

Abstract
Three bioactive compounds were isolated from an organic extract of an ascomycete fungus of the order Chaetothyriales (MSX 47445) using bioactivity-directed fractionation as part of a search for anticancer leads from filamentous fungi. Of these, two were benzoquinones [betulinan A (1) and betulinan C (3)] and the third was a terphenyl compound BTH-II0204-207:A (2). The structures were elucidated using a set of spectroscopic and spectrometric techniques; the structure of the new compound (3) was confirmed via single crystal X-ray diffraction. Compounds (1–3) were evaluated for cytotoxicity against a human cancer cell panel, for antimicrobial activity against Staphylococcus aureus and Candida albicans, and for phosphodiesterase (PDE4B2) inhibitory activities. The putative binding mode of 1–3 with PDE4B2 was examined using a validated docking protocol, and the binding and enzyme inhibitory activities correlated.
Historically, natural products have played an important role in drug discovery. Of the 1355 newly approved drugs worldwide during the time period of 1981–2010, ~50% can be traced to, or were inspired by, natural products.1 Moreover, of the thirteen natural product–derived drugs that were approved in the US between 2005 and 2007, five were the first members of new classes,2 and in 2010, fingolimod, an analogue of the fungal metabolite myriocin, was approved as the first oral drug to reduce multiple sclerosis relapses.3 In July of 2012, carfilzomib, an analogue of the natural product epoxomicin, which was isolated originally from an Actinomycete,4 was approved to treat patients with multiple myeloma.5 In short, natural products remain an invaluable source for novel bioactive leads.
As part of a multidisciplinary project to identify structurally diverse anticancer leads,6,7 the Mycosynthetix library, representing over 55,000 accessions of filamentous fungi, is being examined systematically.8–12 Fungi represent an under explored source for bioactive secondary metabolites. In 1991, the number of fungi was estimated as 1.5 million species,13 while current estimates suggest more than 5.1 million species.14 Regardless, less than 100,000 species have been characterized taxonomically,14 with likely a smaller percentage studied for bioactive secondary metabolites, and only a portion of these have been evaluated for anticancer activity.
An organic fraction of the filamentous fungus MSX 474459, which was isolated from highly decomposed woody debris from a tropical forest in 1990, displayed modest but equipotent cytotoxic activity against a panel of three cancer cell lines: MCF-7, H460, and SF268 (~75% inhibition of cell growth when tested at 20 μg/mL). Hence, this fungus was selected for further study, and three compounds, two benzoquinones (1 and 3) and one terphenyl compound (2), were isolated and characterized. All three compounds were evaluated for cytotoxicity against a human cancer cell panel, for antimicrobial activity against Staphylococcus aureus and Candida albicans, and for their phosphodiesterase (PDE4B2) inhibitory activities; the results with the latter were the most encouraging and led to docking studies.
RESULTS AND DISCUSSION
A solid-phase culture of MSX 47445 was extracted with 1:1 CHCl3-MeOH and partitioned with organic solvents to yield an orange-red extract, which was purified using flash chromatography to yield seven fractions. Of these, fraction 2 was the most cytotoxic against three cancer cell lines, and it was subjected to further purifications using preparative and semipreparative HPLC to yield three compounds (1–3) with > 97% purity as measured by UPLC (Supporting Information Figure S1).
Compound 1 (30.2 mg) was obtained as an orange powder. The molecular formula was determined as C20H16O4 by HRESIMS. The NMR data, in conjunction with HRMS data and UV maxima of 194, 238, and 320 nm, identified 1 as the known compound betulinan A, first described by Lee et al.15 in 1996 from the fungus Lenzites betulina.
Compound 2 (12.1 mg) was obtained as a pale yellow powder. HRESIMS data suggested a molecular formula of C19H16O3. The compound showed distinctive UV maxima at 202, 259, and 315 nm. The NMR data were in agreement with those reported for BTH-II0204-207:A, a terphenyl compound first reported in 2011 by Beggins et al.16 from the pathogenic bacterium Burkholderia pseudomallei.
