HMG-CoA reductase- and EGFR-targeted therapies as a novel therapy for GBM [98].
STAT3 IN GBM RESISTANCE TO EGFR-TARGETED THERAPY
STAT3 and Oncogenesis
STAT3 is a transcription factor that has been shown to induce oncogenesis of normal fibroblasts [102] and cancers of the prostate [103] and skin [104, 105]. STAT3 also transforms mouse bone marrow cells into highly aggressive T cell leukemia in mice [106]. In contrast, activated STAT3 has been shown to suppress c-myc-, but not RasV12-mediated malignant transformation of mouse embryonic fibroblasts [106]. In normal brain, the role of STAT3 as an oncogene appears to depend on genetic status of PTEN and EGFR [37]. In PTEN-proficient mouse astrocytes, STAT3 behaves as a tumor suppressor and conversely, dual-suppression of PTEN and STAT3 leads to their malignant transformation. In contrast to the tumor suppressive role in PTEN-positive astrocytes, STAT3 promotes EGFRvIII-induced glial transformation by forming a complex with EGFRvIII in the nucleus [37]. The oncogenic role of STAT3 in gliomas is further supported by the notion that STAT3 activation is rarely detected in normal brain tissues [81, 107].
STAT3 in Human Cancers
In cancerous cells, STAT3 activation has been consistently shown to associate with more malignant cancer biology and poor prognosis [28, 81, 107-110]. STAT3 is highly activated in malignant gliomas [81, 107] and the extent of STAT3 activation correlates with glioma grade [81]. STAT3 can be activated via phosphorylation at Y705 and/or S727. As described in Fig. (3a), STAT3 Y705 is directly phosphorylated by RTKs (EGFR and EGFRvIII) and non-receptor TK (JAK2). Inactive JAK2 is constitutively bound to G-protein coupled receptors (IL-R, LIF-R, gp130) and autophosphorylates upon receptor activation. In addition, JAK2 can be phosphorylated at Y1007/1008 directly by RTKs (EGFR/EGFRvIII and PDGFR) and non-receptor TK, Src [111, 112]. As described in Fig. (3b), in addition to Y705 phosphorylation, STAT3 can be activated via S727 phosphorylation through EGF- and IL-6-dependent mechanisms [103, 113-116]. Despite both Y705 and S727 phosphorylation can activate the transcriptional activity of STAT3, little information is available with regard to the relationship between p-STAT3 (Y705) and p-STAT3 (S727). Interestingly, a recent study by Qin et al. [103] showed that activation of STAT3 through a phosphomimetic S727 promotes prostate tumorigenesis independent of Y705 phosphorylation.
Fig. 3. Signaling pathways that lead to STAT3 activation.
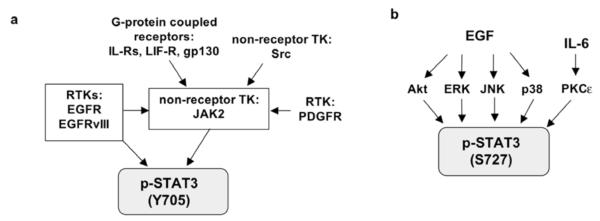
a. STAT3 can be phosphorylated at Y705 directly by RTKs (EGFR and EGFRvIII) and non-receptor TK (JAK2) and becomes activated. Inactive JAK2 is constitutively bound to the G-protein coupled receptors (IL-R, LIF-R, gp130) and becomes auto-phosphorylated at Y1007/1008 upon receptor activation. In addition, JAK2 can be phosphorylated at Y1007/1008 directly by RTKs (EGFR/EGFRvIII and PDGFR) and non-receptor TK, Src.
b. STAT3 activation via S727 phosphorylation can be initiated by stimulation of EGF (via Akt, ERK, JNK and p38) and IL-6 (via PKCε).
As summarized in Fig. (4), activated STAT3 dimerizes and translocates into the nucleus to activate expression of genes that are important for G1 cell cycle progression (cyclin D1), oncogenesis (c-Myc, c-fos and iNOS), anti-apoptosis (Bcl-XL, Mcl-1, pim-1), EMT (TWIST), metastasis (MMP-1/2), angiogenesis (VEGF and iNOS) [28, 117-123] and immuno-suppression (IL-23) [124]. Together, these findings clearly indicate that STAT3 is highly activated in many human cancers, including, malignant gliomas and that it is an important molecule that converges signals of several pathways and mediates many important cellular processes.
