

Abstract
The hepatocyte nuclear factor 4 alpha (HNF4α, NR2A1) is a member of the nuclear receptor (NR) family of transcription factors that use conserved DNA binding domains (DBDs) and ligand binding domains (LBDs)1,2. HNF4α is the most abundant DNA-binding protein in the liver, where some 40% of the actively transcribed genes have a HNF4α response element 1,3,4. These regulated genes are largely involved in the hepatic gluconeogenic program and lipid metabolism3,5,6. In the pancreas too, HNF4α is a master regulator controlling an estimated 11% of islet genes7. HNF4α protein mutations are linked to Maturity Onset of Diabetes in Young 1 (MODY1) and hyperinsulinemic hypoglycemia (HH)8–11. Prior structural analyses of NRs, while productive with individual domains, have lagged in revealing the connectivity patterns of NR domains. Here, we describe the 2.9 Å crystal structure of the multi-domain HNF4α homodimer bound to its DNA response element and coactivator-derived peptides. A convergence zone connects multiple receptor domains in an asymmetric fashion joining distinct elements from each monomer. An arginine target of PRMT1 methylation protrudes directly into this convergence zone and sustains its integrity. A serine target of protein kinase C is also responsible for maintaining domain-domain interactions. These post-translational modifications manifest into changes in DNA binding by communicating through the tightly connected surfaces of the quaternary fold. We find that some MODY1 mutations, positioned on the LBD and hinge regions of the receptor, compromise DNA binding at a distance by communicating through the inter-junctional surfaces of the complex. The overall domain representation of the HNF4α homodimer is different from that of the PPARγ-RXRα heterodimer, even when both NR complexes are assembled on the same DNA element. Our findings suggest that unique quaternary folds and inter-domain connections in NRs could be exploited by small-molecule allosteric modulators that impact distal functions in these polypeptides.
We previously reported the only high resolution structural example of a multi-domain NR complex, that of the PPARγ-RXRα heterodimer on its DNA response element12. To understand the extent of domain integration in other NRs, we analyze here the crystal structure of the complex of HNF4α, an obligate homodimer bound to its DNA element and coactivator derived peptides. HNF4α uses the linear domain arrangement shown in Figure 1A. Our efforts to crystallize the full-length HNF4α were unsuccessful. However, by proteolytically probing its DNA-assembled complex, we identified an extended segment corresponding to the DBD-hinge-LBD portions corresponding to residues 46-368 (Supplementary Figure 1). Cloning, expression and purification of the stable DBD-hinge-LBD multi-domain segment made it possible to obtain well-diffracting crystals of a complex with its consensus response element and coactivator (NcoA2) peptide. Electron density maps for all the inter-domain junctions of the complex are shown in Supplementary Figures 3–5. The response element consists of a direct repeat of AGGTCA half-sites with one base-pair spacing (DR1). The DR1 is the major consensus binding site for both HNF4α and PPARγ-RXRα4,6,13.
Figure 1. Overall organization of the HNF4α homodimer on DNA.

a, Linear depiction of the HNF4α protein domains. b, Electron density map (2fo-fc) for one of the two HNF4α homodimer/DNA complexes of the crystallographic asymmetric unit. c, Superposition of the two independent HNF4α homodimer/DNA complexes in the asymmetric unit. The two homodimeric complexes are colored orange/yellow in one case, and blue/purple in the other. The ligands in the LBD are shown in blue, Zn is shown in grey, and the coactivator peptides are in red/brown. Numbers with prime refer to the upstream positioned subunit. d, The positioning of both LBDs in a complex on top of the upstream DBD fosters high affinity DR1 DNA. e, Contribution of various receptor domains to the DNA binding affinity of HNF4α DNA binding is measured using fluorescent polarization studies with a 5′ FITC-labeled DR1. The x-axis shows concentration of the HNF4α protein, and the y-axis shows the fractional DNA bound. Removal of the AB domain (ΔAB), F-domain (ΔF) or both (ΔABΔF) does not alter the DNA affinity compared to the full-length (FL) receptor. However, removal of the LBD reduces the affinity of the resulting DBD-hinge (DBD) region to a Kd of approximately 6000 nM, whereas the presence of the LBD together with the DBD-hinge (ΔABΔF) allows DNA binding with a Kd of 82 nM (see also Supplementary Table 2).