Compound 3 (6.2 mg) was obtained as an orange powder. The molecular formula was determined as C19H14O3 via HRESIMS, establishing an index of hydrogen deficiency of 13. The UV maxima (198, 235, and 331 nm) and NMR data suggested structural similarity with compound 1, although a key difference was the loss of structural symmetry. Relative to 1, compound 3 also lacked one methoxy moiety, as supported by a 30 amu difference in the HRMS data. 1H NMR data (Table 1) revealed the presence of 10 aromatic protons (δH 7.45–7.52 for H-2′ to H-6′ and δH 7.33–7.42 for H-2″ to H-6″), suggesting two mono substituted benzene rings, one olefinic proton (δH 6.88, H-6), and one methoxy group (δH 3.80, 3-OCH3). The 13C NMR data revealed the presence of 19 carbons, consistent with the molecular formula and indicative of two carbonyls, which were assigned as quinone carbons (δC 187.4 and 183.3, for C-1 and C-4, respectively), four olefinic carbons (δC 132.7, 155.4, 144.5, and 133.0, for C-2, C-3, C-5, and C-6, respectively), and 10 aromatic carbons (δC 130.7, 128.2, 129.0, 128.2, 130.7, 129.4, 128.8, 130.3, 128.8, and 129.4, for C-2′, C-3′, C-4′, C-5′, C-6′, C-2″, C-3″, C4″, C-5″, and C-6″, respectively). Thus far, the spectroscopic data accounted for 12 of the 13 degrees of unsaturation, and hence, the 13th degree completed the quinone ring. COSY data identified two spin systems, which corresponded to the aromatic protons of the two phenyl rings. An HMBC correlation was observed from 3-OCH3 to C-3, indicating the connectivity of the methoxy group. HMBC correlations from H-6 to C-4, C-2, and C-1′ were observed. NOESY correlations were observed from the olefinic proton H-6 to the equivalent C-2′/C-6′ and from the 3-OCH3 to the equivalent C-2″/C-6″ (Figure 1b). The last structure elucidation hurdle was to verify whether the central ring was an ortho or para quinone, but the spectroscopic data were inconclusive, since the observed HMBC and NOESY correlations for the H-3 and the 3-OCH3 were equally valid for either substitution pattern. What increased the dilemma of the substitution pattern were contradictory NMR data that were published by two different research groups for a synthetic17 and a natural18 compound with the same molecular formula (compound 4). Our NMR data were in agreement with those reported by Singh and co-workers, except for one carbon where the 13C NMR data differed by about 12 ppm.18 Sawayama et al.17 reported the synthesis of 4, where clear differences were observed between the NMR data of synthetic and natural 4, and they stated that reexamination of the structure of natural 4 was “underway by Dr. S. B. Singh.” However, since this reexamination has not been reported yet, compound 3 was crystallized from ethyl acetate at room temperature to give monoclinic crystals, and single crystal X-ray diffraction established the structure of 3 with the carbonyl carbons para to each other (Figure 1a). To be consistent with the literature, the trivial name betulinan C was ascribed to 3.
Table 1.
1H (500 MHz) and 13C (125 MHz) NMR Data for Betulinan C (3) in CDCl3
| position | δC, type | δH (J in Hz) |
|---|---|---|
| 1 | 187.4, C | -- |
| 2 | 132.7, C | -- |
| 3 | 155.4, C | -- |
| 4 | 183.3, C | -- |
| 5 | 144.5, C | -- |
| 6 | 133.0, CH | 6.88, s |
| 1′ | 128.8, C | -- |
| 2′, 6′ | 129.4, CH | 7.52, dd (8.0, 1.7) |
| 3′, 5′ | 128.8, CH | 7.45, m |
| 4′ | 130.3, CH | 7.45, m |
| 1″ | 130.0, C | -- |
| 2″, 6″ | 130.7, CH | 7.33, dd (8.0, 1.7) |
| 3″, 5″ | 128.2, CH | 7.42, m |
| 4″ | 129.0, CH | 7.40, m |
| 3-OCH3 | 61.67, CH3 | 3.80, s |
Figure 1.
