

Abstract
Both chronic stress and antidepressant medications have been associated with changes in body weight. In the current study, we investigate mechanisms by which stress and antidepressant medications interact to affect meal patterns. A group of mice was subjected to the chronic social defeat stress model of major depression followed by fluoxetine treatment and was subsequently analyzed for food intake using metabolic cages. Here, we report that chronic social defeat stress increases food intake by specifically increasing meal size, an effect that is reversed by fluoxetine treatment. In an attempt to gain mechanistic insight into changes in meal patterning induced by stress and fluoxetine, fasting serum samples were collected every four hours over a 24-hour period and acyl-ghrelin, leptin, and corticosterone levels were measured. Chronic stress induces a peak in acyl-ghrelin levels just prior to lights off, which is shifted in mice treated with fluoxetine. Taken together, these results indicate that stress increases food intake by decreasing satiation, and that fluoxetine can reverse stress-induced changes in meal patterns.
Keywords: Feeding, Stress, Fluoxetine, Ghrelin, Leptin
Introduction
Multiple studies now indicate that certain mental disorders including major depressive disorder (Anderson, Cohen, Naumova, Jacques, & Must, 2007; Beydoun & Wang, 2010; Cizza, 2011; de Wit, et al., 2010; Gavin, Rue, & Takeuchi, 2010; Merikangas, Mendola, Pastor, Reuben, & Cleary, 2011; Papakostas, et al., 2005; Rivera, et al., 2011) and post-traumatic stress disorder (Chwastiak, Rosenheck, & Kazis, 2011; Coughlin, 2011; Coughlin, Kang, & Mahan, 2011; Dedert, et al., 2010; Perkonigg, Owashi, Stein, Kirschbaum, & Wittchen, 2009; Scott, McGee, Wells, & Oakley Browne, 2008; Vieweg, et al., 2006; Vieweg, et al., 2007; Wilson, 2010) significantly increase the risk of developing obesity. In addition, several medications, including antidepressants, used to treat these psychiatric disorders are also associated with changes in body weight (Serretti & Mandelli, 2010). Despite the association between mental illness, psychotropic medications, and weight changes, relatively little is known about the underlying biological mechanisms that mediate these interactions.
Rodent models of major depression have confirmed the importance of chronic stress in body weight homeostasis (Tamashiro, Nguyen, & Sakai, 2005). The visible burrow system, one of the best-studied models of stress, induces changes in food intake and body composition (Tamashiro, Hegeman, et al., 2007; Tamashiro, Hegeman, & Sakai, 2006; Tamashiro, et al., 2004; Tamashiro, Nguyen, et al., 2007). In this model, group-housed rats form a dominance hierarchy. While dominant male rats maintain normal body weight and food intake, subordinate males significantly reduce their food intake and lose weight during the testing period. Interestingly, during the recovery phase following removal from the visible burrow system, subordinate male rats regain their lost weight mostly as adipose tissue and develop obesity and concurrent elevated leptin levels (Tamashiro, Nguyen, et al., 2007). During the stressful hierarchy-formation phase, subordinate males reduce food intake primarily via decreased meal frequency and meal size. In contrast, during the recovery phase, subordinate males increase food intake via increased meal size (Melhorn, et al., 2010).
Using the chronic social defeat stress (CSDS) model in mice, which shares features with both post-traumatic stress disorder and major depression with co-morbid anxiety (Avgustinovich, Gorbach, & Kudryavtseva, 1997; Krishnan, Berton, & Nestler, 2008), our group has analyzed the effect of psychosocial stress on body weight regulation. In this model, a substantial proportion of mice subjected to repeated social aggression develop behavioral deficits similar to symptoms observed in human mental illness, including social avoidance, changes in locomotor activity, impairments in cognition, hyperphagia, decreased preference for natural reward, increased preference for drug reward, and anxiety-like behaviors (Chuang, Cui, et al., 2010; Cruz, Quadros, Hogenelst, Planeta, & Miczek, 2011; Rygula, et al., 2005; Yu, et al., 2011). These so-called “susceptible” mice (Krishnan, et al., 2007) display multiple metabolic abnormalities including body weight gain, redistribution of body fat into internal organs, and insulin and leptin resistance (Chuang, Cui, et al., 2010; Chuang, Krishnan, et al., 2010). The circulating hormone ghrelin appears to be an important mediator of certain CSDS-induced behavioral and metabolic disturbances. CSDS increases acyl-ghrelin levels, and mice lacking ghrelin receptors exhibit exaggerated social avoidance behavior after CSDS (Lutter, Sakata, et al., 2008). More recently, we have shown that following CSDS, wild-type mice, but not ghrelin receptor-null littermates, display conditioned place preference for high fat diet. Importantly, restoration of ghrelin receptor expression specifically in catecholaminergic neurons reduces social isolation, but at the expense of also restoring conditioned place preference for high fat diet (Chuang, et al., 2011). These findings suggest that ghrelin may be an important mediator linking changes in appetite to mood symptoms.
Because CSDS increases food intake and conditioned place preference for high fat diet in susceptible mice (Chuang, Cui, et al., 2010; Chuang, et al., 2011), it appears to model aspects of depression with atypical features. Therefore, we wanted to determine if antidepressant treatment could reverse the effects of CSDS on food intake in this subpopulation of animals. We chose fluoxetine because it is a widely prescribed antidepressant and because several initial studies reported that fluoxetine suppresses food intake in rodents (Gamaro, Prediger, Lopes, Bassani, & Dalmaz, 2008; Grignaschi & Samanin, 1992; Lightowler, et al., 1996; Wong, Reid, & Threlkeld, 1988), and human studies also suggest that fluoxetine acutely suppresses food intake (Serretti & Mandelli, 2010). For example, during a one-year maintenance trial, fluoxetine treatment was associated with a modest initial weight loss during the first four weeks of treatment (Michelson, et al., 1999).