Fig. 4.
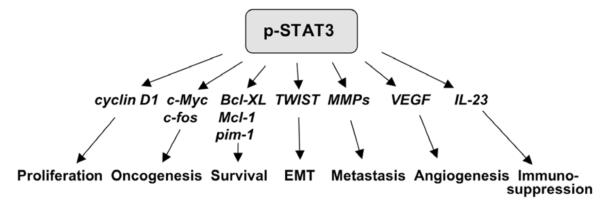
Activated STAT3 modulates expression of many important genes involved in oncogenesis, as well as, various important cellular processes in human cancers.
STAT3/JAK2 Inhibitors
Given the pivotal and central role that STAT3 plays in many human cancers, STAT3 has emerged as a major molecular target for cancer therapy [125]. Several anti-STAT3 agents are under pre-clinical and clinical evaluation, and can be classified into two major categories: (1) direct STAT3 inhibitors and (2) indirect inhibitors that target STAT3’s upstream activating kinase, JAK2. For example, STA-21 small molecule compound directly inhibits STAT3 and targets breast cancer cells in vitro [126]. Platinum compounds, IS3 295 and CPA-7, directly inhibit STAT3’s DNA-binding ability, in which CPA-7 demonstrates anti-metastasis activity toward colon tumors [127]. Another direct STAT3 inhibitor, STAT3 decoy, is consisted of double-stranded decoy oligodeoxynucleotides which closely correspond to the STAT3 binding site within the c-fos promoter. STAT3 decoy shows promising anti-tumor effects in head and neck squamous cell carcinomas [128] and GBM cells [129]. Indirect STAT3 inhibitors JSI-124, AG490 and WP1066 target JAK2 and suppress the growth of GBM cells and/or xenografts [81, 130, 131]. LLL3, a structural analogue of STA-21, showed anti-GBM activity [132]. INCB018424 (Incyte), an orally active small molecular weight JAK2 inhibitor, has been examined in metastatic prostate cancer and the results are forthcoming (clinical trial # NCT00638378). INCB018424 is being evaluated in clinical trials for multiple myeloma (clinical trial # NCT00639002). A phase 0 clinical trial is recruiting head and neck cancer patients to determine the effects of STAT3 decoy (clinical trial # NCT00696176). Three phase I trials are enrolling patients with relapsed/refractory non-Hodgkin’s lymphoma or multiple myeloma (NCT00511082) and with advanced solid tumors (NCT00955812) to evaluate the efficacy of a STAT3 small molecule inhibitor OPB-31121.
EGFR-STAT3 Interactions in GBM Resistance to EGFR-targeted Therapy
EGFR physically interacts and functionally cooperates with STAT3, at both cytoplasmic and nuclear levels. At the cytoplasmic level, via the two docking autophosphorylated tyrosines (Y1068 and Y1086), cell-surface EGFR interacts with STAT3 SH2 domain [133]. This interaction leads to phosphorylation of STAT3 at Y705 and its activation. Cell-surface EGFRvIII also interacts with and phosphorylates STAT3. Importantly, we and others showed in cancers of breast, colon and skin that cell-surface EGFR cooperates with STAT3 to induce expression of TWIST (to facilitate EMT), VEGF (to promote angiogenesis) and Eme1 endonuclease (to reduce drug-induced DNA damage) [121, 123, 134]. In primary breast carcinomas, co-expression of EGFR and activated STAT3 (Y705) is frequent, 39% [123]. In primary gliomas, the extent of concurrent EGFR/EGFRvIII expression and STAT3 activation was also a frequent event and positively correlates with glioma grade [81]. The few reports with respect to the ability of EGFR versus EGFRvIII to activate STAT3 in GBM have shown rather inconsistent results. For example, a study reported that the PI3K pathway is dominant over the MAPK and STAT3 pathways in GBM with a high level of EGFRvIII expression [23]. However, another study reports that STAT3 is more activated in EGFRvIII-carrying GBM than those with EGFR and mixed expression [135].
