

Abstract
Craniofacial and neural tissues develop in concert throughout pre- and postnatal growth. FGFR-related craniosynostosis syndromes, such as Apert syndrome (AS), are associated with specific phenotypes involving both the skull and the brain. We analyzed the effects of the FGFR P253R mutation for Apert syndrome using the Fgfr2+/P253R mouse to evaluate the effects of this mutation on these two tissues over the course of development from day of birth (P0) to postnatal day 2 (P2). Three-dimensional magnetic resonance microscopy and computed tomography images were acquired from Fgfr2+/P253R mice and unaffected littermates at P0 (N=28) and P2 (N=23). 3D coordinate data for 23 skull and 15 brain landmarks were statistically compared between groups. Results demonstrate that the Fgfr2+/P253R mice show reduced growth in the facial skeleton and the cerebrum, while the height and width of the neurocranium and caudal regions of the brain show increased growth relative to unaffected littermates. This localized correspondence of differential growth patterns in skull and brain point to their continued interaction through development and suggest that both tissues display divergent postnatal growth patterns relative to unaffected littermates. However, the change in the skull-brain relationship from P0 to P2 implies that each tissue affected by the mutation retains a degree of independence, rather than one tissue directing the development of the other.
Keywords: Apert syndrome, craniosynostosis, suture, mouse, skull, brain, development, fibroblast growth factor receptor 2
INTRODUCTION
Apert syndrome (AS) is an autosomal dominant disorder occurring in approximately 15 out of 1 million live births characterized by coronal craniosynostosis, midfacial deficiency, syndactyly of the hands and feet, CNS abnormalities, and other anomalies, [Cohen et al., 1992]. More than 99% of all cases of AS are associated with one of two mutations of fibroblast growth factor receptor 2 (FGFR2) [Park et al., 1995; Wilkie et al., 1995]. Studies indicate that approximately one-third of individuals with Apert syndrome have missense mutations of the Pro253Arg amino acid, while the remaining individuals have a mutation in the neighboring amino acid, Ser252Trp [von Gernet et al., 2000; Wilkie et al., 1995]. These gain-of-function mutations modify the ligand-binding affinity and alter the specificity of FGFR2 toward a number of FGF ligands affecting many of the processes mediated by the FGF/FGFR signaling pathway including osteogenesis and central nervous system development [Eswarakumar et al., 2002; Ibrahimi et al., 2001; Marcucio et al., 2011; Wilke et al., 1997; Yu et al., 2000].
The constellation of phenotypic anomalies observed in AS results from alteration of the FGF/FGFR signaling pathway during development, including those of the skull and central nervous system (CNS). CNS anomalies include megalencephaly [Cohen and Kreiborg 1990; 1991] increased intracranial volume [Gosain et al., 1995; Posnick et al., 1995], ventriculomegaly [Pooh et al., 1999; Renier et al., 2000; Tokumaru et al., 1996; Yacubian-Fernandes et al., 2004], and dysmorphology of the corpus callosum and other CNS structures [Cohen and Kreiborg 1990; 1991; de Leon et al., 1987; Posnick et al., 1995]. Skull anomalies include premature coronal suture synostosis [Cohen 2000; Cohen and Kreiborg, 1996], midfacial hypoplasia, and a midline defect of the anterior fontanelle [Cohen 2000], among other anomalies. Longitudinal analyses of individuals with AS suggest that the size of the anterior fontanelle defect diminishes over time as bony islands form within it [Kreiborg and Cohen, 1990]. Intracranial volume in individuals with AS does not differ from unaffected individuals at birth, but is significantly larger after 3.5 months of age and continues to be significantly larger throughout the lifespan [Gosain et al., 1995]. The shape of the head, however, is not uniformly affected. There is an overall reduction of head circumference, while there is an increase in height (i.e., turribrachycephaly) with growth in individuals with AS [Cohen and Kreiborg 1993a].
A Fgfr2+/P253R Apert syndrome inbred mouse [Wang et al., 2010] has been developed and analyzed to fully characterize the specific contributions of the Pro253Arg mutation of the FGFR2 gene to phenotypes observed in individuals with AS. In an analysis of the morphology of the brain at postnatal day 0 (P0) in Fgfr2+/P253R mice, gross asymmetry of the overall brain, changes in the form of the corpus callosum, and enlargement of the ventricles were noted in those mice carrying the P253R mutation relative to unaffected littermates [Aldridge et al., 2010]. Additionally, there are varying degrees of coronal craniosynostosis, ranging from partial closure of one suture to complete fusion of both coronal sutures in Fgfr2+/P253R mice at birth, while all Fgfr2+/P253R P0 mice show synostosis of the zygomatic-maxillary and premaxilla-maxillary sutures [Martínez-Abadias et al., 2010; Wang et al., 2010]. Analysis of the overall form of the skull showed reduction rostrocaudally and increase dorsoventrally in the Fgfr2+/P253R mice relative to unaffected littermates [Martínez-Abadias et al., 2010; Wang et al., 2010].