In the current study, we tested the hypothesis that fluoxetine treatment will normalize food intake in mice subjected to CSDS. We exposed mice to CSDS or control handling conditions. Mice displaying social avoidance were randomized to receive either fluoxetine or placebo. Multiple behavioral and metabolic parameters were then measured, including meal patterning and circulating levels of serum hormones known to regulate food intake and body weight homeostasis.
Methods
Animals and housing
Animals were housed in the UT Southwestern vivarium in a temperature-controlled environment on a 12 h light/dark cycle (lights on 0600) and maintained on regular chow (4% fat diet #7001, Harlan-Teklad, Madison, WI).
Ethics statement
All animal procedures were approved and carried out in accordance with both the UT Southwestern Institutional Animal Care and Use Committee (IACUC) guidelines and “Principles of laboratory animal care” (NIH publication No. 86-23, revised 1985)
Chronic social defeat stress and antidepressant treatment
The CSDS paradigm used in the current study was recently reported (Lutter, Krishnan, et al., 2008). Eight-week-old male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were group-housed and acclimated to the vivarium 1 week prior to the commencement of social defeat. Mice were exposed to a different CD1 strain aggressor mouse (Charles River Laboratories, Wilmington, MA) each day prior to lights out (1800) for 5 min over a total of 10 days. After 5 min of physical contact, test mice were separated from the aggressor and placed across a plastic separator with holes, where they remained in sensory contact with the CD1 aggressor for the remainder of the 24 hours, but physical contact was prevented. Control mice were handled daily in the palm of the hand for 30 seconds and housed in equivalent cages with members of the same strain. After the last defeat, all mice were housed individually and a social interaction task was performed to measure the behavioral consequences of chronic defeat stress.
The social interaction task involved placing mice in a new arena (44 cm × 44 cm) with a small animal cage (5 cm × 10 cm) at one end. Their movements were tracked for 2.5 min in the absence of another mouse, followed by 2.5 min in the presence of a caged, unfamiliar CD1 mouse. Social interaction was quantified by comparing the amount of time the test mouse spent in the interaction zone near the small animal cage in the presence vs. in the absence of the target CD1 mouse, as determined using Ethovision 3.0 software (Noldus, Leesburg, VA). Thirty-two out of 120 mice subjected to CSDS did not display social avoidance (defined as an interaction ratio < 100%) and were removed from further study. Five out of 120 control mice displayed social avoidance and were removed from the study. Twenty-four hours following social interaction testing, the remaining mice were implanted with either a fluoxetine or placebo pellet in the dorsal interscapular region under brief isoflurane anesthesia. Custom-made continuous release pellets (Innovative Research of America, Sarasota, FL) were used to deliver a chronic dose of fluoxetine (15 mg/kg/day) for 20 days. Previous studies showed that pellets prepared in this manner produce physiologically relevant concentrations of fluoxetine 20 days after implantation (Covington, et al., 2009).
Metabolic Studies
Experiments were conducted in the UT Southwestern Metabolic Phenotyping Core using metabolic cages (TSE Systems, Chesterfield, MO). A representative cohort of mice from all four groups of the experimental population was pair matched for body weight and lean body mass (Supplemental Figure 1B–C) to correct for presumed body composition-related differences in food intake. These mice were transferred to the metabolic phenotyping core for 4 days of acclimation and then placed in metabolic cages for 4 days with access to regular chow. Meal analysis over a 48-hour period was assessed using Lab Master Software (TSE Systems, Chesterfield, MO).
Serum hormone levels
Blood samples were collected across the circadian cycle every four hours starting one hour after lights on. Animals from each time point were fasted four hours prior to sacrifice by cervical dislocation in order to control for the effects of food intake on leptin and ghrelin levels. Because of the relatively large amount of serum needed to measure ghrelin levels, we could only obtain the amount of blood required for hormone analysis by collecting blood samples as a terminal procedure and not through serial blood draws. Trunk blood was immediately collected in EDTA-coated tubes (BD Biosciences, San Jose, CA) and was further processed by the addition of 4-(hydroxymercuri)benzoic acid sodium salt (#55540, Sigma Aldrich, St. Louis, MO) to preserve neuropeptide hormones. A fraction of blood sample was set aside and acidified by the addition of 0.1 N HCl in order to stabilize acyl-ghrelin. Serum levels of leptin and corticosterone were measured as previously described (Chuang, Cui, et al., 2010; Chuang, Krishnan, et al., 2010), with intra- and inter-assay variabilities of 5% and 12% for leptin, and 3% and 5% for corticosterone, respectively. Levels of acyl-ghrelin were quantified using a commercially available enzyme immunoassay kit (#10006307, Cayman Chemical, Ann Arbor, MI), with an intra- and inter-assay variability of 4% and 9%, respectively. All hormone levels were analyzed in duplicate.
Statistical analyses
Data are reported as mean±SEM. Statistical analyses were performed using Student’s t-test, repeated measures analysis of variance (RMANOVA) or two way ANOVA followed by Bonferroni post hoc tests. All statistical analyses were performed using Prism (v 5.0, GraphPad Software Inc., San Diego, CA) software. Statistical significance was defined as p<0.05.