At the nuclear level, EGFR interacts with STAT3 to activate expression of iNOS gene in carcinomas of breast and epidermoid [28]. Nuclear EGFRvIII interacts with STAT3 in normal astrocytes and such interaction contributes to their malignant transformation into glioma [37]. It is speculated that nuclear EGFRvIII-STAT3 interaction involves the tyrosine kinase function of EGFRvIII, albeit the exact effect of nuclear EGFRvIII on STAT3 remains unknown. Most recently, we found nuclear EGFR-EGFRvIII and nuclear STAT3 cooperate to activate expression of pro-inflammatory gene, COX-2, in malignant gliomas [38]. Together, these findings indicate that EGFR/EGFRvIII and STAT3 pathways significantly interact at multiple levels, leading to gene activation and more aggressive tumor behaviors.
The multi-level interactions between EGFR and STAT3 emerge as a potential mechanism underlying the resistance of GBM to EGFR-targeted therapy. In primary specimens and cancer cell lines, STAT3 activation is paradoxically sustained when EGFR is inhibited [28, 81, 136]. The STAT3-activating kinase, JAK2, is activated in GBM cell lines and combined inhibition of JAK2 and EGFR/EGFRvIII abolishes STAT3 activation and synergistically suppresses the growth of EGFR- and EGFRvIII-expressing cell lines of breast cancer [28] and epidermoid carcinoma [28, 137], and GBM [81]. These encouraging in vitro observations provide a rationale to evaluate the efficacy of combination of EGFR and STAT3/JAK2 inhibitors in targeting GBM in vivo.
CONCLUDING REMARKS
GBM is the most common brain cancer in adults and unfortunately, is also the most aggressive type and the least responsive to various therapies. Overexpression of EGFR and/or EGFRvIII is frequently found in GBM and is generally associated with more malignant phenotype and poor clinical outcome. Consequently, EGFR-targeted therapy emerges as a promising anti-GBM therapy. However, the clinical efficacy of EGFR-targeted therapy has been only modest in GBM patients. Although intrinsic drug resistance is known to be a major obstacle for EGFR-targeted therapy, the underlying mechanisms are still poorly understood, despite extensive investigations are being conducted to shed light on these mechanisms. Experiences drawn from clinical trials indicate that mono and combination EGFR-targeted therapies encountered many challenges, including, insufficient penetration through BBB, drug inactivated induced by concurrent uses of EIAEDs, drug efflux at BBB, inability to inhibit tumoric EGFR kinase activity, and the lack of a consistent association between biomarkers and patient response. Therefore, it remains an important task to better our understanding of the complex and interactive nature of the EGFR- and EGFRvIII-mediated signaling networks, to identify the alternative signaling pathways that GBMs activate while the EGFR activity is inhibited by EGFR-targeted agents, and to identify other underlying mechanisms in order to improve the observed modest efficacy of EGFR-targeted therapy in GBM patients.
ACKNOWLEDGEMENTS
This study was supported by NIH grants (5K01-CA118423 and 2P50-NS020023-26), the Elsa U Pardee Foundation and the Pediatric Brain Tumor Foundation (to H.-W. L).
ABBREVIATIONS
- AA
anaplastic astrocytoma
- AO
anaplastic oligodendroglioma
- BBB
blood-brain barrier
- BCRP
breast cancer resistance protein
- BCNU
bis-chloronitrosourea
- B-Myb
B-myeloblastosis viral oncogene homolog
- CNS
central nervous system
- c-Met
c-mesenchymal-epithelial transition factor
- COX-2
cyclooxygenase-2
- DNA-PK
DNA-dependent protein kinase
- EIAED
enzyme-inducing antiepileptic drug
- EFS
event-free survival
- EGFR
epidermal growth factor receptor
- EGFRvIII
epidermal growth factor receptor variant III
- EMT
epithelial-mesenchymal transition
- E2F1
electro-acoustic 2 factor 1
- GLI1
glioma-associated oncogene homologue 1
- GBM
glioblastoma multiforme
- HGF
hepatocyte growth factor
- HMG-CoA
3-hydroxy-3-mehylglutaryl CoA
- IL-6
interleukin-6
- iNOS
inducible nitric oxide synthase
- JAK2
Janus-activated kinase 2
- Mcl-1
myeloid cell leukemia sequence 1
- MMP
matrix metalloproteinase
- MRP
multidrug resistance protein
- mTOR
mammalian target of rapamycin
- NSCLC
non-small cell lung cancer
- OS
overall survival
- PCNA
proliferating cell nuclear antigen
- PDGFR
platelet-derived growth factor receptor
- PFS
progression-free survival
- PR
partial response
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- RTK
receptor tyrosine kinase
- SD
stable disease
- STAT3
signal transducer and activator of transcription 3
- STAT35
signal transducer and activator of transcription 5
- TGF-α
tumor transforming factor-α
- TMZ
temozolomide
- VEGF
vascular endothelial growth factor
REFERENCES