X-ray diffraction data was collected to 2.9 Å resolution and the structure refined (see Supplementary Table 1). The crystal asymmetric unit contains two independent representations of the HNF4α homodimer/DNA/peptide complex. The electron density map from one complex, and the comparison of the two complexes is in Figures 1B–C. The two representations are nearly identical, with RMSD of less than 2.0 A over all their atoms. The LBD and DBD portions match their previously determined isolated structures (Supplementary Figure 6–7). Both DBDs are in register with their half-sites, interacting with the major grooves (Supplementary Figure 8–9). Helix-12 of the LBDs is in the active conformation and a coactivator LXXLL peptide is bound to each LBD.
The HNF4α homodimer shows a striking and complex pattern of interfacial junctions. A central zone incorporates surfaces from both LBDs, the DBD of the upstream subunit, and the hinge region of the downstream subunit. This domain convergence zone suggests a path of communication between the conserved domains through their coupled surfaces (Figure 2A). The LBDs, symmetrical in their mutual interactions when viewed in isolation, cooperate in a highly asymmetric fashion to straddle the surface of only the upstream DBD (Figure 1d). As a result, the overall complex appears partitioned towards the upstream half of the DR1, and adopts a highly asymmetrical organization for a homodimeric transcription factor. A previous study suggested that HNF4α homodimers could bind asymmetrically to their DNA response elements6. The resulting quaternary arrangement creates precisely the correct DBD to DBD distances needed to match the geometric constraints of the two AGGTCA half-sites and their intervening spacer. At the same time, the quaternary organization renders both LBD pockets and their coactivator interacting surfaces unencumbered, allowing free access to both ligands and LLXXLL elements, respectively.
Figure 2. Domain-domain contacts of HNF4α.

a, Circle indicates the convergence center where four domains (both LBDs, the upstream DBD, the downstream hinge) come together. b, Arg-91, located on the surface of the DBD, inserts deeply into a pocket at the base of the LBD-LBD surface. Numbers with prime refer to the upstream positioned subunit. c, Ser-78, positioned on the back-side of the DNA recognition helix, also fosters the convergence of the DBD and the LBDs.
The interface that forms between the upstream subunit’s DBD and the downstream subunit’s hinge region is one important domain-domain interface of the complex, and reminiscent of an interaction we described previously for the PPARγ-RXRα complex12. The resulting arrangement places the two DBDs in a solid head-to-tail arrangement that extends their combined footprint to perfectly match their DR1 contact surface. The manner by which two LBDs cooperate to interact with the upstream DBD is particularly evocative, suggesting the physical integration of all three domains may be required for high affinity DNA-binding (Figure 1d). Measuring first the DNA binding affinity of the HNF4α that only contains its DBD and hinge portions, we observed very weak binding to DR1 with a Kd of approximately 6000 nM. When the LBD portion of the receptor is contained within the polypeptide, the complex displayed a 75-fold enhanced affinity for DR1, with a Kd of approximately 80 nM (see Figure 1E and Supplementary Table 2). These results are consistent with our observation that the LBD and DBD modules are physically and functionally integrated to establish high-affinity DNA binding. These findings are consistent with previous study that showed the LBD enhances the half-life of the HNF4α DNA binding complex significantly 14.
We next measured the DNA binding contributions of the N-terminal (AB region) and C-terminal (F region) portions of the polypeptide, both of which were removed from our crystallization construct (Figure 1A). We found little contribution from these segments to the overall affinity of the complex for DNA, when examined individually or in combination (Figure 1D). The proteolytically sensitive nature of these regions, even in the DNA binding complex of HNF4α also suggests they are poorly ordered and not involved in DNA binding (Supplementary Figure 1a–c). We additionally prepared the isolated AB and F domain fragments of HNF4α and tested their ability to bind to the rest of the homodimeric-DNA complex. However, we detected no appreciable binding of these receptor portions with the rest of the complex (Supplementary Figure 1d).