Results
Eight-week-old male C57BL/6J mice were subjected to either 10 days of CSDS or control handling conditions (Figure 1A). After this 10-day period, defeated mice that displayed social avoidance (“CSDS” mice, Supplemental Figure 1A) and nonstressed controls were randomized into two equal groups that received a subcutaneous pellet releasing a 20-day supply of either fluoxetine or placebo. This protocol resulted in four groups of mice with a large number: control-placebo (n = 41), control-fluoxetine (n = 40), CSDS-placebo (n = 39), and CSDS-fluoxetine (n = 37). As previously reported (Berton, et al., 2006), chronic fluoxetine treatment reversed social avoidance 29 days after the last bout of social stress (Supplemental Figure 1A). We then sought to determine whether chronic social stress affects meal patterning, and if fluoxetine influences food intake in these mice. However, there has been much uncertainty on how to interpret measures of food intake when differences in body composition exist (Butler & Kozak, 2010; Tschop, et al., 2011). Therefore, to rigorously answer this question, a subset of mice from all four groups was pair matched for body weight and lean body mass (Supplemental Figure 1B–C) to correct for presumed body composition-related differences in food intake, and food intake was measured over a 4-day period using metabolic cages.
Figure 1. Effects of chronic social stress and fluoxetine on meal patterning.

A. Eight-week-old male C57BL/6J mice were subjected to ten days of chronic social defeat stress (CSDS) and tested for social interaction. Animals displaying social avoidance along with nonstressed controls showing a social preference were randomized into two equal groups that received pellets releasing a 20-day supply of fluoxetine or placebo. A subset of mice from each experimental group (Supplemental Figure 1A) was pair-matched for body weight and monitored in metabolic cages 4 days after social interaction testing (days 33–36). Meal patterns were analyzed over 48 hours on days 34–36. B. Total chow consumed in the light phase (no effect) and dark phase (significant effect of defeat, F1,18 = 9.41). C. Meal number in the light phase (no effect) and dark phase (significant defeat X fluoxetine interaction, F1,18 = 9.11). D. Meal size in the light phase (no effect) and dark phase (significant defeat X fluoxetine interaction, F1,18 = 12.49). E. Satiety ratio calculated during the light phase (no effect) and dark phase (no effect). N = 5–6/group. *p< 0.05, **p < 0.01, ***p < 0.001. Data presented as mean ± S.E.M. D, Day.
As shown in Figure 1, there were no differences noted in total chow consumed, meal number, meal size, or satiety ratio among groups during the light phase when mice are typically inactive. In contrast, mice exposed to CSDS plus placebo demonstrated an increase in total chow consumed during the dark phase (Fig. 1B). Meal pattern analysis revealed that this increase was the result of fewer meals (Fig. 1C) and increased meal size (Fig. 1D). The satiety ratio (g of food per meal/intermeal interval) was the same in all groups indicating that CSDS increases food intake primarily by impairing satiation (the process of intrameal termination) during the dark phase without affecting satiety (the length of appetite suppression after a meal). The effects of fluoxetine on meal patterning were restricted to the CSDS-exposed mice, for which fluoxetine normalized CSDS-induced changes in total chow consumed, meal number, and meal size (Fig. 1B–D).
The metabolic cage data indicate that CSDS increases food intake in susceptible mice, and that fluoxetine preferentially reduces hyperphagia in these mice. Furthermore, CSDS appears to increase food intake only during the dark phase, suggesting a diurnal effect. For further mechanistic insight into the effects of CSDS and fluoxetine on meal patterning, we next analyzed levels of circulating hormones after CSDS. Acyl-ghrelin, leptin, and corticosterone levels have previously been demonstrated to be disturbed in susceptible mice following CSDS (Chuang, Cui, et al., 2010; Krishnan, et al., 2007; Lutter, Sakata, et al., 2008). However, the measurements in these previous studies were made at a single time point, and thus likely were not reflective of time-of-day differences in appetitive behavior, as detected in the current study. Because the current study was designed to collect samples over a 24-hour period, we were therefore able to measure serum hormone levels across the circadian cycle.
Consistent with our previous observations (Lutter, Sakata, et al., 2008), acyl-ghrelin levels are increased in susceptible mice exposed to CSDS in the late light phase just prior to lights off (Fig. 2A, ZT9), with trends towards increased levels in this group during the preceding two timepoints. Fluoxetine treatment normalizes acyl-ghrelin levels after CSDS at ZT9. A significant peak of acyl-ghrelin expression at ZT17 was noted only in the CSDS-fluoxetine group (the functional significance of this peak will be addressed in the Discussion). We next analyzed leptin levels, previously shown to be reduced after CSDS (Chuang, Cui, et al., 2010), and found significant reductions in leptin in both CSDS groups at ZT5, ZT9, and ZT13. Fluoxetine treatment did not significantly alter leptin levels in either control or CSDS groups (Fig. 2B). Chronic social stress reduced morning levels of corticosterone in both CSDS groups (Fig. 2C), consistent with previous observations (Krishnan, et al., 2007). Fluoxetine also caused a significant reduction of corticosterone levels at ZT21 in both control and CSDS groups. No other effects of CSDS or fluoxetine were noted in corticosterone levels at other time points.
Figure 2. Serum hormone levels.
