

Abstract
We report on a pedigree with a pair of brothers each with minor anomalies, developmental delay and autistic-symptoms who share an unbalanced translocation (not detectable by karyotype). The unbalanced translocation involves a 7.1 Mb loss of the terminal portion of 10q, and a 4.2Mb gain of 11q. One of the brothers also developed a cerebellar juvenile pilocytic astrocytoma. The father was found to be a balanced carrier and the couple had a previous miscarriage. We demonstrate that the breakpoint for the triplicated region from chromosome 11 is adjacent to two IgLON genes, namely Neurotrimin (NTM) and Opioid Binding/Cell Adhesion Molecule-like (OPCML). These genes are highly similar neural cell adhesion molecules that have been implicated in synaptogenesis and oncogenesis respectively. The children also have a 10q deletion and are compared to other children with the 10q deletion syndrome which generally does not involve autism spectrum disorders or cancer. Together these data support a role for NTM and OPCML in developmental delay and potentially in cancer susceptibility.
Keywords: autism spectrum disorder, cerebellar juvenile pilocytic astrocytoma, unbalanced translocation, t(10;11), NTM, OPCML
INTRODUCTION
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental conditions characterized by abnormalities in social interactions and communication, as well as restricted interests and repetitive behavior. The development of ASD is etiologically complex; however genetic factors have been shown to play a major role. The genetic anomalies implicated in ASDs are exceedingly heterogeneous; amongst these different kinds of genetic anomalies, genomic structural imbalances are prominent [Abrahams and Geschwind, 2008; Folstein and Rosen-Sheidley, 2001].
In this report, we examine an unbalanced translocation involving deletion of chromosome 10q and duplication of 11q with development of ASD and of cerebellar juvenile pilocytic astrocytoma. Distal chromosome 10q deletions represent a recurrent syndrome with characteristic features including craniofacial dysmorphisms, congenital heart and urogenital defects, growth retardation and development delay [Tanabe et al., 1999; Wulfsberg et al., 1989]. As autism spectrum disorders and oncogenesis are generally not considered a part of the 10q deletion syndrome, the associated 11q duplication may be potentially implicated in these processes in our patients. To this end, immediately adjacent to the breakpoint on chromosome 11q are two neuronal cell adhesion molecules of the IgLON group (a subgroup of the immunoglobulin superfamily of cell adhesion molecules) [Schofield et al., 1989; Struyk et al., 1995]. In particular, Neurotrimin (NTM) has been implicated in Alzheimer disease [Liu et al., 2007] and in influencing intelligence [Pan et al., 2011], and Opioid-Binding Protein/Cell Adhesion Molecule (OPCML) in the epigenetics of tumor progression [Cui et al., 2008]. The association of cancer with ASD is an important topic [Butler et al., 2005; Tabares-Seisdedos and Rubenstein, 2009] as each may have genetic liabilities. To our knowledge there have been no previously reported cases involving the particular loci we report in this case.
CLINICAL REPORTS
The two brothers were age 7 and 5 years old respectively at the time of presentation. Their parents are healthy individuals who are nonconsanguineous. Neither of the parents or anyone in the family has been diagnosed with ASD or intellectual disability. The parents have had a previous pregnancy that ended in a miscarriage.
The older brother was born healthy after a normal pregnancy and weighed approximately 7 lb at birth. His developmental milestones were delayed. Within his first month, he was noted to have difficulty with latching on during feeding, and he struggled with nursing. At approximately 3 months of age he was noted to have difficulty fixing and tracking visually. He was eventually diagnosed with alternating esotropia for which he had corrective surgery with partial improvement in vision. His motor skills showed considerable delay secondary to generalized hypotonia. He was unable to sit without support until 18 months and was unable to walk independently until 3 years. He was also noted to have fine motor delays such as difficulty grasping a pencil appropriately.
Language development was also delayed. At 14 months, characteristics of verbal apraxia were noted. Therapy was initiated and at 23 months he could string together some vowels and consonants. At this time although he had age-appropriate receptive language skills, his expressive language scores fell 2 SDs below the mean. His first clear words did ot come until 3 years, after which his language development advanced rapidly.