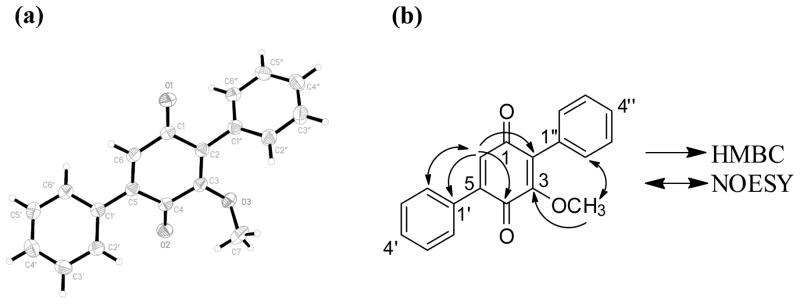
(a) X-ray crystallographic structure with 50% probability ellipsoids. (b) key HMBC and NOESY correlations of 3.
Compounds structurally related to 1–3 have been identified as phosphosdiesterase (PDE) inhibitors. Terferol (5), which was isolated from Streptomyces showdoensis SANK 65080, possessed inhibitory activity against cyclic adenosine 3′,5′-monophosphate phosphodiesterase (cAMP-PDE) and cyclic guanosine 3′,5′-monophosphate phosphodiesterase (cGMP-PDE).19 The concentrations of 5 required for 50% inhibition of cAMP-PDE and cGMP-PDE were 0.82 and 0.96 μM, respectively.19 Moreover, Biggins et al.16 evaluated two terferol related compounds, BTH-II0204-207:A (2) and BTH-II0204-207:C, for PDE inhibition activity against 11 PDE families. The latter was inactive, while 2 showed activity against PDE11 as well as four out of the five PDE4s that were examined. PDE4 is an essential regulator of the secondary messenger cAMP in numerous cell types, and the reduction in cAMP degradation by several inhibitors, such as rolipram, piclamilast, roflumilast, cilomilast, and tetomilast, has suggested a broad range of clinical applications for the treatment of asthma and chronic obstructive pulmonary disease (COPD),20,21 some types of brain tumors,22,23 and other inflammatory diseases.24 In 2011, roflumilast (Daliresp) was approved by the U.S. FDA as the first selective PDE4 inhibitor to reduce COPD exacerbations.25 Moreover, abnormal regulation of cAMP and/or cGMP metabolism upon altered expression and activity of PDE isoforms has been implicated in the pathogenesis of various types of cancer, including prostate cancer, colon cancer, hematological malignancies, melanoma, and brain tumors.26,27 Based on these reports, the effect of 1–3 on the activity of recombinant human PDE4B228 were evaluated; PDE4B is the predominant isoform present in human monocytes and neutrophils and is involved mainly in inflammation.29 Of these, 3 was the most potent with an IC50 value of 17 μM, followed by compounds 2 and 1 with IC50 values of 31 and 44 μM, respectively (Figure 2; Table 2).
Figure 2.

Plots of the effect of compounds 1–3 and rolipram (positive control) on PDE4B2 activity. Substrate Conc. = 100 nM (cAMP).
Table 2.