Characterizations of skull and brain morphology at a single age (i.e., P0) provide a snapshot of the consequences of the FGFR2 mutations on the skull and the brain at a single developmental time point. Throughout growth, there is a continual interaction between tissues via biochemical and biomechanical mechanisms, such that the growth of the skull and the brain influence each other (Fig 1) [Mao et al., 2003; Marcucio et al., 2011; Opperman, 2000; Parsons et al., 2011; Richtsmeier et al., 2006; Yu and Ornitz, 2001]. By analyzing the magnitude and direction of growth-related changes in the skull and brain in Fgfr2+/P253R mice we can begin to form a picture of the timing and location of developmental contributions to phenotypes observed in AS.
Figure 1.

Three-dimensional reconstructions of the brain and skull of a postnatal day 0 unaffected mouse (A) and a postnatal day 2 unaffected mouse (B) illustrating the relationship between the brain and skull. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
By examining age-related change of the skull and brain in an inbred Fgfr2+/P253R model for AS at early developmental stages we can capture changes in the skull and brain that are most relevant to the age-related changes in individuals with AS at the time during which they develop, rather than after they have occurred. Postnatal day 2 (P2) mice roughly correspond to 10 month-old infants, in terms of body-to-brain ratios [Kobayashi, 1963]. Documenting age-related change from P0 to P2 allows correlation of findings in mice with clinically and developmentally analogous changes in individuals with AS prior to 1 year of age.
This study is the first to quantitatively examine growth of the skull and brain concurrently during this critical period of early postnatal development (from P0-P2) in Fgfr2+/P253R mice and their unaffected littermates. To do this, several quantitative analyses comparing Fgfr2+/P253R mice and unaffected (Fgfr2+/+) littermates are performed: 1) comparison of brain form at P0; 2) comparison of skull form at P0; 3) comparison of brain form at P2; 4) comparison of skull form at P2; 5) comparison of growth patterns of the brain from P0 to P2; 6) comparison of growth patterns of the skull from P0 to P2 in Fgfr2+/P253R mice and unaffected littermates; and 7) combined brain-skull analyses. For all analyses, we test the null hypothesis that Fgfr2+/P253R mice and unaffected littermates do not differ in form or growth.
MATERIALS AND METHODS
Breeding the Apert Fgfr2+ P253R mouse model
Fgfr2+/P253R mice and their unaffected littermates were bred on an inbred C57BL/6J background to minimize variation due to genetic differences [Wang et al., 2010]. P0 and P2 mice were euthanized by inhalation anesthetics and fixed in 4% paraformaldehyde. Genotyping of tail DNA was completed using polymerase chain reaction to identify mutant and unaffected mice. The care and use of mice for this study were in compliance with the animal welfare guidelines approved by the Mount Sinai School of Medicine Animal Care and Use Committee. Our sample consisted of Fgfr2+/P253R (N=15) and their unaffected littermates (N=13) from six litters at P0, and Fgfr2+/P253R (N=7) and their unaffected littermates (N=13) from eight litters at P2.
Magnetic Resonance Microscopy and Micro Computed Tomography Imaging Protocols
Whole, fixed mouse heads were submerged in 2% Magnevist (Bayer Health Care, Wayne, NJ) phosphor-buffered solution for 10 days prior to scanning [Aldridge et al., 2010]. Magnetic resonance microscopy (MRM) images of the heads of P0 and P2 mice were acquired at the High Field MRI facility at Pennsylvania State University (www.imaging.psu.edu/facilities/high-field). To prevent the animals from drying out and to minimize magnetic susceptibility artifacts during scanning, the specimens were immersed in a flourinert liquid, FC-43 (3M, St. Paul, MN). A vertical 14.1 Tesla Varian (Varian Inc., Palo Alto, CA) imaging system was used to acquire the MRM images of the P0 and P2 mouse heads. Postprocessing of the MRM images was completed with Matlab (The MathWorks, Inc., Natick, MA). Standard imaging experiments were performed with an isotropic resolution of 80 μm. By zero-filling each direction by a factor of two, the pixel resolution of the standard experiment was 40 (μm)3. Micro-computed tomography (μ-CT) images of the mice were acquired at the Center for Quantitative Imaging at Pennsylvania State University (www.cqi.psu.edu) using an HD-600 OMNI-X high resolution X-ray computed tomography system (Bio-Imaging Research Inc., Lincolnshire, IL) following previously established protocols [Hill et al., 2007; Martínez-Abadias et al., 2010]. Pixel sizes ranged from 15 to 20 μm and slice thicknesses ranged from 16 to 25 μm. μ-CT image data were reconstructed on 1024x1024 pixel grids as 16-bit TIFFs, and then reduced to 8-bit TIFFs for landmark collection and analysis. Based on hydroxyapatite phantoms imaged with the specimens, the minimum thresholds used to create bone isosurfaces ranged from 70 to 100 mg/cm3 partial density of hydroxyapatite.