In terms of behavior, the patient was noted to be very sensitive to certain textures. He also developed fixations and obsessional behavior. He also had intermittent motor tics, including blinking. No history of seizures or other neurological conditions were noted. No dysmorphic features noted on physical exam.
With regard to cognitive and adaptive assessments, results of the Differential Ability Scales-II (DAS-II) indicated that his overall cognitive abilities fell in the low-average range (GCA score of 89, 23rd centile); however, he exhibited a high degree of variability amongst his domain scores. He obtained a Verbal Cluster score of 102 (average) and his performance in the Verbal Comprehension and the Naming Vocabulary subtests were at the 31st and 76th centile respectively (suggesting that his expressive vocabulary is stronger than his receptive language skills). His nonverbal reasoning cluster score was 95, with a score of 44 and 51 in picture similarities and matrices respectively (suggesting that his nonverbal reasoning skills are comparable to those of same-age peers). On the spatial subsets, he obtained a spatial cluster of 79 (low range) and he scored 36 (8th centile) and 40 (16th centile) in the pattern construction and copying subsets respectively.
On the PPVT-4, the patient’s single-word receptive language fell in the average range (score=101, 53rd centile), and on the Vineland-II his adaptive behavior composite fell in the moderately low range. (Communication, Receptive Communication and Expressive Communication were all in the adequate range, while Daily Living and Socialization were in the moderately low range, and Motor Skills were in the low range.) The patient was also administered the ADOS-Module 3, which showed that he used complex language primarily involving short sentences with the occasional use of unusual language, and some repetitive questioning. His eye contact was limited, as was spontaneous gesturing and range of facial expression. He also engaged in some atypical social interactions (like belching out loud). He had a limited ability to describe emotions. He exhibited some sensory-seeking behaviors including feeling the texture of a book, picking at the label on a water-bottle, and visually inspecting/peering at a toy. Results of the above evaluations, in addition to the ADI-R based on parental report, indicated that his presentation was consistent with autism.
The younger brother (who was 5 at the time of examination) exhibited strabismus and GERD at age 3 to 4 months along with intermittent episodes of tonic stiffening. He was also noted to have decreased fine motor skills, apraxia, difficulty with motor planning and mild axial and appendicular hypotonia. There was some concern regarding torticollis, and he was given a tentative diagnosis of Sandifer syndrome.
At 3 years 7 months an MRI revealed a left cerebellar juvenile pilocytic astrocytoma. This was removed shortly thereafter, with significant improvement. After the surgery he was able to run downhill (something he was unable to do in the past). Also, prior to the surgery there were episodes of intermittent eye fluttering and unresponsiveness (EEG did not show any seizure activity), which improved after the surgery. His fine motor skill also improved, but some problems with motor planning persisted.
Subsequent to the surgery, the patient exhibited OCD symptoms and tics similar to his brother. The OC-spectrum symptoms continued for months in his fifth year and up to the time of this report. Diagnoses being considered by his child psychiatrist included Tourette syndrome and PDD-NOS. The child was not available for standardized testing.
Head circumference for the older brother was at the 24th centile at 28 months and for the younger brother was at 25th centile at 12 months. Neither had a broad nasal bridge, hypertelorism, malformed ears or any other dysmorphic features.
MATERIALS AND METHODS
Microarray analysis
Genome-wide clinical microarray analysis carried out on the younger brother revealed two imbalances: a ~7.1 Mb loss of terminal region of long arm of chromosome 10 (from 128,130,813 bp to 135,286,082 bp) and an ~4.0 Mb gain of the terminal region of chromosome 11 (from 130,413,338 bp to 134,432,324 bp) (data not shown).
Karyotype
Cytogenic analysis carried out on the older brother and on the parents revealed an apparently normal male karyotype with no consistent chromosome abnormality or rearrangement evident (data not shown).
FISH
Targeted FISH analysis was carried out on metaphase cells from cultured blood using centromeric probes along with the telomere probes to identify chromosomes. The result in the younger brother of this FISH analysis showed one signal for the probe specific to the 10q telomere, consistent with a deletion of this region observed by microarray analysis, and three signals for the probe specific to the 11q telomere, consistent with the gain of this region observed by microarray analysis: one hybridization signal was observed on each normal chromosome 11 and the third hybridization signal was on the long arm of a C-group chromosome. Inverted DAPI staining demonstrated that the extra hybridization signal for 11q is on 10q. Therefore the loss of the long arm of chromosome 10 and the gain of the long arm of chromosome 11 are the result of a simple unbalanced translocation between these two chromosomes.