PDE4B2 Inhibition Activity and Docking Results of Compounds 1–3
| compound | PDE4B2 inhibition IC50 (μM) | Docking score (kcal/mol) | Docking score rank |
|---|---|---|---|
| betulinan A (1) | 44 | −8.071 | 4 |
| BTH-II0204-207:A (2) | 31 | −8.277 | 3 |
| betulinan C (3) | 17 | −8.732 | 2 |
| roliprama | 0.4 | −11.396 | 1 |
postive control
Molecular docking and other computational approaches are being used increasingly to explore the ligand-binding interactions of PDE4 inhibitors.30–33 As such, compounds 1–3 were docked into the crystal structure of human PDE4B using Glide Extra Precision.34,35 The docking protocol was verified by testing its ability to reproduce the experimental binding mode of co-crystallized rolipram (Supporting Information Figure S4). To this end, rolipram bound to the crystal structure was removed from the binding pocket and docked back into the cofactor binding site; the root-mean-square deviation between the predicted conformation and the observed X-ray crystallographic data was 1.1 Å, indicating the capability of the docking protocol to reproduce the binding mode of rolipram (Supporting Information Figure S4). Compounds 1–3 were docked into the cAMP binding site of PDE4B. The docking scores calculated with Glide correlated with the biological activity (Table 2); compound 3 displayed the highest activity (IC50 value of 17 μM) and also the top-ranked docking score (−8.732 kcal/mol). In contrast, compound 1 had the lowest activity (IC50 value of 44 μM) and showed the lowest docking score (−8.071 kcal/mol). Finally, the pyrrolidinone rolipram was included, not only for the docking protocol validation, but also as a positive control in the enzymatic assay; rolipram was top ranked in both docking score and in vitro activity.
Compounds 1–3 and rolipram displayed a similar binding mode (Figures 3 and S5). The two predicted hydrogen bonds between the free amino group of Gln443 and the cyclopentyloxy and methoxyphenyl groups of rolipram were in agreement with the observations derived from the crystallographic structure of PDE4B in complex with rolipram. As shown, Glide found a similar hydrogen bond with Gln443 and the carboxyl group for the most active compound 3 (Figures 3c and 3d); favorable π interactions with Phe446 in the binding pocket were also observed. Compounds 1 and 2 did not show hydrogen bonds with Gln433, but similar π interactions were predicted (Figures 3a, 3b and S5). Taken together, these observations suggested that the binging modes predicted with Glide for compounds 1–3 were reasonable.
Figure 3.

Binding conformation of 1 (a), 2 (b) and 3 (c) predicted by Glide. Crystallographic rolipram (maroon) is shown as a reference with hydrogen bonds displayed as yellow/black dashes. Nonpolar hydrogen atoms are omitted. (d) Two-dimensional interaction map of the optimized docking model of compound 3 in the cAMP binding pocket of PDE4B. Amino acid residues within 4.5 Å of the ligand are displayed. Blue arrows indicate hydrogen bonding to amino acid side chain atoms.
Compounds 1–3 were assayed for cytotoxicity and antimicrobial activity. When tested against the three cancer cell lines MCF-7, H460, and SF268 (Supporting Information Table S1), compounds 2 and 3 showed moderate cytotoxicity while 1 was inactive. Compounds 2 and 3 were equipotent against S. aureus with MIC values of 25 μg/mL, while none of the compounds showed activity against C. albicans.
In conclusion, three compounds (1–3) were isolated and characterized from the fungus MSX 47445. The structure of the new paraquinone, 3, was assigned unequivocally by NMR and single crystal X-ray diffraction. The effect of compounds 1–3 on the activity of PDE4B was assessed both in vitro and in silico; compound 3 was the most potent, being approximately a half order of magnitude less potent than the positive control, rolipram. Further studies are ongoing to expand the knowledge base of this class of compounds, particularly given their compact structures.