Each of the HNF4α LBDs displays electron density for a trapped a fatty acid, consistent in size with a myristic acid derived from E. coli, where the protein was expressed (Supplementary Figure 2)15,16. The fatty acid is believed to lend structural integrity to the HNF4α/γ subfamily. Linoleic acid has been shown to be an exchangeable and potential endogenous ligand of HNF4α, although this molecule does not confer significant transcriptional activity17. A stabilizing fatty acid, or a silent molecule that cannot switch on and off receptor activity, raises the question of how HNF4α activity is otherwise regulated.
The activities of NRs can be regulated by a variety of post-translational modifications (PTMs)18. In the case of HNF4agr;, two PTMs are well-described for their ability to regulate receptor properties 19,20. These modifications control the receptor’s ability to bind DNA, and by extension its ability to regulate gene expression. Here, we identify the quaternary sites of these PTMs within HNF4α for the first time. The first site, Arg-91, is a target of PRMT1, an enzyme that adds up to two methyl groups to the arginine side-chain. Arg-91 methylation produces a marked enhancement in the DNA binding activity of HNF4α19. The second site Ser-78, is phosphorylated by protein kinase C (PKC) which disrupts the ability of HNF4α to bind DNA20. Therefore, taken together, these two PTMs act as on and off switches for regulating the receptor activity.
Arg-91 methylation substantially enhances DNA-affinity, but is not positioned to directly influence DNA binding from its location on the DBD farthest from the DNA. Figure 2B shows how its side-chain deeply protrudes into the LBD-LBD cooperating surface that we described above as the receptor’s multi-domain convergence center. There is a cavity directly above the side-chain of Arg-91 to accommodate the two extra methyl groups, and the extension of the side-chain through methylation would more firmly “glue” the DBD junctional interface with both LBDs. Therefore, this PTM acts to allosterically bias the receptor to bind DNA, by stabilizing the inter-domain junctions associated with the final productive DNA complex.
We next analyzed the location of Ser-78, the site of PKC phosphorylation in a number of NRs20. Along with HNF4α, other NRs including FXR, RAR, VDR, PPARα, PXR and TR2are similarly targeted by PKC, which in each case phosphorylates a similarly positioned serine on the DBD. Curiously, this serine always resides on the “wrong side” of the DNA recognition helix, as is the case in HNF4α, where it seemingly cannot participate directly in DNA binding (Figure 2C)20,21. Yet Ser-78 phosphorylation nevertheless weakens receptor-DNA binding substantially20. Our structure indicates that Ser-78 is positioned to engage the receptor’s interfacial connections so as to reduce DNA binding allosterically. Figure 2C suggests how an added phosphate group on this residue would create clash, both in size and charge, with nearby Tyr-319, a residue that physically connects the receptor LBD with the DBD through Ser-78. Phosphorylation would compromise the integrity of the quaternary fold needed for efficient DNA binding. Allosteric mechanisms of this type cannot be understood using the isolated crystal structures of DBDs or LBDs alone, as both Arg-91 and Ser-78 would appear to be too far from DNA-binding surface from that analysis. The current analysis of the domain organization, however, shows the unique positioning of these residues being consistent with their ability to impact the receptor’s DNA binding function allosterically.