Targeted FISH analysis was also carried out on the parents. The father was found to be a balanced carrier, and the mother was found to be normal (Fig 1).
Figure 1.
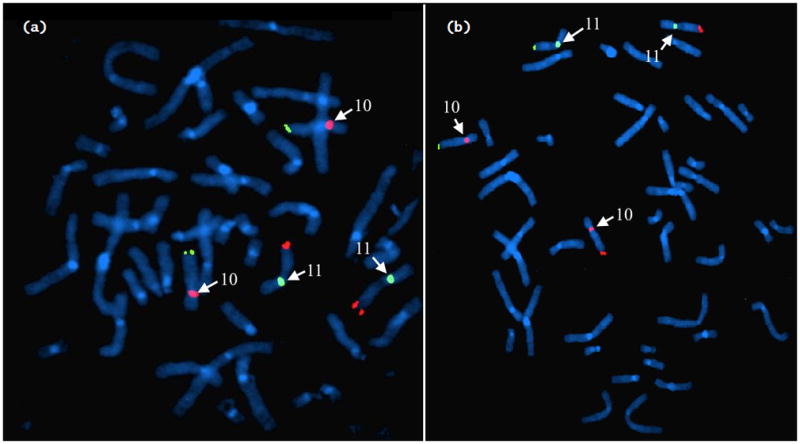
FISH analysis of parents (a: mother, b: father) using centromeric probes along with telomeric probes to identify chromosomes. Probes for chromosome 10 show up pink for the centromere and green for the telomere; for chromosome 11 they show up green for the centromere and red for the telomere (centromere probes labeled). The mother’s FISH shows a normal arrangement, with 2 chromosomes 10 (pink centromere + green telomeres) and 2 chromosomes 11 (green centromere + red telomeres). The father’s FISH shows the translocation: one chromosome 10 and one chromosome 11 with the expected pink and green and green and red pattern. The other chromosomes 10 and 11 have a pink centromere with red telomere, and green centromere with green telomere respectively.
Custom fine-tiling array CGH
Custom array comparative genomic hybridization (aCGH) was performed. The total distance covered was 8,638,924 (126,840,823 to 135,524,747) and 5,538,388 (129,408,128 to 134,946,516) base pairs on chromosomes 10 and 11 respectively. There was a probe after every 350 base pairs. This was performed on DNA obtained from both brothers to accurately determine the breakpoint of the translocation. The breakpoint on chromosome 10 was at position 128,138,744, and breaks the C10orf90 gene between exon 14 and exon 15 such that C10orf90 (exons 1–14) and all subsequent genes are lost to the deletion (Human NCBI build 37/Hg19) (Fig 2). The triplicated breakpoint started at position 130,898,884 on chromosome 11, approximately 340 kb away from the transcription start site for NTM approximately (Fig 2).
Figure 2.
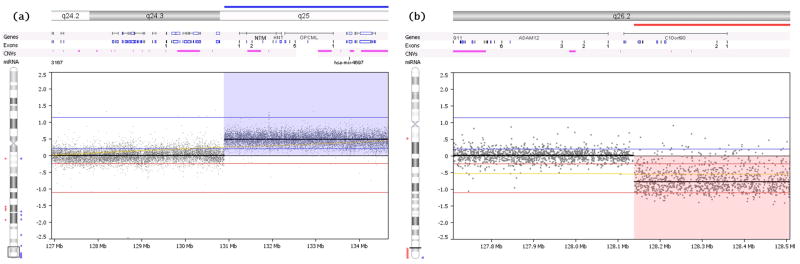
Custom fine-tiling array comparative genomic analysis (aCGH) showing the breakpoint in the chromosomes (a: chromosome 10, b: chromosome 11). The breakpoint in chromosome 10 is adjacent to the NTM and OPCML genes. The breakpoint on chromosome 11 breaks through the C10orf90 gene and leaves a small portion of the 3′-prime portion of C10orf90 gene on the remaining portion of 10q. The deletion and the duplication both extend to the end of the telomere.