- [1].Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat. Clin. Practice. 2006;2:494–503. doi: 10.1038/ncpneuro0289. quiz 491 p following 516. [DOI] [PubMed] [Google Scholar]
- [2].Selznick LA, Shamji MF, Fecci P, Gromeier M, Friedman AH, Sampson J. Molecular strategies for the treatment of malignant glioma--genes, viruses, and vaccines. Neurosurg. Rev. 2008;31:141–155. doi: 10.1007/s10143-008-0121-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- [4].Sandsmark DK, Pelletier C, Weber JD, Gutmann DH. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol. Histopathol. 2007;22:895–903. doi: 10.14670/HH-22.895. [DOI] [PubMed] [Google Scholar]
- [5].Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JHY, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- [6].Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A Novel Splice Variant of GLI1 That Promotes Glioblastoma Cell Migration and Invasion. Cancer Res. 2009;69:6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Friedman HS, Bigner DD. Glioblastoma multiforme and the epidermal growth factor receptor. N. Engl. J. Med. 2005;353:1997–1999. doi: 10.1056/NEJMp058186. [DOI] [PubMed] [Google Scholar]
- [8].Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol. Cancer Ther. 2007;6:1909–1919. doi: 10.1158/1535-7163.MCT-07-0047. [DOI] [PubMed] [Google Scholar]
- [9].Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin. Immunol. 2008;20:267–75. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Karpel-Massler G, Schmidt U, Unterberg A, Halatsch M-E. Therapeutic Inhibition of the Epidermal Growth Factor Receptor in High-Grade Gliomas: Where Do We Stand? Mol. Cancer Res. 2009;7:1000–1012. doi: 10.1158/1541-7786.MCR-08-0479. [DOI] [PubMed] [Google Scholar]
- [11].Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur. J. Cancer. 2001;3(Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- [13].Yamazaki H, Ohba Y, Tamaoki N, Shibuya M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn. J. Cancer Res. 1990;81:773–779. doi: 10.1111/j.1349-7006.1990.tb02644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA. 1992;89:4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS. Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- [18].Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- [19].Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Carpenter G, Liao HJ. Trafficking of receptor tyrosine kinases to the nucleus. Exp. Cell. Res. 2009;315:1556–1566. doi: 10.1016/j.yexcr.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cohen S, Carpenter G, King L., Jr. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J. Biol. Chem. 1980;255:4834–4842. [PubMed] [Google Scholar]
- [22].Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, White FM. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA. 2007;104:12867–12872. doi: 10.1073/pnas.0705158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Prigent SA, Nagane M, Lin H, Huvar I, Boss GR, Feramisco JR, Cavenee WK, Huang HS. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J. Biol. Chem. 1996;271:25639–25645. doi: 10.1074/jbc.271.41.25639. [DOI] [PubMed] [Google Scholar]
- [25].Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol. Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- [26].Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang WC. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–4351. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell. Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- [28].Lo H-W, Hsu S-C, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih J-Y, Hung M-C. Nuclear Interaction of EGFR and STAT3 in the Activation of iNOS/NO Pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- [29].Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell. Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- [30].Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- [31].Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother. Oncol. 2005;76:157–161. doi: 10.1016/j.radonc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- [32].Cao H, Lei ZM, Bian L, Rao CV. Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta. Endocrinology. 1995;136:3163–3172. doi: 10.1210/endo.136.7.7540549. [DOI] [PubMed] [Google Scholar]
- [33].Klein C, Gensburger C, Freyermuth S, Nair BC, Labourdette G, Malviya AN. A 120 kDa nuclear phospholipase Cgamma1 protein fragment is stimulated in vivo by EGF signal phosphorylating nuclear membrane EGFR. Biochemistry. 2004;43:15873–15883. doi: 10.1021/bi048604t. [DOI] [PubMed] [Google Scholar]