We next asked whether some MODY1 and HH linked point mutations are similarly positioned in sensitive inter-junctional surfaces (Figure 3A). For R76W and R80W mutations (in HH), there is a simple explanation for receptor dysfunction, as this pair of arginine residues directly contacts the AGGTCA half-sites (Supplementary Figures 9 and 10). V255M alters a residue that points into the LBD pocket, the only residue doing so among all the MODY1 and HH mutations. We found a number of mutations lie at the sensitive domain-domain junctions of the complex. Sites such as R127W, D126Y, D126H, and R125W locate to the downstream hinge region where it forms domain-domain arrangements with the upstream DBD (Supplementary Figures 4 and 10). Mutational changes in this hinge site would misalign the interaction between domain-domain surfaces required to bridge the two DBDs into register with their successive AGGTCA half-sites. Indeed, we find these mutant proteins had substantially compromised DNA affinity (Supplementary Figure 10). This loss of DNA binding also translates to a reduction in the transcriptional activity 22. We next examined MODY1 mutations I314F, R324H, and their adjacent residues (R322A, Q318A, D316A and N315A), which were found to be on the LBD and at the multi-domain convergence center of the complex (Figure 3B–D). These mutations reduced the DNA affinity and transcriptional activity of the receptor (Figure 3D, and Supplementary Figure 11).
Figure 3. Disease-linked mutations in HNF4α.

a, Summary of MODY1 and HH point mutations identified clinically in human populations. b,c, The MODY1 mutations, in many cases (residues in red) map to the “convergence center” of the receptor domains (blue circle in b). d, DNA affinity measurements of the WT and mutant receptors, as described in Figure 1e. See also Supplementary Figure 10 for studies of other disease-linked mutations.
Our examination of PTMs and MODY1 mutations show that changes introduced in the LBD, the hinge region, or in the DBD away from the DNA interface, still impact the DNA binding properties of the receptor at a distance, by communicating through the inter-domain junctions of the quaternary fold. It is interesting to note the subtlety of a single PTM or a single amino-acid mutational change, and the large distance with which these signals travel across the polypeptide to modulate DNA-binding. Therefore, the domain convergence center should be appropriately viewed as both a sensitive center for receiving signals, and an allosteric transmission system for propagating signals. At the same time, it is important to note that the two subunits of the homodimer are in altogether different environments due to the asymmetric nature of the two subunits. PTM sites such as Ser-78 or Arg-91 significantly influence the complex only if they occur in the upstream DBD. In the same way, some MODY1 mutations would appear to be damaging if located in one, but not the other, subunit of the homodimer. Du to the α in the liver and pancreas, the loss of even a fractional population of functional homodimers caused by heterozygous mutations is disease-causing.
Since both the HNF4α homodimer and PPARγ-RXRα complexes target DR1, we asked if their quaternary architectures were related. The common DR1 is expected establish a similar DBD-DBD spacing in these complexes. Figures 4A–B present the PPARγ-RXRα heterodimer and the HNF4α homodimer in an identical way, based on the layout of their common DR1 sequences. Figure 4C shows the superposition of these complexes when their DR1s are aligned to match. Indeed, the DBDs occupy nearly identical positions in both DR1 complexes. Nevertheless, the higher order quaternary arrangements are distinct for these two complexes (Figure 4). In HNF4α, the LBDs are biased toward the upstream DBD, while in the PPARγ-RXRα complex is biased toward the downstream RXR DBD. Moreover, PPARγ-RXRα complex has its own type of domain convergence center, which is not identical to what we see in the HNF4α complex.
Figure 4. Comparison the HNF4α homodimer and the PPARγ-RXRα heterodimer complexes on DR1 DNA.
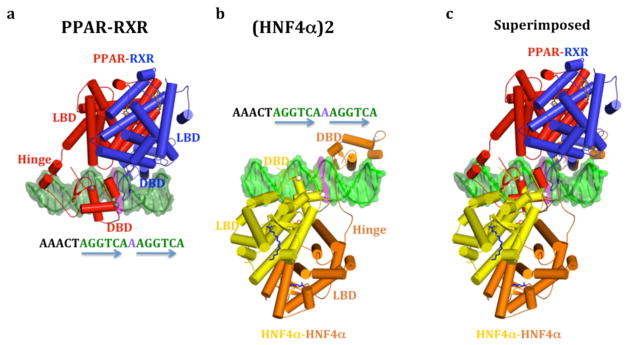
a, The PPARγ-RXRα heterodimer on DR1, b, The HNF4α homodimer on DR1, and c) their overlap when the DR1 sequences are superimposed, showing the distinct domain-domain arrangements in these two complexes. The two complexes are shown in an identical fashion with respect to the DNA sequence facing the viewer.