DISCUSSION
We present a pedigree with an unbalanced translocation involving a 7.1Mb loss of the terminal portion of 10q and a 4.2 Mb gain of 11q, associated with development of ASD and cerebellar juvenile pilocytic astrocytoma.
Loss of the terminal part of 10q (10q deletion syndrome) has been reported before, and has a diverse phenotypic presentation. Facial features include microcephaly, a broad nasal bridge, hypertelorism, strabismus, malformed or low-set ears, thin upper lip vermilion, downslanting palpebral fissures, and a short neck. Other features include cardiac and urogenital anomalies, developmental and motor delay, and neurodevelopmental deficits [Scigliano et al., 2004].
Some of the features in the children we report are consistent with symptoms reported in terminal 10q deletion syndrome. These include neurodevelopmental delay, delay in motor development and strabismus. ASD has been reported in only one case of del(10q26.2-q26.3) [Yatsenko et al., 2009]. Somatic deletion of portions of 10q has been discovered to arise in glioblastomas [Steck et al., 1999], although cancer has never been reported to be associated with germ-line 10q deletion syndrome. Several genes within the 10q deletion interval may play role in cancer. These include: O6-Methylguanine-DNA Methyltransferase gene (MGMT), inactivation of which has been implicated in development of various cancers including glioblastoma multiforme [Lotfi et al., 2011]; Early B-cell Factor 3 gene (EBF3), a putative tumor suppressor gene the silencing of which has been associated with development of various brain, colorectal, breast, liver, and bone tumors [Zhao et al., 2006]; Dihydropyrimidinase-like 4 gene (DPYSL4), a novel apoptosis-inducible factor controlled by p53 in response to DNA damage [Kimura et al., 2011], and Inositol Polyphosphate-5-Phosphatase A gene (INPP5A) which has been associated with cutaneous squamous cell carcinoma [Sekulic et al., 2010].
The breakpoint for the triplicated region on chromosome 11 was adjacent to Neurotrimin (NTM) and Opioid Binding Protein/Cell Adhesion Molecule-like (OPCML) which are both from the IgLON gene cluster, and encode similar neural cell adhesion molecules. The OPCML gene has been implicated as a tumor suppressor gene in ovarian cancer, being frequently silenced as an early event in ovarian cancer cell lines [Czekierdowski et al., 2006; Sellar et al., 2003]. A more recent study has shown that OPCML mRNA down-regulation occurs in a majority of intracranial tumors and glioma cell lines [Reed et al., 2007]. Recent studies have also implicated SNPs in the OPCML gene in schizophrenia [O’Donovan et al., 2008; Panichareon et al., 2012]; however no studies have linked it with other developmental disorders or ASD. A recent association study showed that NTM and 11q25 in general is associated with intelligence quotient [Pan et al., 2011]. Studies have shown that NTM is involved in axonal fasciculation of specific cerebellar systems and may also be involved in the formation of excitatory synapses and their stabilization into adulthood [Chen et al., 2001].
Chromosome 10 Open Reading Frame 90 gene (C10orf90) which was broken in our pedigree, may have a role in primary cilium assembly and centriole formation [Knorz et al., 2010]. This gene has been previously reported in a deletion syndrome associated with craniofacial dysmorphism, various degrees of MR and growth failure [Yatsenko et al., 2009]. Disruption of the C10orf90 gene was disrupted at the chromosomal breakpoint; however, the 5′ region of the gene is lost to the deletion, thereby this break likely affected this gene by haploinsufficiency only and may be considered a candidate with the other 10q genes via haploinsufficiency.
In conclusion, this pedigree presents with developmental delays, autism spectrum disorder and cancer. The pedigree has some similarities to cases of 10q deletion as previously reported in the literature; however the pedigree also has additional features of ASD and cerebellar juvenile pilocytic astrocytoma. The topic of cancer arising in children with genetic causes for developmental delay is critical as many cellular mechanisms are known to concurrently cause each condition. Although there are many candidate genes, we postulate that the NTM and OPCML genes based on increased gene dosage may have a major role to play in the presentation in this pedigree. Further cases will need to be ascertained in order to establish this at a statistical level.