Landmark Data Collection Protocols
Three-dimensional landmark coordinate data were collected from the MRM images of the brain and μ-CT images of the skull for each mouse. Landmark coordinate data were collected for 15 points on the brain [Aldridge et al., 2010] and 23 points on the skull [Martínez-Abadias et al., 2010] (Table I, Fig 2). Landmarks for both the brain and skull were digitized twice by the same observer to minimize measurement error following previously published protocols [Aldridge et al., 2007; Richtsmeier et al., 1995]. After checking for gross errors, the two trials were averaged for analysis.
Table I.
Anatomical definitions of the landmarks collected from MRM and μ-CT images of mice at P0 and P2. Landmarks are illustrated in Figure 2. Definitions of skull landmarks can be found at www.getahead.psu.edu.
| Brain Landmark(s) | Definition | Skull Landmark(s) | Abbreviation at www.getahead.psu.edu |
|---|---|---|---|
| B1, B2 | Most superolateral point of intersection of olfactory bulb with anterior frontal lobe surface (bilateral) | S1 | lnsla |
| S2 | lnslp | ||
| B3, B4 | Most caudolateral point on the occipital lobe surface (bilateral) | S3, S4 | lflac, rflac (bilateral) |
| B5, B6 | Most lateral point on the cerebellar surface (bilateral) | S5, S6 | lzyt, rzyt (bilateral) |
| B7 | Most caudal point on the cerebellar surface | S7, S8 | lpto, rpto (bilateral) |
| B8 | Genu of corpus callosum | S9, S10 | lfbc, rfbc (bilateral) |
| B9 | Anterior commissure | S11, S12 | lpst, rpst (bilateral) |
| B10 | Splenium of corpus callosum | S13, S14 | locc, rocc (bilateral) |
| B11 | Intersection of pons with most caudal aspect of the ventral cerebral surface | S15 | opi |
| B12, B13 | Origin of the middle cerebral artery from Circle of Willis on ventral cerebral surface (bilateral) | S16, S17 | laalf, raalf (bilateral) |
| S18 | ethma | ||
| B14, B15 | Centroid of the head of caudate nucleus (bilateral) | S19, S20 | lpalf, rpalf (bilateral) |
| S21 | cpsh | ||
| S22 | amsph | ||
| S23 | bas |
Figure 2.

Anatomical locations of the landmarks collected from the brain and skull of P0 and P2 mice, illustrated on 3-D reconstructions of MRM and μ-CT scans of a P2 unaffected mouse. A) Brain, dorsal view. B) Midsagittal slice. C) Brain, right lateral view. D) Horizontal slice. E) Skull, dorsal view. F) Skull, right lateral view. G) Skull, ventral view. Definitions for landmark abbreviations are provided in Table 1 and are further defined at www.getahead.psu.edu.
Morphometric analyses of landmark coordinate data
Overall size comparison
We statistically compared overall size of the skull and of the brain in Fgfr2+/P253R and unaffected mice at both P0 and P2. Size of the brain was measured as the geometric mean of all neural linear distances estimated using 3D neural landmarks, and skull size as the geometric mean of all skull linear distances estimated from 3D skull landmarks [Darroch and Mosimann, 1985; Falsetti et al., 1993; Jungers et al., 1995]. These measures were then statistically compared using ANOVA, with genotype, age, and genotype*age as factors in IBM SPSS 19.0 (IBM, New York).