EXPERIMENTAL SECTION
General Experimental Procedures
UV and IR spectra were acquired on a Varian Cary 100 Bio UV-Vis spectrophotometer and a Perkin-Elmer Spectrum One with Universal ATR attachment, respectively. NMR experiments were conducted in either CDCl3, acetone-d6 or DMSO-d6 with TMS as a reference via a JEOL ECA-500, operating at 500 MHz for 1H and 125 MHz for 13C. HRESIMS was performed on a Thermo LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source. UPLC was carried out on a Waters Acquity system with data collected and analyzed using Empower software. HPLC was carried out using a Varian Prostar HPLC system equipped with ProStar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2). For preparative HPLC, a Phenomenex Synergi Max-RP 80 (4 μm; 250 × 21.2 mm) column was used at a 21 mL/min flow rate, while for the semi-preparative HPLC, a Phenomenex Gemini-NX C18 (4 μm; 250 × 10 mm) column was used at a 4.7 mL/min flow rate. For UPLC, a Waters BEH C18 column (1.7 μm; 50 × 2.1 mm) was used with a 0.6 mL/min flow rate. Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf using a 40 g Silica Gold column and monitored by UV and evaporative light-scattering detectors. X-ray crystallography data were acquired using a Bruker APEX CCD diffractometer (MoKᾱ radiation, graphite monochromator). All other reagents and solvents were obtained from Fisher Scientific and were used without further purification.
Producing Organism and Fermentation
Mycosynthetix fungal strain 47445 was isolated from highly decomposed woody debris in 1990. The growth conditions were as described previously9,12 and outlined in the supplementary materials. For molecular identification, the internal transcribed spacer regions 1 & 2 and 5.8S nrDNA (ITS) were sequenced, since this region of the ribosomal RNA operon has been proposed as a barcode marker for fungi.36 Detailed methodology for DNA extraction, PCR amplification, sequencing, and phylogenetic analyses are outlined in the supplementary materials. The combined ITS and LSU sequence was deposited in the GenBank (accession no JX310275). The analyses of both the rRNA regions (ITS and D1/D2 of the LSU) suggested that MSX 47445 was a member of the Chaetothyriales, Ascomycota and shares phylogenetic affinities with the mitosporic fungus Cyphellophora sp.
Extraction and Isolation
To the large-scale solid fermentation culture of MSX 47445, 500 mL of 1:1 MeOH-CHCl3 were added. The culture was chopped with a spatula and shaken overnight (~16 h) at ~100 rpm at rt. The sample was filtered with vacuum, and the remaining residues were washed with 100 mL of 1:1 MeOH-CHCl3. To the filtrate, 900 mL CHCl3 and 1500 mL H2O were added; the mixture was stirred for 2 h and then transferred into a separatory funnel. The bottom layer was drawn off and evaporated to dryness. The dried organic extract was re-constituted in 300 mL of 1:1 MeOH-CH3CN and 200 mL of hexanes. The biphasic solution was stirred for an hour and then transferred to a separatory funnel. The MeOH-CH3CN layer was drawn off and evaporated to dryness under vacuum. The defatted material (1.2 g, orange red) was dissolved in a mixture of CHCl3-MeOH, adsorbed onto Celite 545, and fractionated via flash chromatography using a gradient solvent system of hexane-CHCl3-MeOH at a 40 mL/min flow rate and 53.3 column volumes over 63.9 min to afford seven fractions. Fraction 2 eluted with 100% CHCl3 (~247 mg) was subjected to preparative HPLC using an isocratic system of 55:45 CH3CN-H2O over 30 min at a flow rate of 4.7 mL/min to yield seven sub-fractions. Sub-fraction 5 yielded compound 1 (30.2 mg), which eluted at ~22.5 min. Sub-fraction 2 was subjected to semipreprative HPLC and yielded compounds 2 (12.1 mg) and 3 (6.2 mg), which eluted at 9.5 and 19.0 min, respectively. UPLC was used to evaluate the purity of 1–3 using a gradient solvent system that initiated with 20:80 CH3CN-H2O to 100% CH3CN over 4.5 min; all compounds were >97% pure (Supporting Information Figure S1).
Betulinan C (3): orange powder; UV (MeOH) λmax (log ε) 330 (3.62), 235 (4.14), 203 (4.32) nm; IR (diamond) vmax 1661, 1640, 1593, 1330, 1267, 1090, 1072, 935, 889, 849, 809, 776, 766 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1; HRESIMS m/z 291.1017 [M + H]+ (calcd for C19H14O3 291.1016).