Acknowledgments
FUNDING
EMM is a recipient of the NIMH grant, 5K23MH080954-04, and he holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. This research was funded in part by a Mahoney Pilot Research Grant from the Brown Institute for Brain Science.
Footnotes
COMPETING INTERESTS
The authors report no biomedical financial interests and no conflict of interests.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gil O, Ren YQ, Zanazzi G, Salzer JL, Hillman DE. Neurotrimin expression during cerebellar development suggests roles in axon fasciculation and synaptogenesis. J Neurocytol. 2001;30(11):927–937. doi: 10.1023/a:1020673318536. [DOI] [PubMed] [Google Scholar]
- Cui Y, Ying Y, van Hasselt A, Ng KM, Yu J, Zhang Q, Jin J, Liu D, Rhim JS, Rha SY, Loyo M, Chan AT, Srivastava G, Tsao GS, Sellar GC, Sung JJ, Sidransky D, Tao Q. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS One. 2008;3(8):e2990. doi: 10.1371/journal.pone.0002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czekierdowski A, Czekierdowska S, Szymanski M, Wielgos M, Kaminski P, Kotarski J. Opioid-binding protein/cell adhesion molecule-like (OPCML) gene and promoter methylation status in women with ovarian cancer. Neuro Endocrinol Lett. 2006;27(5):609–613. [PubMed] [Google Scholar]
- Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2(12):943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Kimura J, Kudoh T, Miki Y, Yoshida K. Identification of dihydropyrimidinase-related protein 4 as a novel target of the p53 tumor suppressor in the apoptotic response to DNA damage. Int J Cancer. 2011;128(7):1524–1531. doi: 10.1002/ijc.25475. [DOI] [PubMed] [Google Scholar]
- Knorz VJ, Spalluto C, Lessard M, Purvis TL, Adigun FF, Collin GB, Hanley NA, Wilson DI, Hearn T. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol Biol Cell. 2010;21(21):3617–3629. doi: 10.1091/mbc.E10-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Arias-Vasquez A, Sleegers K, Aulchenko YS, Kayser M, Sanchez-Juan P, Feng BJ, Bertoli-Avella AM, van Swieten J, Axenovich TI, Heutink P, van Broeckhoven C, Oostra BA, van Duijn CM. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet. 2007;81(1):17–31. doi: 10.1086/518720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi M, Afsharnezhad S, Raziee HR, Ghaffarzadegan K, Sharif S, Shamsara J, Lary S, Behravan J. Immunohistochemical assessment of MGMT expression and p53 mutation in glioblastoma multiforme. Tumori. 2011;97(1):104–108. doi: 10.1177/030089161109700118. [DOI] [PubMed] [Google Scholar]
- O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini JL, Spencer CC, Howie B, Leung HT, Hartmann AM, Möller HJ, Morris DW, Shi Y, Feng G, Hoffmann P, Propping P, Vasilescu C, Maier W, Rietschel M, Zammit S, Schumacher J, Quinn EM, Schulze TG, Williams NM, Giegling I, Iwata N, Ikeda M, Darvasi A, Shifman S, He L, Duan J, Sanders AR, Levinson DF, Gejman PV, Cichon S, Nöthen MM, Gill M, Corvin A, Rujescu D, Kirov G, Owen MJ, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Cloninger CR. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- Pan Y, Wang KS, Aragam N. NTM and NR3C2 polymorphisms influencing intelligence: family-based association studies. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(1):154–160. doi: 10.1016/j.pnpbp.2010.10.016. [DOI] [PubMed] [Google Scholar]
- Panichareon B, Nakayama K, Thurakitwannakarn W, Iwamoto S, Sukhumsirichart W. OPCML gene as a schizophrenia susceptibility locus in Thai population. J Mol Neurosci. 2012;46(2):373–377. doi: 10.1007/s12031-011-9595-2. [DOI] [PubMed] [Google Scholar]