- [34].Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol. Biol. Cell. 2007;18:1064–1072. doi: 10.1091/mbc.E06-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lo H-W, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung M-C. Novel prognostic value of nuclear EGF receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- [36].Liao HJ, Carpenter G. Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor. Cancer Res. 2009;69:6179–6183. doi: 10.1158/0008-5472.CAN-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lo HW, Cao X, Zhu H, Ali-Osman F. COX-2 is a Novel Transcriptional Target of the Nuclear EGFR-STAT3 and EGFRvIII-STAT3 Signaling Axes. Mol Cancer Res. 2010 doi: 10.1158/1541-7786.MCR-09-0391. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Quan AL, Barnett GH, Lee SY, Vogelbaum MA, Toms SA, Staugaitis SM, Prayson RA, Peereboom DM, Stevens GH, Cohen BH, Suh JH. Epidermal growth factor receptor amplification does not have prognostic significance in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2005;63:695–703. doi: 10.1016/j.ijrobp.2005.03.051. [DOI] [PubMed] [Google Scholar]
- [40].Zhou YH, Tan F, Hess KR, Yung WK. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: relationship to tumor grade and survival. Clin. Cancer Res. 2003;9:3369–3375. [PubMed] [Google Scholar]
- [41].Newcomb EW, Cohen H, Lee SR, Bhalla SK, Bloom J, Hayes RL, Miller DC. Survival of patients with glioblastoma multiforme is not influenced by altered expression of p16, p53, EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 1998;8:655–667. doi: 10.1111/j.1750-3639.1998.tb00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005;3:38. doi: 10.1186/1479-5876-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Aldape KD, Ballman K, Furth A, Buckner JC, Giannini C, Burger PC, Scheithauer BW, Jenkins RB, James CD. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004;63:700–707. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- [44].Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, Rimm D, Burtness BA. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin. Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- [45].Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, Huo LF, Miller S, Hung MC. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol. Carcinog. 2009;48:610–617. doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moy B, Kirkpatrick P, Kar S, Goss P. Lapatinib. Nature reviews. 2007;6:431–432. doi: 10.1038/nrd2332. [DOI] [PubMed] [Google Scholar]
- [47].Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ, Kang X. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27:3355–3366. [PubMed] [Google Scholar]
- [48].Rivera F, Vega-Villegas ME, Lopez-Brea MF, Marquez R. Current situation of Panitumumab, Matuzumab, Nimotuzumab and Zalutumumab. Acta oncologica (Stockholm, Sweden) 2008;47:9–19. doi: 10.1080/02841860701704724. [DOI] [PubMed] [Google Scholar]
- [49].Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE, 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004;22:133–142. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- [50].Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crino L, Brandes AA. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br. J. Cancer. 2007;96:1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, Sathornsumetee S, Herndon JE, 2nd, Dowell JM, McLendon RE, Provenzale JM, Sampson JH, Smith RP, Swaisland AJ, Ochs JS, Lyons P, Tourt-Uhlig S, Bigner DD, Friedman HS, Rich JN. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin. Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- [52].Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J. Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- [53].Prados MD, Yung WK, Wen PY, Junck L, Cloughesy T, Fink K, Chang S, Robins HI, Dancey J, Kuhn J. Phase-1 trial of gefitinib and temozolomide in patients with malignant glioma: a North American brain tumor consortium study. Cancer Chemother. Pharmacol. 2008;61:1059–1067. doi: 10.1007/s00280-007-0556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schwer AL, Damek DM, Kavanagh BD, Gaspar LE, Lillehei K, Stuhr K, Chen C. A phase I dose-escalation study of fractionated stereotactic radiosurgery in combination with gefitinib in patients with recurrent malignant gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2008;70:993–1001. doi: 10.1016/j.ijrobp.2007.07.2382. [DOI] [PubMed] [Google Scholar]