The structural comparison indicates that the DNA response element type is not the only driver of quaternary structure in NRs. Receptor organization appears to be highly dependent on the constellation of non-conserved amino-acids on these LBD surfaces, and the length and sequence of the hinge segments, which are unique to NR members. We also note that DNA recognition is not identical in these two complexes. The PPAR uses its hinge region to recognize an additional six base-pair segment located upstream to the DR1 core element, establishing the polarity of subunits in that heterodimer. HNF4α subunits do not use their hinge regions for DNA recognition, nor do they contact sequences outside the core DR1.
Our crystallographic studies with both NR complexes argue against the notion of a “common architecture” for the full-length NRs23. Our findings also dispel the view that NR polypeptides are arrays of “domains-on-a-string”, each of which confers its own independent function without physical and functional integration. The repertoire of quaternary structures in the NR family is likely to be diverse, even though both the DBDs and LBDs are conserved. This expectation stems from the fact that neither hinge regions, nor LBD surface residues are conserved in the NR family, yet these features are the key drivers of quaternary folding. The multiple response element configurations employed in the NR family are another driver of quaternary organization.
Mounting evidence points to the importance of inter-domain communication in the NR family. For estrogen receptors, the activities of ligands are influenced by the response elements, and DNA can also influence coactivator binding24. In the glucocorticoid receptors, small conformational changes in the DBD propagate across the receptor to influence the LBD, and in the androgen receptor too there evidence of DBD to LBD communication25,26. Our findings reveal that PTMs can modulate the inter-domain connections in the quaternary fold. It has been reported that certain PPARγ ligands can selectively block the Cdk5 phosphorylation of PPARγ2, indicating communications between the LBD pocket and the site of phosphorylation27,28. Ser-273 in PPARγ is positioned within a domain-domain junction of the PPARγ-RXRα complex (Supplementary Figure 12). From its position, the phosphorylation state of Ser-273 can communicate to the PPARγ ligand binding pocket and to the DNA reading heads of the PPARγ-RXRα heterodimer.
For HNF4α, small molecules, directed to the sensitive inter-junctional junctions sites may prove to be beneficial for treating MODY1 patients where the DNA binding properties have been mutationally compromised. To find these molecules, high throughput screening efforts must target the complete architecture of this receptor and not just the isolated LBD. We point out two locations in the quaternary structure of the HNF4α complex that appear accessible for the binding of small-molecule allosteric modulators (Supplementary Figure 13). An expanded understanding of the physical connectivity between LBDs, DBDs and other domains in the NR family should expand and better guide the discovery of receptor modulators with therapeutic value.
METHODS
Expression, purification and crystallization
HNF4α proteins used in this study reference the NCBI sequence HNF4 NM_178849. All HNF4α (human) constructs including FL (1-464), ΔABΔF (46-368), ΔF (1-368), ΔAB (46-464), and DBD (46-126), and others were expressed from pET46 Ek/LIC vector in BL21(DE3) E. coli cells (Novagen). HNF4α ΔABΔF (human) construct was used in crystallization experiments. Cells were induced with 0.5 mM IPTG at 17 °C for 16 hours, and lysed in 20 mM Tris (pH 7.5), 500 mM NaCl, 20 mM imidazole and 10% glycerol. Purification used involved His-Bind resin (Novagen), SP-Sepharose column (GE Healthcare) and Gel filtration (Superdex 200). Purified HNF4α was then combined with oligonucleotide (DR1) in 1:1.5 molar ratio. The oligonucleotide strands 5′-GGAACTAGGTCAAAGGTCAG-3′ and 5′ CCTGACCTTTGACCTAGTTC 3′ were purified and annealed. Coactivator peptide (EKHKILHRLLQDSY) was also added in 3X molar ratio. A final Gel filtration step was carried out to remove any excess DNA. Crystals were grown with 5–10% PEG 3350, 25 mM MgCl2, 25 mM NH4CL, 10 mM DTT, and 0.1 M MES pH.6.5. at 4 °C.