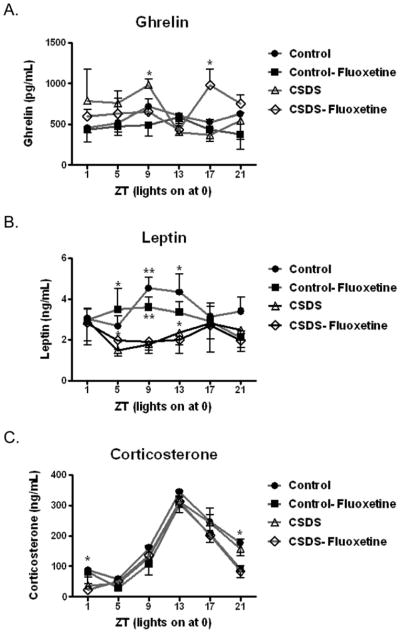
Twenty-four hours after social interaction retesting (Day 40), mice were fasted for 4 hours and sacrificed every 4 hours over a 24-hour period (ZT0 = lights on). Serum was collected and tested for A. acyl-ghrelin (no primary effect of group or time of day; significant effect of fluoxetine, F1,16 = 5.94 at ZT9 and significant defeat X fluoxetine interaction, F1,16 = 4.54 at ZT17). B. leptin (primary effect of group F3,109 = 4.85; significant effect of defeat, F1,23 = 4.91 at ZT5, significant effect of defeat, F1,17 = 18.79 at ZT9 and significant effect of defeat, F1,17 = 5.37 at ZT13) and C. corticosterone (primary effect of time of day F5,124 = 48.06; significant effect of defeat, F1,21 = 5.05 at ZT1 and significant effect of fluoxetine, F1,20 = 6.17 at ZT21). N = 6–8/group/timepoint. *p< 0.05, **p < 0.01, ***p < 0.001. Data presented as mean ± S.E.M.
Discussion
Though the link between stress and body weight change has long been appreciated, less is known about the neurobiological mechanisms that underlie the effect of antidepressant treatment on body weight changes. We hypothesized that fluoxetine treatment would reverse the hyperphagia observed in mice following CSDS. In the current study, we report several novel observations on the association of stress, antidepressants, and food intake among stress-susceptible animals.
Stress and Satiation
Previously we reported that mice displaying depression-like behavior after CSDS exhibit pronounced hyperphagia (Chuang, Cui, et al., 2010; Chuang, Krishnan, et al., 2010). Here we find that CSDS increases food intake primarily by increasing meal size, suggesting a specific deficit in satiation or the processes that terminates a meal. This finding is consistent with previous work in the visible burrow system in which subordinate male rats increase food intake during the recovery phase by increasing meal size (Melhorn, et al., 2010). In contrast, other stress models lacking a social component, such as the chronic unpredictable stress and chronic restraint stress models, report a decrease in food intake in response to stress (Chiba, et al., 2012; Varga, et al., 2011). It may be the case that psychosocial stress, rather than more neutral environmental stressors, uniquely contributes to the development of hyperphagia. For this reason, our key finding of enhanced meal size has several potential clinical implications. First, it indicates that behavioral techniques aimed at improving satiation and not satiety (how long one’s appetite remains suppressed after a meal) should be the focus of treatment of obesity in patients with mental disorders (Blundell & MacDiarmid, 1997; Green, Delargy, Joanes, & Blundell, 1997). Secondly, it begins to delineate the neural circuits that control different aspects of food intake following exposure to stress. Ghrelin signaling directly on catecholaminergic neurons affects mood symptoms and conditioned place preference for high fat diet following CSDS (Chuang, et al., 2011). In contrast, changes in satiation may be regulated by the coordinated action of the vagus nerve and gut hormones, which serve as peripheral sensors of energy intake to control meal size (Berthoud, 2008).
Fluoxetine and Satiation
Our observation that meal size is restored to control levels in CSDS animals treated with chronic fluoxetine carries several implications for the role of serotonin in satiation mechanisms. Serotonin has been demonstrated to be an important regulator of feeding in rodents. Administration of serotonin receptor agonists activates pro-opiomelanocortin (POMC)-expressing and inhibits agouti-related peptide/neuropeptide Y (AgRP/NPY)-expressing neurons in the arcuate nucleus of the hypothalamus, resulting in a suppression of food intake (Heisler, et al., 2006; Lam, et al., 2008). Additionally, animals lacking melanocortin 4 receptors (MC4R), as well as transgenic mice overexpressing the MC4R antagonist agouti, are unresponsive to the anorectic effects of augmented serotonin signaling (Heisler, et al., 2006; Lam, et al., 2008), which suggests an essential role for melanocortin signaling in the regulation of food intake by serotonin. These studies provide a general framework through which to interpret our finding that fluoxetine suppresses meal size; however, it is unclear why we do not observe a similar reduction among control mice. In nonstressed rats, it was also reported that daily food intake in the terminal phase of a chronic fluoxetine regimen was unaltered by the drug (Gamaro, et al., 2008). The mechanism explaining this discrepancy between control and CSDS animals may involve important neural adaptations to repeated social stress that sensitize feeding circuits to the appetite-suppressing effects of fluoxetine. This is a plausible hypothesis in the context of CSDS, which is known to induce long-lasting disturbances in limbic circuitry responsible for orchestrating both emotional and appetitive behaviors (Berton, et al., 2006; Tsankova, et al., 2006). As an example, various models of both acute and chronic stress, including CSDS, result in increased activity of mesocorticolimbic dopaminergic circuitry (Krishnan, et al., 2007; Lammel, Ion, Roeper, & Malenka, 2011; Valenti, Lodge, & Grace, 2011), a circuit which is thought to coordinate motivated behaviors associated with feeding (Palmiter, 2007). Moreover, in the CSDS model, hyperactivity of ventral tegmental area (VTA) dopaminergic neurons is restored to control, nonstressed levels by a chronic fluoxetine regimen similar to the one we utilize here (Cao, et al., 2010). Of note is the fact that dopamine neuron firing among nonstressed animals is unaffected by fluoxetine (Cao, et al., 2010), again pointing to a phenotype-specific effect of this antidepressant. Interestingly, ghrelin receptors expressed on presynaptic dopaminergic neurons of the VTA are known to provide excitatory input to this neuronal population (Abizaid, et al., 2006), and infusion of ghrelin directly into the VTA results in increased food intake (Naleid, Grace, Cummings, & Levine, 2005). It is possible that, in the CSDS model, fluoxetine normalizes the stress-induced decrease in satiation by reducing acyl-ghrelin levels, which in turn function to reverse the neuronal adaptations associated with CSDS. While this possibility has yet to be fully examined, we speculate that CSDS alters the basic function of satiation circuits in such a way as to render them plastic to modulation by fluoxetine.