X-ray Crystallography
Crystallographic data for compound 3 has been deposited with the Cambridge Crystallographic Data Centre, deposition number 904704. Compound’s 3 crystals were grown in ethyl acetate at rt. X-ray crystal structure analysis of 3 were as follows: formula C19H13O3, MW = 290.31, block-shaped yellow crystal, a = 14.6693 (18) Å, b = 7.3806 (9) Å, c = 14.3582 (18) Å, β = 115.259 (1)°, T = 193 (2) K, Z = 4, monoclinic, space group P2(1)/c, GOF = S = 1.043, V = 1405.9 (3) Å3, R1 (3088 reflections, I>2σ(I)) = 0.0521, wR2 (all 3719 reflections) = 0.1476, λ = 0.71073 Å.
Cytotoxicity Assay
The cytotoxicity measurements against the MCF-737 human breast carcinoma (Barbara A. Karmanos Cancer Center), NCI-H46038 human large cell lung carcinoma (HTB-177, American Type Culture Collection (ATCC), and SF-26839 human astrocytoma (NCI Developmental Therapeutics Program) cell lines were performed as described previously.40,41
Antimicrobial Assay
The compounds were screened for antimicrobial activity using an agar plate diffusion assay as described previously.8
Phosphodiesterase Inhibitor Assay
The PDE inhibitor assay was performed at BPS Bioscience Inc. as described previously.13 Detailed experimental procedures are provided in the Supporting Information.
Molecular Modeling
Compounds 1–3 were prepared using the LigPrep 2.4 module of Maestro 9.1 (Schrödinger, LLC). The crystal structure of human PDE4B in complex to the inhibitor rolipram was retrieved from the Protein Data Bank (PDB entry 1RO6).42 Docking was performed with the cAMP catalytic domain using Glide (Grid-Based Ligand Docking with Energetics; Schrödinger, LLC) program, version 5.6.35 The Protein Preparation Wizard module of Maestro was used to prepare the protein.43 During protein preparation, H2O molecules were deleted. For docking, the scoring grids were centered on the crystal structure of rolipram using the default bounding sizes. All structures were docked and scored using Glide.35 The best docked poses were selected as the ones with the lowest Glide Score; the more negative the Glide Score, the more favorable the binding. 2D interactions maps were generated with Discovery Studio 3.1 from Accelrys Software Inc.
Supplementary Material
Acknowledgments
This research was supported by program project grant P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. The high resolution mass spectrometry data were acquired at the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro.
Footnotes
Dedicated to Dr. Lester A. Mitscher, of the University of Kansas, for his pioneering work on the discovery of bioactive natural products and their derivatives.
Notes
The authors declare no competing financial interest.
Information about the producing organism and its fermentation, the experimental protocol for phosphodiesterase inhibitor assay, UPLC chromatograms of compounds 1–3, 1H and 13C NMR spectra for compound 3, plots of the effect of compounds 1–3 and rolipram on PDE4B2 activity, Phylogram of the most likely tree, comparison between the binding position of rolipram within the crystal structure and the binding mode predicted by Glide, and cytotoxicity and antimicrobial activities of compounds 1–3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey AL. Drug Discovery Today. 2008;13:894–901. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Strader CR, Pearce CJ, Oberlies NH. J Nat Prod. 2011;74:900–907. doi: 10.1021/np2000528. [DOI] [PubMed] [Google Scholar]
- 4.Hanada M, Sugawara K, Kaneta K, Toda S, Nishiyama Y, Tomita K, Yamamoto H, Konishi M, Oki T. J Antibiot. 1992;45:1746–1752. doi: 10.7164/antibiotics.45.1746. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Department of Health and Human Services U.S. Food and Drug Administration. FDA approves Kyprolis for some patients with multiple myeloma. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312920.htm.