Data collection and structure determination
Diffraction data was collected at the Argonne National Laboratory SBC-CAT 19ID beamline and the structure solved by molecular replacement. X-ray data were collected at a wavelength of 0.9793 Å, at 100 degrees Kelvin. The backbone dihedral (Ramachandran) angles for the amino-acids in the final coordinates conform to preferred/allowed/disallowed statistics of (%) 90.5/8.2/1.3. The details of X-ray diffraction data collection, structure solution and refinement are in Supplementary Table 1.
Fluorescence polarization assay
All the mutants used in DNA binding assays were prepared using QuikChange Site-Directed Mutagenesis (Stratagene). The DNA strands 5′-GGAACTAGGTCAAAGGTCAG-3′ and 5′ CCTGACCTTTGACCTAGTTC were annealed to make the DR1 for binding studies. For binding assay, 2nM fluoresceinated DNA (5′ end conjugation on the top strand) was incubated with purified HNF4α protein for two hours at room temperature. Protein concentration was varied by serial dilution in binding buffer (20mM Tris-HCl, pH 7.8, 20mM NaCl, 8% glycerol, 10mM DTT). The fluorescence polarization signals were recorded using 96-well black polystyrene plates on FlexStation 3. The data were later converted to fluorescence anisotropy values and normalized. The Kd of each construct and mutants were calculated by fitting the curve in KaleidaGraph 4.1.
Interactions of the A/B and F domains with the HNF4α receptor complex
For the study shown in Supplementary Figure 1D, the AB domain (residues 1-45) and F domain (369-465) of human HNF4α were expressed from pET46 Ek/LIC vector in BL21(DE3) E. coli cells (Novagen). Each protein was purified using a HisTrap™ column, followed by size exclusion chromatography on a HiLoad™ 16/60 Superdex™ 200 column. The final samples were prepared in 10mM phosphate (pH=7.0) and 100mM NaCl buffer for fluorescein labeling. 10μg of each peptide, HNF4α-AB, HNF4α-F and PGC-1α LXXLL-motif peptide (AEEPSLLKKLLAY, synthetically made and purchased from AnaSpec) were incubated with fluorescein-5-EX succinimidyl ester at molar ratio of 1:2 in 100mM potassium phosphate (pH=7.0) coupling buffer at 37°C for 60 minutes, quenched by adding 100mM Tris (pH=8) and incubated for 30 minutes at room temperature. The labeled peptides were purified using Sephadex™ G-15 column for fluorescence polarization measurements. The HNF4αΔABΔF-DNA complex was prepared as described in the crystallization method section for these peptide binding assays. 5nM of each fluorescein-labeled peptide was incubated with HNF4α-DNA complex of serially-diluted concentrations for 2 hours at room temperature. Similar fluorescence polarization signals recording and data processing were conducted as described in the method section for fluoresceinated DNA binding assays.
Transcription Reporter Assays
For the transcriptional reporter studies in Supplementary Figure 11, we employed both HEK293T and COS-7 cells that were seeded in 24-well plates and one day later transfected with 400 ng of the pCMV-Tag1-HNF4α (46-368) wild-type or mutant plasmid, 100 ng of apoCIII-pTKLuc reporter (a generous gift from Dr. Daniel Kelly at SBMRI) and 10 ng of pRL (control Renilla luciferase) using jetPEI regent (Polyplus) according to the manufacturer’s protocol. Luciferase activity was measured 48 h after transfection using the Dual-Glo Luciferase Assay System (Promega) and data were normalized by the relative ratio of firefly and Renilla luciferase activity.