Regulation of ghrelin and leptin levels by CSDS and fluoxetine
Ghrelin and leptin levels have previously been shown to be increased and decreased, respectively, in rodent models of stress including chronic unpredictable stress and CSDS (Asakawa, et al., 2001; Chuang, Cui, et al., 2010; Chuang, et al., 2011; Garza, Guo, Zhang, & Lu, 2011; Kristenssson, et al., 2006; Lu, Kim, Frazer, & Zhang, 2006; Lutter, Sakata, et al., 2008; Ochi, et al., 2008; Patterson, Ducharme, Anisman, & Abizaid, 2010; Rouach, et al., 2007). Here, our findings indicate that acyl-ghrelin, but not leptin, levels are altered after fluoxetine exposure following CSDS (Fig. 2A and 2B). It should be noted that serum hormone levels were measured from the overall pool of mice and not the sub-group selected for metabolic cage monitoring, although similar levels of hyperphagia were observed in both groups, suggesting that the groups are likely comparable.
Fluoxetine treatment after CSDS is associated with reduced ghrelin levels at ZT 9, but with the appearance of a peak at ZT 17. One possible mechanism for this action was reported by the Inui Laboratory, who demonstrated that selective serotonin reuptake inhibitors, including fluoxetine, decrease acyl-ghrelin levels via central activation of the 5-HT2c receptor (Fujitsuka, et al., 2009). Our data suggest that 5-HT2c receptor signaling may be important for the timing of the acyl-ghrelin peak and that fluoxetine treatment shifts the peak until late in the dark phase (the potential implications of this shift on feeding behaviors will be discussed below). We have previously shown that ghrelin receptor signaling is required for the increased food intake and conditioned place preference for high fat diet induced by CSDS (Chuang, et al., 2011; Lutter, Sakata, et al., 2008). Further studies will be necessary to determine if fluoxetine normalizes satiation after CSDS via changes in ghrelin signaling or through a distinct mechanism.
Regulation of meal patterns by CSDS-induced changes in leptin and ghrelin
We also show that changes in leptin and acyl-ghrelin levels only occur at specific times of the day. After CSDS, leptin levels are significantly lower than in control mice during the late light/early dark phase. Meanwhile, CSDS induces a peak in ghrelin levels in late light phase at ZT9. This well-timed increase in the relative ratio of ghrelin/leptin may explain why feeding differences are only observed in the dark phase. Recent work from the Sternson Laboratory identified a ‘flip-flop’ memory circuit in AgRP/NPY neurons in the arcuate nucleus of the hypothalamus (Yang, Atasoy, Su, & Sternson, 2011). In their model, acyl-ghrelin acts on pre-synaptic neurons to increase glutamate release onto AgRP/NPY neurons in a feed-forward loop. This excitatory neurotransmission increases feeding responses for hours, even after withdrawal of ghrelin, until leptin terminates feeding by acting on POMC-expressing neurons to inhibit AgRP/NPY neuronal activity via opioid release. Our data suggest that CSDS reduces leptin levels from ZT5 to ZT17, creating a 12-hour ‘window’ in which hyperphagia can occur. However, feeding does not happen until late in the light phase when acyl-ghrelin levels peak. The feeding bout would then continue through the early dark phase until leptin levels rise at ZT17 in the late dark phase. This model would also explain why a shift in the peak of acyl-ghrelin levels in the CSDS-fluoxetine group from ZT9 to ZT17 is not associated with increased food intake because leptin levels have returned to baseline and no longer permit activation of AgRP/NPY neurons.
Potential Clinical Relevance
One limitation of this study is that our findings only apply to the specific sub-groups that were selected for analysis. Specifically, we included mice susceptible to social avoidance after exposure to CSDS in addition to mice that displayed reversal of social avoidance by treatment with the antidepressant fluoxetine. This decision was made to achieve greater relevance for patients who present in clinical situations. Typically patients presenting for treatment have experienced mood-related symptoms induced by periods of stress and frequently receive antidepressant medications. Therefore, our treatment groups most accurately reflect actual clinical scenarios. Further studies that analyze metabolic parameters in mice ‘unsusceptible’ to the effects of CSDS as well as avoidant mice that do not respond to antidepressant treatment may be warranted to determine the metabolic consequences of these states.
Conclusions
Together these observations support a model in which stress pathways reduce satiation to increase meal size. Additionally, antidepressant treatment may normalize food intake in tandem with mood symptoms among subjects exposed to psychosocial stress. These insights may yield more effective treatments for stress-induced obesity.
Supplementary Material
A. Social interaction results of animals in study population. (B–F) Additional parameters of mice monitored in metabolic cages. B. Body weight (no significant difference), C. Lean body mass (no significant effect), D. adipose tissue (significant effect of defeat, F1,18 = 6.31), E. Locomotor activity during light phase (significant effect of defeat, F1,18 = 6.19 and significant effect of fluoxetine, F1,18 = 7.12), and F. Locomotor activity during dark phase (significant effect of defeat, F1,18 = 5.41). N = 5–6/group. *p< 0.05, **p < 0.01, ***p < 0.001. Data presented as mean ± S.E.M.
Highlights.