- 6.Orjala J, Oberlies NH, Pearce CJ, Swanson SM, Kinghorn AD. In: Bioactive Compounds from Natural Sources. Natural Products as Lead Compounds in Drug Discovery. 2. Tringali C, editor. Taylor & Francis; London, UK: 2012. pp. 37–63. [Google Scholar]
- 7.El-Elimat T, Zhang X, Jarjoura D, Moy FJ, Orjala J, Kinghorn AD, Pearce CJ, Oberlies NH. ACS Med Chem Lett. 2012;3:645–649. doi: 10.1021/ml300105s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayers S, Ehrmann BM, Adcock AF, Kroll DJ, Carcache de Blanco EJ, Shen Q, Swanson SM, Falkinham JO, 3rd, Wani MC, Mitchell SM, Pearce CJ, Oberlies NH. J Pept Sci. 2012;18:500–510. doi: 10.1002/psc.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayers S, Graf TN, Adcock AF, Kroll DJ, Matthew S, Carcache de Blanco EJ, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2011;74:1126–1131. doi: 10.1021/np200062x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Matthew S, Carcache de Blanco EJ, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. J Antibiot. 2012;65:3–8. doi: 10.1038/ja.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayers S, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Wani MC, Darveaux BA, Pearce CJ, Oberlies NH. Tetrahedron Lett. 2011;52:5128–5230. doi: 10.1016/j.tetlet.2011.07.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sy-Cordero AA, Graf TN, Adcock AF, Kroll DJ, Shen Q, Swanson SM, Wani MC, Pearce CJ, Oberlies NH. J Nat Prod. 2011;74:2137–2142. doi: 10.1021/np2004243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawksworth DL. Mycol Res. 1991;95:641–655. [Google Scholar]
- 14.Blackwell M. Am J Bot. 2011;98:426–438. doi: 10.3732/ajb.1000298. [DOI] [PubMed] [Google Scholar]
- 15.Lee IK, Yun BS, Cho SM, Kim WG, Kim JP, Ryoo IJ, Koshino H, Yoo ID. J Nat Prod. 1996;59:1090–1092. doi: 10.1021/np960253z. [DOI] [PubMed] [Google Scholar]
- 16.Biggins JB, Liu X, Feng Z, Brady SF. J Am Chem Soc. 2011;133:1638–1641. doi: 10.1021/ja1087369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawayama Y, Tsujimoto T, Sugino K, Nishikawa T, Isobe M, Kawagishi H. Biosci, Biotechnol, Biochem. 2006;70:2998–3003. doi: 10.1271/bbb.60389. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Ondeyka JG, Herath KB, Guan Z, Collado J, Pelaez F, Leavitt PS, Gurnett A, Nare B, Liberator P, Singh SB. J Nat Prod. 2006;69:710–712. doi: 10.1021/np0505418. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa F, Enokita R, Naito A, Iijima Y, Yamazaki M. J Antibiot. 1984;37:6–9. doi: 10.7164/antibiotics.37.6. [DOI] [PubMed] [Google Scholar]
- 20.Claveau D, Chen SL, O’Keefe S, Zaller DM, Styhler A, Liu S, Huang Z, Nicholson DW, Mancini JA. J Pharmacol Exp Ther. 2004;310:752–760. doi: 10.1124/jpet.103.064691. [DOI] [PubMed] [Google Scholar]
- 21.Press NJ, Banner KH. Prog Med Chem. 2009;47:37–74. doi: 10.1016/S0079-6468(08)00202-6. [DOI] [PubMed] [Google Scholar]
- 22.Goldhoff P, Warrington NM, Limbrick DD, Jr, Hope A, Woerner BM, Jackson E, Perry A, Piwnica-Worms D, Rubin JB. Clin Cancer Res. 2008;14:7717–7725. doi: 10.1158/1078-0432.CCR-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farias CB, Lima RC, Lima LO, Flores DG, Meurer L, Brunetto AL, Schwartsmann G, Roesler R. Oncology. 2008;75:27–31. doi: 10.1159/000151616. [DOI] [PubMed] [Google Scholar]
- 24.Jeon YH, Heo YS, Kim CM, Hyun YL, Lee TG, Ro S, Cho JM. Cell Mol Life Sci. 2005;62:1198–1220. doi: 10.1007/s00018-005-4533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.U.S. Department of Health and Human Services U.S. Food and Drug Administration. [accessed June 2, 2012];Approval Package For: Application Number: 022522orig1s000. 2011 http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022522Orig1s000Approv.pdf.