Supplementary Material
Footnotes
Accession Code
The PDB ID code is 4IQR
See separate file provided that accompanies this manuscript
Author Contributions
V.C. expressed, purified and crystallized the complex. P.H. along with V.C. solved and refined the structure and carried out the mutational binding studies. Y.K. collected, processed and reduced the X-ray diffraction data and assisted with the molecular replacement search in structure determination. N.P. along with V.C. made the expression constructs for crystallization and for DNA binding studies. D.W. carried out the transcription assays. F.R. supervised the work and wrote the manuscript.
Author information
The authors declare no competing financial interests.
References
- 1.Sladek FM, Zhong WM, Lai E, Darnell JE., Jr Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4:2353–2365. doi: 10.1101/gad.4.12b.2353. [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 3.Bolotin E, et al. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology. 2010;51:642–653. doi: 10.1002/hep.23357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallerman O, et al. Molecular interactions between HNF4a, FOXA2 and GABP identified at regulatory DNA elements through ChIP-sequencing. Nucleic Acids Res. 2009;37:7498–7508. doi: 10.1093/nar/gkp823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon JC, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 6.Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res. 2012;40:5343–5356. doi: 10.1093/nar/gks190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odom DT, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryffel GU. Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol. 2001;27:11–29. doi: 10.1677/jme.0.0270011. [DOI] [PubMed] [Google Scholar]
- 9.Ellard S, Colclough K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat. 2006;27:854–869. doi: 10.1002/humu.20357. [DOI] [PubMed] [Google Scholar]
- 10.Kapoor RR, et al. Hyperinsulinaemic hypoglycaemia. Arch Dis Child. 2009;94:450–457. doi: 10.1136/adc.2008.148171. [DOI] [PubMed] [Google Scholar]
- 11.Flanagan SE, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162:987–992. doi: 10.1530/EJE-09-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandra V, et al. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang G, Lee U, Sladek FM. Proposed mechanism for the stabilization of nuclear receptor DNA binding via protein dimerization. Mol Cell Biol. 1997;17:6546–6554. doi: 10.1128/mcb.17.11.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wisely GB, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure. 2002;10:1225–1234. doi: 10.1016/s0969-2126(02)00829-8. [DOI] [PubMed] [Google Scholar]
- 16.Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem. 2002;277:37973–37976. doi: 10.1074/jbc.C200420200. [DOI] [PubMed] [Google Scholar]
- 17.Yuan X, et al. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One. 2009;4:e5609. doi: 10.1371/journal.pone.0005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weigel NL, Moore NL. Steroid receptor phosphorylation: a key modulator of multiple receptor functions. Mol Endocrinol. 2007;21:2311–2319. doi: 10.1210/me.2007-0101. [DOI] [PubMed] [Google Scholar]
- 19.Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24:233–243. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun K, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol. 2007;21:1297–1311. doi: 10.1210/me.2006-0300. [DOI] [PubMed] [Google Scholar]
- 21.Gineste R, et al. Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol Endocrinol. 2008;22:2433–2447. doi: 10.1210/me.2008-0092. [DOI] [PubMed] [Google Scholar]
- 22.Lu P, et al. Structural basis of natural promoter recognition by a unique nuclear receptor, HNF4alpha. Diabetes gene product. J Biol Chem. 2008;283:33685–33697. doi: 10.1074/jbc.M806213200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rochel N, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat Struct Mol Biol. 2011;18:564–570. doi: 10.1038/nsmb.2054. [DOI] [PubMed] [Google Scholar]
- 24.Hall JM, McDonnell DP, Korach KS. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol. 2002;16:469–486. doi: 10.1210/mend.16.3.0814. [DOI] [PubMed] [Google Scholar]
- 25.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helsen C, et al. Evidence for DNA-binding domain--ligand-binding domain communications in the androgen receptor. Mol Cell Biol. 2012;32:3033–3043. doi: 10.1128/MCB.00151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi JH, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi JH, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.