Chronic social defeat stress increases meal size in mice
Fluoxetine reverses stress-induced increase in meal size
Fluoxetine shifts stress-induced increases in plasma acyl-ghrelin levels
Intrameal satiation is regulated in a model of depression and by an antidepressant
Acknowledgments
Support. National Institutes of Health: K08 MH084058-1A1 (ML), R01 MH082876 (CAM), R01 MH085298 (JMZ), UL1RR024923, RL1 DK081182, RL1 DK081185, T32-MH076690-05 (EN), and the Medical Scientist Training Program (JK). National Alliance for Research in Schizophrenia and Depression (NARSAD): Young Investigator Award (ML). The University of Texas Southwestern Medical Center: The Disease Oriented Clinical Scholars Program (ML) and the Summer Undergraduate Research Fellowship Program (AK).
We thank Edgaro Falcón, Sadé Spencer, Angela Walker, and Michelle Sidor for technical assistance.
Abbreviations
- CSDS
Chronic Social Defeat Stress
- ZT
Zeitgeber Time
Footnotes
Conflict of Interest: None (JK, J-CC, ESN, AK, AGG, SM, JMZ, ML). CAM has received honoraria from GlaxoSmithKline, Pfizer, and Servier, consulting fees from Orphagen Pharmaceuticals, and research funding from GlaxoSmithKline and Pfizer for unrelated projects.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschop MH, Gao XB, Horvath TL. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SE, Cohen P, Naumova EN, Jacques PF, Must A. Adolescent obesity and risk for subsequent major depressive disorder and anxiety disorder: prospective evidence. Psychosom Med. 2007;69:740–747. doi: 10.1097/PSY.0b013e31815580b4. [DOI] [PubMed] [Google Scholar]
- Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Fujimiya M, Katsuura G, Makino S, Fujino MA, Kasuga M. A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology. 2001;74:143–147. doi: 10.1159/000054680. [DOI] [PubMed] [Google Scholar]
- Avgustinovich DF, Gorbach OV, Kudryavtseva NN. Comparative analysis of anxiety-like behavior in partition and plus-maze tests after agonistic interactions in mice. Physiol Behav. 1997;61:37–43. doi: 10.1016/s0031-9384(96)00303-4. [DOI] [PubMed] [Google Scholar]
- Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil. 2008;20(Suppl 1):64–72. doi: 10.1111/j.1365-2982.2008.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Beydoun MA, Wang Y. Pathways linking socioeconomic status to obesity through depression and lifestyle factors among young US adults. J Affect Disord. 2010;123:52–63. doi: 10.1016/j.jad.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell JE, MacDiarmid JI. Fat as a risk factor for overconsumption: satiation, satiety, and patterns of eating. J Am Diet Assoc. 1997;97:S63–69. doi: 10.1016/s0002-8223(97)00733-5. [DOI] [PubMed] [Google Scholar]
- Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59:323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JL, Covington HE, 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han MH. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–16458. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39:112–119. doi: 10.1016/j.pnpbp.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Chuang JC, Cui H, Mason BL, Mahgoub M, Bookout AL, Yu HG, Perello M, Elmquist JK, Repa JJ, Zigman JM, Lutter M. Chronic social defeat stress disrupts regulation of lipid synthesis. J Lipid Res. 2010;51:1344–1353. doi: 10.1194/jlr.M002196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Krishnan V, Yu HG, Mason B, Cui H, Wilkinson MB, Zigman JM, Elmquist JK, Nestler EJ, Lutter M. A beta3-adrenergic-leptin-melanocortin circuit regulates behavioral and metabolic changes induced by chronic stress. Biol Psychiatry. 2010;67:1075–1082. doi: 10.1016/j.biopsych.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Perello M, Sakata I, Osborne-Lawrence S, Savitt JM, Lutter M, Zigman JM. Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest. 2011;121:2684–2692. doi: 10.1172/JCI57660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwastiak LA, Rosenheck RA, Kazis LE. Association of psychiatric illness and obesity, physical inactivity, and smoking among a national sample of veterans. Psychosomatics. 2011;52:230–236. doi: 10.1016/j.psym.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizza G. Major depressive disorder is a risk factor for low bone mass, central obesity, and other medical conditions. Dialogues Clin Neurosci. 2011;13:73–87. doi: 10.31887/DCNS.2011.13.1/gcizza. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SS. Post-traumatic Stress Disorder and Cardiovascular Disease. Open Cardiovasc Med J. 2011;5:164–170. doi: 10.2174/1874192401105010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SS, Kang HK, Mahan CM. Selected Health Conditions Among Overweight, Obese, and Non-Obese Veterans of the 1991 Gulf War: Results from a Survey Conducted in 2003–2005. Open Epidemiol J. 2011;4:140–146. doi: 10.2174/1874297101104010140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, Quadros IM, Hogenelst K, Planeta CS, Miczek KA. Social defeat stress in rats: escalation of cocaine and “speedball” binge self-administration, but not heroin. Psychopharmacology (Berl) 2011;215:165–175. doi: 10.1007/s00213-010-2139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit LM, Fokkema M, van Straten A, Lamers F, Cuijpers P, Penninx BW. Depressive and anxiety disorders and the association with obesity, physical, and social activities. Depress Anxiety. 2010;27:1057–1065. doi: 10.1002/da.20738. [DOI] [PubMed] [Google Scholar]