- Reed JE, Dunn JR, du Plessis DG, Shaw EJ, Reeves P, Gee AL, Warnke PC, Sellar GC, Moss DJ, Walker C. Expression of cellular adhesion molecule ‘OPCML’ is down-regulated in gliomas and other brain tumours. Neuropathol Appl Neurobiol. 2007;33(1):77–85. doi: 10.1111/j.1365-2990.2006.00786.x. [DOI] [PubMed] [Google Scholar]
- Schofield PR, McFarland KC, Hayflick JS, Wilcox JN, Cho TM, Roy S, Lee NM, Loh HH, Seeburg PH. Molecular characterization of a new immunoglobulin superfamily protein with potential roles in opioid binding and cell contact. EMBO J. 1989;8(2):489–495. doi: 10.1002/j.1460-2075.1989.tb03402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scigliano S, Gregoire MJ, Schmitt M, Jonveaux PH, LeHeup B. Terminal deletion of the long arm of chromosome 10. Clin Genet. 2004;65(4):294–298. doi: 10.1111/j.1399-0004.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- Sekulic A, Kim SY, Hostetter G, Savage S, Einspahr JG, Prasad A, Sagerman P, Curiel-Lewandrowski C, Krouse R, Bowden GT, Warneke J, Alberts DS, Pittelkow MR, DiCaudo D, Nickoloff BJ, Trent JM, Bittner M. Loss of inositol polyphosphate 5-phosphatase is an early event in development of cutaneous squamous cell carcinoma. Cancer Prev Res (Phila) 2010;3(10):1277–1283. doi: 10.1158/1940-6207.CAPR-10-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellar GC, Watt KP, Rabiasz GJ, Stronach EA, Li L, Miller EP, Massie CE, Miller J, Contreras-Moreira B, Scott D, Brown I, Williams AR, Bates PA, Smyth JF, Gabra H. OPCML at 11q25 is epigenetically inactivated and has tumor suppressor function in epithelial ovarian cancer. Nat Genet. 2003;34(3):337–343. doi: 10.1038/ng1183. [DOI] [PubMed] [Google Scholar]
- Steck PA, Lin H, Langford LA, Jasser SA, Koul D, Yung WK, Pershouse MA. Functional and molecular analyses of 10q deletions in human gliomas. Genes Chromosomes Cancer. 1999;24(2):135–143. doi: 10.1002/(sici)1098-2264(199902)24:2<135::aid-gcc6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Struyk AF, Canoll PD, Wolfgang MJ, Rosen CL, D’Eustachio P, Salzer JL. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J Neurosci. 1995;15(3 Pt 2):2141–2156. doi: 10.1523/JNEUROSCI.15-03-02141.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabares-Seisdedos R, Rubenstein JL. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. 2009;14(6):563–589. doi: 10.1038/mp.2009.2. [DOI] [PubMed] [Google Scholar]
- Tanabe S, Akiba T, Katoh M, Satoh T. Terminal deletion of chromosome 10q: clinical features and literature review. Pediatr Int. 1999;41(5):565–567. doi: 10.1046/j.1442-200x.1999.01105.x. [DOI] [PubMed] [Google Scholar]
- Wulfsberg EA, Weaver RP, Cunniff CM, Jones MC, Jones KL. Chromosome 10qter deletion syndrome: a review and report of three new cases. Am J Med Genet. 1989;32(3):364–367. doi: 10.1002/ajmg.1320320319. [DOI] [PubMed] [Google Scholar]
- Yatsenko SA, Kruer MC, Bader PI, Corzo D, Schuette J, Keegan CE, Nowakowska B, Peacock S, Cai WW, Peiffer DA, Gunderson KL, Ou Z, Chinault AC, Cheung SW. Identification of critical regions for clinical features of distal 10q deletion syndrome. Clin Genet. 2009;76(1):54–62. doi: 10.1111/j.1399-0004.2008.01115.x. [DOI] [PubMed] [Google Scholar]
- Zhao LY, Niu Y, Santiago A, Liu J, Albert SH, Robertson KD, Liao D. An EBF3-mediated transcriptional program that induces cell cycle arrest and apoptosis. Cancer Res. 2006;66(19):9445–9452. doi: 10.1158/0008-5472.CAN-06-1713. [DOI] [PubMed] [Google Scholar]