- [55].Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, 2nd, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol. 2009 Jun 28; doi: 10.1007/s11060-009-9950-0. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin. Cancer Res. 2007;13:3731–3737. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- [57].Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M, Kapadia A, Rabbitt J, Page MS, Fedoroff A, Xie D, Kelley SK. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro. Oncol. 2006;8:67–78. doi: 10.1215/S1522851705000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Brown PD, Krishnan S, Sarkaria JN, Wu W, Jaeckle KA, Uhm JH, Geoffroy FJ, Arusell R, Kitange G, Jenkins RB, Kugler JW, Morton RF, Rowland KM, Jr., Mischel P, Yong WH, Scheithauer BW, Schiff D, Giannini C, Buckner JC. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008;26:5603–5609. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].de Groot JF, Gilbert MR, Aldape K, Hess KR, Hanna TA, Ictech S, Groves MD, Conrad C, Colman H, Puduvalli VK, Levin V, Yung WK. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J. Neurooncol. 2008;90:89–97. doi: 10.1007/s11060-008-9637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Krishnan S, Brown PD, Ballman KV, Fiveash JB, Uhm JH, Giannini C, Jaeckle KA, Geoffroy FJ, Nabors LB, Buckner JC. Phase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: results of North Central Cancer Treatment Group protocol N0177. Int. J. Radiat. Oncol. Biol. Phys. 2006;65:1192–1199. doi: 10.1016/j.ijrobp.2006.01.018. [DOI] [PubMed] [Google Scholar]
- [62].Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- [63].Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L, Eisenhauer E. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2009 Jun 5; doi: 10.1007/s00280-009-1041-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [64].Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, D’Hondt L, Strauven T, Chaskis C, In’t Veld P, Michotte A, De Greve J. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009;20:1596–1603. doi: 10.1093/annonc/mdp032. [DOI] [PubMed] [Google Scholar]
- [65].Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- [66].Heimberger AB, Learn CA, Archer GE, McLendon RE, Chewning TA, Tuck FL, Pracyk JB, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Brain Tumors in Mice Are Susceptible to Blockade of Epidermal Growth Factor Receptor (EGFR) with the Oral, Specific, EGFR-Tyrosine Kinase Inhibitor ZD1839 (Iressa) Clin. Cancer Res. 2002;8:3496–3502. [PubMed] [Google Scholar]
- [67].Cappuzzo F, Ardizzoni A, Soto-Parra H, Gridelli C, Maione P, Tiseo M, Calandri C, Bartolini S, Santoro A, Crino L. Epidermal growth factor receptor targeted therapy by ZD 1839 (Iressa) in patients with brain metastases from non-small cell lung cancer (NSCLC) Lung Cancer (Amsterdam, Netherlands) 2003;41:227–231. doi: 10.1016/s0169-5002(03)00189-2. [DOI] [PubMed] [Google Scholar]
- [68].Hofer S, Frei K, Rutz HP. Gefitinib accumulation in glioblastoma tissue. Cancer Biol. Ther. 2006;5:483–484. doi: 10.4161/cbt.5.5.2653. [DOI] [PubMed] [Google Scholar]
- [69].Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, Kuhn JG, Wilson R, Nowak NJ, Cowell JK, DeAngelis LM, Wen P, Gilbert MR, Chang S, Yung WA, Prados M, Holland EC. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin. Cancer Res. 2005;11:7841–7850. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- [70].Ozvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist. Updat. 2005;8:15–26. doi: 10.1016/j.drup.2005.02.002. [DOI] [PubMed] [Google Scholar]
- [71].Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein-mediated drug resistance. Mol. Cancer Ther. 2004;3:1119–1125. [PubMed] [Google Scholar]
- [72].Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin. Cancer Res. 2006;12:2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- [73].Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JH. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1−/−/Mdr1a/1b−/− (triple-knockout) and wild-type mice. Mol. Cancer Ther. 2008;7:2280–2287. doi: 10.1158/1535-7163.MCT-07-2250. [DOI] [PubMed] [Google Scholar]
- [74].Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, John-Williams LS, Koch KM, Serabjit-Singh CJ. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethy l]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metabol Dispos.: the biological fate of chemicals. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- [75].Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metabol Dispos.: the biological fate of chemicals. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- [76].Eller JL, Longo SL, Kyle MM, Bassano D, Hicklin DJ, Canute GW. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56:155–162. doi: 10.1227/01.neu.0000145865.25689.55. discussion 162. [DOI] [PubMed] [Google Scholar]
- [77].Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009;11:448–458. doi: 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- [80].Marie Y, Carpentier AF, Omuro AM, Sanson M, Thillet J, Hoang-Xuan K, Delattre JY. EGFR tyrosine kinase domain mutations in human gliomas. Neurology. 2005;64:1444–1445. doi: 10.1212/01.WNL.0000158654.07080.B0. [DOI] [PubMed] [Google Scholar]