- Dedert EA, Becker ME, Fuemmeler BF, Braxton LE, Calhoun PS, Beckham JC. Childhood traumatic stress and obesity in women: The intervening effects of PTSD and MDD. J Trauma Stress. 2010 doi: 10.1002/jts.20584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitsuka N, Asakawa A, Hayashi M, Sameshima M, Amitani H, Kojima S, Fujimiya M, Inui A. Selective serotonin reuptake inhibitors modify physiological gastrointestinal motor activities via 5-HT2c receptor and acyl ghrelin. Biol Psychiatry. 2009;65:748–759. doi: 10.1016/j.biopsych.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Gamaro GD, Prediger ME, Lopes J, Bassani MG, Dalmaz C. Fluoxetine alters feeding behavior and leptin levels in chronically-stressed rats. Pharmacol Biochem Behav. 2008;90:312–317. doi: 10.1016/j.pbb.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Garza JC, Guo M, Zhang W, Lu XY. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3beta/beta-catenin signaling. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AR, Rue T, Takeuchi D. Racial/ethnic differences in the association between obesity and major depressive disorder: findings from the Comprehensive Psychiatric Epidemiology Surveys. Public Health Rep. 2010;125:698–708. doi: 10.1177/003335491012500512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SM, Delargy HJ, Joanes D, Blundell JE. A satiety quotient: a formulation to assess the satiating effect of food. Appetite. 1997;29:291–304. doi: 10.1006/appe.1997.0096. [DOI] [PubMed] [Google Scholar]
- Grignaschi G, Samanin R. Role of serotonin and catecholamines in brain in the feeding suppressant effect of fluoxetine. Neuropharmacology. 1992;31:445–449. doi: 10.1016/0028-3908(92)90082-z. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Jobst EE, Sutton GM, Zhou L, Borok E, Thornton-Jones Z, Liu HY, Zigman JM, Balthasar N, Kishi T, Lee CE, Aschkenasi CJ, Zhang CY, Yu J, Boss O, Mountjoy KG, Clifton PG, Lowell BB, Friedman JM, Horvath T, Butler AA, Elmquist JK, Cowley MA. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51:239–249. doi: 10.1016/j.neuron.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Berton O, Nestler E. The use of animal models in psychiatric research and treatment. Am J Psychiatry. 2008;165:1109. doi: 10.1176/appi.ajp.2008.08071076. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Kristenssson E, Sundqvist M, Astin M, Kjerling M, Mattsson H, Dornonville de la Cour C, Hakanson R, Lindstrom E. Acute psychological stress raises plasma ghrelin in the rat. Regul Pept. 2006;134:114–117. doi: 10.1016/j.regpep.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Lam DD, Przydzial MJ, Ridley SH, Yeo GS, Rochford JJ, O’Rahilly S, Heisler LK. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology. 2008;149:1323–1328. doi: 10.1210/en.2007-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightowler S, Wood M, Brown T, Glen A, Blackburn T, Tulloch I, Kennett G. An investigation of the mechanism responsible for fluoxetine-induced hypophagia in rats. Eur J Pharmacol. 1996;296:137–143. doi: 10.1016/0014-2999(95)00704-0. [DOI] [PubMed] [Google Scholar]
- Lu XY, Kim CS, Frazer A, Zhang W. Leptin: a potential novel antidepressant. Proc Natl Acad Sci U S A. 2006;103:1593–1598. doi: 10.1073/pnas.0508901103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter M, Krishnan V, Russo SJ, Jung S, McClung CA, Nestler EJ. Orexin signaling mediates the antidepressant-like effect of calorie restriction. J Neurosci. 2008;28:3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhorn SJ, Krause EG, Scott KA, Mooney MR, Johnson JD, Woods SC, Sakai RR. Meal patterns and hypothalamic NPY expression during chronic social stress and recovery. Am J Physiol Regul Integr Comp Physiol. 2010;299:R813–822. doi: 10.1152/ajpregu.00820.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas AK, Mendola P, Pastor PN, Reuben CA, Cleary SD. The association between major depressive disorder and obesity in US adolescents: results from the 2001–2004 National Health and Nutrition Examination Survey. J Behav Med. 2011 doi: 10.1007/s10865-011-9340-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson D, Amsterdam JD, Quitkin FM, Reimherr FW, Rosenbaum JF, Zajecka J, Sundell KL, Kim Y, Beasley CM., Jr Changes in weight during a 1-year trial of fluoxetine. Am J Psychiatry. 1999;156:1170–1176. doi: 10.1176/ajp.156.8.1170. [DOI] [PubMed] [Google Scholar]
- Naleid AM, Grace MK, Cummings DE, Levine AS. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides. 2005;26:2274–2279. doi: 10.1016/j.peptides.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Ochi M, Tominaga K, Tanaka F, Tanigawa T, Shiba M, Watanabe T, Fujiwara Y, Oshitani N, Higuchi K, Arakawa T. Effect of chronic stress on gastric emptying and plasma ghrelin levels in rats. Life Sci. 2008;82:862–868. doi: 10.1016/j.lfs.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Palmiter RD. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007;30:375–381. doi: 10.1016/j.tins.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Papakostas GI, Petersen T, Iosifescu DV, Burns AM, Nierenberg AA, Alpert JE, Rosenbaum JF, Fava M. Obesity among outpatients with major depressive disorder. Int J Neuropsychopharmacol. 2005;8:59–63. doi: 10.1017/S1461145704004602. [DOI] [PubMed] [Google Scholar]
- Patterson ZR, Ducharme R, Anisman H, Abizaid A. Altered metabolic and neurochemical responses to chronic unpredictable stressors in ghrelin receptor-deficient mice. Eur J Neurosci. 2010;32:632–639. doi: 10.1111/j.1460-9568.2010.07310.x. [DOI] [PubMed] [Google Scholar]