- 26.Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F, Schermuly RT. Expert Opin Invest Drugs. 2010;19:117–131. doi: 10.1517/13543780903485642. [DOI] [PubMed] [Google Scholar]
- 27.Sengupta R, Sun T, Warrington NM, Rubin JB. Trends Pharmacol Sci. 2011;32:337–344. doi: 10.1016/j.tips.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang P, Wu P, Ohleth KM, Egan RW, Billah MM. Mol Pharmacol. 1999;56:170–174. doi: 10.1124/mol.56.1.170. [DOI] [PubMed] [Google Scholar]
- 29.Houslay MD, Schafer P, Zhang KYJ. Drug Discovery Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 30.Kumar D, Patel G, Vijayakrishnan L, Dastidar SG, Ray A. Chem Biol Drug Des. 2011;79:810–818. doi: 10.1111/j.1747-0285.2011.01304.x. [DOI] [PubMed] [Google Scholar]
- 31.Kranz M, Wall M, Evans B, Miah A, Ballantine S, Delves C, Dombroski B, Gross J, Schneck J, Villa JP, Neu M, Somers DO. Bioorg Med Chem. 2009;17:5336–5341. doi: 10.1016/j.bmc.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 32.Mpamhanga CP, Chen B, McLay IM, Ormsby DL, Lindvall MK. J Chem Inf Model. 2005;45:1061–1074. doi: 10.1021/ci050044x. [DOI] [PubMed] [Google Scholar]
- 33.Dym O, Xenarios I, Ke H, Colicelli J. Mol Pharmacol. 2002;61:20–25. doi: 10.1124/mol.61.1.20. [DOI] [PubMed] [Google Scholar]
- 34.Glide, version 5.6. Schrodinger, LLC; New York, NY: 2011. [Google Scholar]
- 35.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 36.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W Fungal Barcoding Consortium. . Proc Natl Acad Sci USA. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soule HD, Vazguez J, Long A, Albert S, Brennan M. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 38.Carney DN, Gazdar AF, Bunn PA, Jr, Guccion JG. Stem Cells. 1982;1:149–164. [PubMed] [Google Scholar]
- 39.Rosenblum ML, Gerosa MA, Wilson CB, Barger GR, Pertuiset BF, de Tribolet N, Dougherty DV. J Neurosurg. 1983;58:170–176. doi: 10.3171/jns.1983.58.2.0170. [DOI] [PubMed] [Google Scholar]
- 40.Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, Falkinham JO, Wani MC, Oberlies NH. J Nat Prod. 2005;68:173–178. doi: 10.1021/np0496587. [DOI] [PubMed] [Google Scholar]
- 41.Li C, Lee D, Graf TN, Phifer SS, Nakanishi Y, Riswan S, Setyowati FM, Saribi AM, Soejarto DD, Farnsworth NR, Falkinham JO, 3rd, Kroll DJ, Kinghorn AD, Wani MC, Oberlies NH. J Nat Prod. 2009;72:1949–1953. doi: 10.1021/np900572g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. J Mol Biol. 2004;337:355–365. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Schrödinger Suite 2010 Protein Preparation Wizard, Epik version 2.1, Impact version 5.6, Prime version 2.2. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.