- [81].Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively Activated STAT3 Frequently Coexpresses with Epidermal Growth Factor Receptor in High-Grade Gliomas and Targeting STAT3 Sensitizes Them to Iressa and Alkylators. Clin. Cancer Res. 2008;14:6042–6054. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- [83].Ringerike T, Blystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J. Cell. Sci. 2002;115:1331–1340. doi: 10.1242/jcs.115.6.1331. [DOI] [PubMed] [Google Scholar]
- [84].Dimitroulakos J, Lorimer IA, Goss G. Strategies to enhance epidermal growth factor inhibition: targeting the mevalonate pathway. Clin. Cancer Res. 2006;12:4426s–4431s. doi: 10.1158/1078-0432.CCR-06-0089. [DOI] [PubMed] [Google Scholar]
- [85].Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- [86].Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- [87].Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- [88].Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, James CD. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol. Cancer Ther. 2007;6:1167–1174. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- [89].Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian Target of Rapamycin Inhibition Promotes Response to Epidermal Growth Factor Receptor Kinase Inhibitors in PTEN-Deficient and PTEN-Intact Glioblastoma Cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- [90].Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000;275:8806–8811. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- [91].Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, Laterra J. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol. Cancer Res. 2008;6:139–150. doi: 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lal B, Goodwin CR, Sang Y, Foss CA, Cornet K, Muzamil S, Pomper MG, Kim J, Laterra J. EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol. Cancer Ther. 2009;8:1751–1760. doi: 10.1158/1535-7163.MCT-09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- [94].Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y, Matsuzawa Y. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br. J. Cancer. 2001;84:886–891. doi: 10.1054/bjoc.2000.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Carlberg M, Dricu A, Blegen H, Wang M, Hjertman M, Zickert P, Hoog A, Larsson O. Mevalonic acid is limiting for N-linked glycosylation and translocation of the insulin-like growth factor-1 receptor to the cell surface. Evidence for a new link between 3-hydroxy-3-methylglutaryl-coenzyme a reductase and cell growth. J. Biol. Chem. 1996;271:17453–17462. doi: 10.1074/jbc.271.29.17453. [DOI] [PubMed] [Google Scholar]
- [96].Sassano A, Platanias LC. Statins in tumor suppression. Cancer Lett. 2008;260:11–19. doi: 10.1016/j.canlet.2007.11.036. [DOI] [PubMed] [Google Scholar]
- [97].Larner J, Jane J, Laws E, Packer R, Myers C, Shaffrey M. A phase I-II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am. J. Clin. Oncol. 1998;21:579–583. doi: 10.1097/00000421-199812000-00010. [DOI] [PubMed] [Google Scholar]
- [98].Cemeus C, Zhao T, Barrett G, Lorimer I, Dimitroulakos J. Lovastatin enhances gefitinib activity in glioblastoma cells irrespective of EGFRvIII and PTEN status. J. Neurooncol. 2008;90:9–17. doi: 10.1007/s11060-008-9627-0. [DOI] [PubMed] [Google Scholar]
- [99].Mantha AJ, McFee KE, Niknejad N, Goss G, Lorimer IA, Dimitroulakos J. Epidermal growth factor receptor-targeted therapy potentiates lovastatin-induced apoptosis in head and neck squamous cell carcinoma cells. J. Cancer Res. Clin. Oncol. 2003;129:631–641. doi: 10.1007/s00432-003-0490-2. [DOI] [PubMed] [Google Scholar]
- [100].Mantha AJ, Hanson JE, Goss G, Lagarde AE, Lorimer IA, Dimitroulakos J. Targeting the mevalonate pathway inhibits the function of the epidermal growth factor receptor. Clin. Cancer Res. 2005;11:2398–2407. doi: 10.1158/1078-0432.CCR-04-1951. [DOI] [PubMed] [Google Scholar]
- [101].Huether A, Hopfner M, Baradari V, Schuppan D, Scherubl H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem. Pharmacol. 2005;70:1568–1578. doi: 10.1016/j.bcp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- [102].Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- [103].Qin HR, Kim HJ, Kim JY, Hurt EM, Klarmann GJ, Kawasaki BT, Duhagon Serrat MA, Farrar WL. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008;68:7736–7741. doi: 10.1158/0008-5472.CAN-08-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Pedranzini L, Leitch A, Bromberg J. Stat3 is required for the development of skin cancer. J. Clin. Invest. 2004;114:619–622. doi: 10.1172/JCI22800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ecker A, Simma O, Hoelbl A, Kenner L, Beug H, Moriggl R, Sexl V. The dark and the bright side of Stat3: proto-oncogene and tumor-suppressor. Front Biosci. 2009;14:2944–2958. doi: 10.2741/3425. [DOI] [PubMed] [Google Scholar]