- Perkonigg A, Owashi T, Stein MB, Kirschbaum C, Wittchen HU. Posttraumatic stress disorder and obesity: evidence for a risk association. Am J Prev Med. 2009;36:1–8. doi: 10.1016/j.amepre.2008.09.026. [DOI] [PubMed] [Google Scholar]
- Rivera M, Cohen-Woods S, Kapur K, Breen G, Ng MY, Butler AW, Craddock N, Gill M, Korszun A, Maier W, Mors O, Owen MJ, Preisig M, Bergmann S, Tozzi F, Rice J, Rietschel M, Rucker J, Schosser A, Aitchison KJ, Uher R, Craig IW, Lewis CM, Farmer AE, McGuffin P. Depressive disorder moderates the effect of the FTO gene on body mass index. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.45. [DOI] [PubMed] [Google Scholar]
- Rouach V, Bloch M, Rosenberg N, Gilad S, Limor R, Stern N, Greenman Y. The acute ghrelin response to a psychological stress challenge does not predict the post-stress urge to eat. Psychoneuroendocrinology. 2007;32:693–702. doi: 10.1016/j.psyneuen.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Rygula R, Abumaria N, Flugge G, Fuchs E, Ruther E, Havemann-Reinecke U. Anhedonia and motivational deficits in rats: impact of chronic social stress. Behav Brain Res. 2005;162:127–134. doi: 10.1016/j.bbr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Scott KM, McGee MA, Wells JE, Oakley Browne MA. Obesity and mental disorders in the adult general population. J Psychosom Res. 2008;64:97–105. doi: 10.1016/j.jpsychores.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71:1259–1272. doi: 10.4088/JCP.09r05346blu. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Hegeman MA, Nguyen MM, Melhorn SJ, Ma LY, Woods SC, Sakai RR. Dynamic body weight and body composition changes in response to subordination stress. Physiol Behav. 2007;91:440–448. doi: 10.1016/j.physbeh.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashiro KL, Hegeman MA, Sakai RR. Chronic social stress in a changing dietary environment. Physiol Behav. 2006;89:536–542. doi: 10.1016/j.physbeh.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Nguyen MM, Fujikawa T, Xu T, Yun Ma L, Woods SC, Sakai RR. Metabolic and endocrine consequences of social stress in a visible burrow system. Physiol Behav. 2004;80:683–693. doi: 10.1016/j.physbeh.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Nguyen MM, Ostrander MM, Gardner SR, Ma LY, Woods SC, Sakai RR. Social stress and recovery: Implications for body weight and body composition. Am J Physiol Regul Integr Comp Physiol. 2007 doi: 10.1152/ajpregu.00371.2007. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Nguyen MM, Sakai RR. Social stress: from rodents to primates. Front Neuroendocrinol. 2005;26:27–40. doi: 10.1016/j.yfrne.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tschop MH, Speakman JR, Arch JR, Auwerx J, Bruning JC, Chan L, Eckel RH, Farese RV, Jr, Galgani JE, Hambly C, Herman MA, Horvath TL, Kahn BB, Kozma SC, Maratos-Flier E, Muller TD, Munzberg H, Pfluger PT, Plum L, Reitman ML, Rahmouni K, Shulman GI, Thomas G, Kahn CR, Ravussin E. A guide to analysis of mouse energy metabolism. Nat Methods. 2011;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Lodge DJ, Grace AA. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci. 2011;31:4280–4289. doi: 10.1523/JNEUROSCI.5310-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga J, Domokos A, Barna I, Jankord R, Bagdy G, Zelena D. Lack of vasopressin does not prevent the behavioural and endocrine changes induced by chronic unpredictable stress. Brain Res Bull. 2011;84:45–52. doi: 10.1016/j.brainresbull.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Vieweg WV, Fernandez A, Julius DA, Satterwhite L, Benesek J, Feuer SJ, Oldham R, Pandurangi AK. Body mass index relates to males with posttraumatic stress disorder. J Natl Med Assoc. 2006;98:580–586. [PMC free article] [PubMed] [Google Scholar]
- Vieweg WV, Julius DA, Bates J, Quinn JF, 3rd, Fernandez A, Hasnain M, Pandurangi AK. Posttraumatic stress disorder as a risk factor for obesity among male military veterans. Acta Psychiatr Scand. 2007;116:483–487. doi: 10.1111/j.1600-0447.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- Wilson DR. Health consequences of childhood sexual abuse. Perspect Psychiatr Care. 2010;46:56–64. doi: 10.1111/j.1744-6163.2009.00238.x. [DOI] [PubMed] [Google Scholar]
- Wong DT, Reid LR, Threlkeld PG. Suppression of food intake in rats by fluoxetine: comparison of enantiomers and effects of serotonin antagonists. Pharmacol Biochem Behav. 1988;31:475–479. doi: 10.1016/0091-3057(88)90376-0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146:992–1003. doi: 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Guo M, Garza J, Rendon S, Sun XL, Zhang W, Lu XY. Cognitive and neural correlates of depression-like behaviour in socially defeated mice: an animal model of depression with cognitive dysfunction. Int J Neuropsychopharmacol. 2011;14:303–317. doi: 10.1017/S1461145710000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Social interaction results of animals in study population. (B–F) Additional parameters of mice monitored in metabolic cages. B. Body weight (no significant difference), C. Lean body mass (no significant effect), D. adipose tissue (significant effect of defeat, F1,18 = 6.31), E. Locomotor activity during light phase (significant effect of defeat, F1,18 = 6.19 and significant effect of fluoxetine, F1,18 = 7.12), and F. Locomotor activity during dark phase (significant effect of defeat, F1,18 = 5.41). N = 5–6/group. *p< 0.05, **p < 0.01, ***p < 0.001. Data presented as mean ± S.E.M.