- [107].Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, Kong LY, Fuller GN, Hiraoka N, Priebe W, Sawaya R, Heimberger AB. The incidence, correlation with tumorinfiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 2008;14:8228–8235. doi: 10.1158/1078-0432.CCR-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lo H-W, Hsu S-C, Xia W, Cao X, Shih J-Y, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung M-C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Alvarez JV, Mukherjee N, Chakravarti A, Robe P, Zhai G, Chakladar A, Loeffler J, Black P, Frank DA. A STAT3 gene expression signature in gliomas is associated with a poor prognosis. Translational Oncogenomics. 2007;2:97–103. doi: 10.4137/tog.s1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin. Cancer Res. 2007;13:1362–1366. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- [111].Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- [112].Fu XY. From PTK-STAT signaling to caspase expression and apoptosis induction. Cell. Death Differ. 1999;6:1201–1208. doi: 10.1038/sj.cdd.4400613. [DOI] [PubMed] [Google Scholar]
- [113].Aziz MH, Manoharan HT, Church DR, Dreckschmidt NE, Zhong W, Oberley TD, Wilding G, Verma AK. Protein kinase C{varepsilon} interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 2007;67:8828–8838. doi: 10.1158/0008-5472.CAN-07-1604. [DOI] [PubMed] [Google Scholar]
- [114].Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- [116].Wen Z, Darnell JE., Jr Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–2067. doi: 10.1093/nar/25.11.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA. 2001;98:7319–7324. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Karni R, Jove R, Levitzki A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene. 1999;18:4654–4662. doi: 10.1038/sj.onc.1202835. [DOI] [PubMed] [Google Scholar]
- [119].Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, Hirano T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity. 1999;11:709–719. doi: 10.1016/s1074-7613(00)80145-4. [DOI] [PubMed] [Google Scholar]
- [120].Sinibaldi D, Wharton W, Turkson J, Bowman T, Pledger WJ, Jove R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene. 2000;19:5419–5427. doi: 10.1038/sj.onc.1203947. [DOI] [PubMed] [Google Scholar]
- [121].Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- [122].Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22:319–329. doi: 10.1038/sj.onc.1206122. [DOI] [PubMed] [Google Scholar]
- [123].Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Heimberger AB, Priebe W. Small molecular inhibitors of p-STAT3: novel agents for treatment of primary and metastatic CNS cancers. Recent Pat. CNS Drug Discov. 2008;3:179–188. doi: 10.2174/157488908786242489. [DOI] [PubMed] [Google Scholar]
- [126].Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, Sebti S, Yu H, Jove R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004;3:1533–1542. [PubMed] [Google Scholar]
- [128].Xi S, Gooding WE, Grandis JR. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene. 2005;24:970–979. doi: 10.1038/sj.onc.1208316. [DOI] [PubMed] [Google Scholar]
- [129].Gu J, Li G, Sun T, Su Y, Zhang X, Shen J, Tian Z, Zhang J. Blockage of the STAT3 signaling pathway with a decoy oligonucleotide suppresses growth of human malignant glioma cells. J. Neurooncol. 2008;89:9–17. doi: 10.1007/s11060-008-9590-9. [DOI] [PubMed] [Google Scholar]
- [130].Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, Kondo Y. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–2444. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- [131].Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- [132].Fuh B, Sobo M, Cen L, Josiah D, Hutzen B, Cisek K, Bhasin D, Regan N, Lin L, Chan C, Caldas H, DeAngelis S, Li C, Li PK, Lin J. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer. 2009;100:106–112. doi: 10.1038/sj.bjc.6604793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–3930. [PubMed] [Google Scholar]
- [134].Vigneron A, Gamelin E, Coqueret O. The EGFR-STAT3 oncogenic pathway up-regulates the Eme1 endonuclease to reduce DNA damage after topoisomerase I inhibition. Cancer Res. 2008;68:815–825. doi: 10.1158/0008-5472.CAN-07-5115. [DOI] [PubMed] [Google Scholar]
- [135].Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006;65:1181–1188. doi: 10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- [136].Albanell J, Rojo F, Baselga J. Pharmacodynamic studies with the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839. Semin. Oncol. 2001;28:56–66. doi: 10.1016/s0093-7754(01)90283-0. [DOI] [PubMed] [Google Scholar]
- [137].Dowlati A, Nethery D, Kern JA. Combined inhibition of epidermal growth factor receptor and JAK/STAT pathways results in greater growth inhibition in vitro than single agent therapy. Mol. Cancer Ther. 2004;3:459–463. [PubMed] [Google Scholar]