Form comparison within each age group
Landmark coordinate data were analyzed using Euclidean Distance Matrix Analysis [Lele and Richtsmeier, 2001], a coordinate system invariant method for the statistical comparison of form and change in form due to growth. This approach defines the form of an object as that characteristic that remains invariant under any translation, rotation, or reflection of the object [Richtsmeier et al., 2002]. EDMA converts landmark coordinate data into a matrix of all possible linear distances among landmarks and tests for differences between groups using a nonparametric bootstrapping method. We have previously established statistically significant differences in the form of the brain and skull at P0 in this mouse model [see Aldridge et al., 2010; Martínez-Abadias et al., 2010; Wang et al., 2010].
To estimate differences in skull form, EDMA first estimates all possible unique linear distances among landmarks for each individual (the form matrix or FM) and then estimates the means of all linear distances for each group (the mean FM), the difference between groups as the ratio of like-linear distances for the two groups (form difference matrix, or FDM), and confidence intervals for the inter-group difference for each linear distance using a non-parametric bootstrapping procedure to statistically evaluate the similarity of linear distances between groups. This procedure allows localization of differences in form to particular linear distances and landmarks [Lele and Richtsmeier 2001]. Additionally, tests for differences in global form of anatomical regions were evaluated at each age by testing a null hypothesis of similarity in form [Lele and Richtsmeier 2001]. Subsets of landmarks were identified to represent specific regions of the skull: face, cranial vault, and cranial base (Table II). These three regions of the skull are embryologically and phylogenetically distinct units, which have been described frequently as partially distinct developmental modules [e.g., Cheverud 1982; 1989; 1995; Hallgrímsson et al., 2004; 2007; Martínez-Abadias et al 2011]. Each subset was analyzed separately and the null hypothesis of similarity in form for a subset (i.e., developmental unit) was rejected if p≤0.05. The same procedures were followed using neural landmarks to compare form of the brain (Table II).
Table II.
Landmark subsets for analyses. Landmarks are illustrated in Figure 2.
| Anatomical Region | # of landmarks | Landmarks in subset | |
|---|---|---|---|
| Brain | Cerebral surface | 7 | B1, B2, B3, B4, B11, B12, B13 |
| Subcortical structures | 5 | B8, B9, B10, B14, B15 | |
| Cerebellum | 3 | B5, B6, B7 | |
|
| |||
| Skull | Face | 6 | S1, S2, S3, S5, S16, S19 |
| Neurocranium | 8 | S7, S8, S9, S10, S11, S12, S13, S14 | |
| Cranial Base | 4 | S15, S21, S22, S23 | |
|
| |||
| PCA | Braincase, Brain | 30 | S2, S3, S4, S7, S8, S9, S10, S11, S12, S13, S14, S15, S21, S22, S23 |
| B1, B2, B3, B4, B5, B6, B7, B8, B9, B10, B11, B12, B13, B14, B15 | |||
Growth pattern (P0→P2) comparison
To analyze patterns of skull growth in Fgfr2+/P253R mice and unaffected littermates from P0 to P2, P0 mice were directly compared to P2 mice within each genotype group. Growth patterns were estimated for each group as the relative change in the lengths of linear distances using methods similar to those described for the form analyses above and detailed in Richtsmeier and Lele [1993]. Differences in growth between the two groups were estimated as a ratio of change experienced by each sample (Fgfr2+/P253R mice and unaffected mice) between P0 and P2. As with the analyses of form at P0 and P2, two non-parametric tests were used to determine whether growth patterns differ between groups. Local differences in growth for specific linear distances were evaluated using non-parametric bootstrapping confidence intervals (10,000 bootstrapped steps) for each linear distance. Confidence intervals that did not include 1.0 indicated that growth for that linear distance differed significantly between Fgfr2+/P253R mice and unaffected littermates from P0 to P2. Growth differences for whole anatomical regions described by subsets of landmarks were tested statistically for a null hypothesis of similarity in growth patterns using 10,000 bootstrapped steps (p≤0.05) [Richtsmeier et al., 1993]. The same procedures were followed to analyze patterns of growth of the brain.
Combined brain-skull comparisons
Regression of size measures
To assess the relationship between brain size and skull size we performed three sets of regression analyses at P0 and at P2. We regressed brain size and skull size to determine whether their relationship is statistically significant 1) in unaffected mice at both ages, 2) in mutant mice at both ages, and 3) in all mice at both ages.
Principal components analysis
To analyze morphological variation and the specific contributions of the skull and brain linear distances to differences in overall head morphology, we performed principal components analyses (PCA) for select linear distances representing the brain and the neurocranium. The 105 linear distances estimated among the fifteen brain landmarks and 105 linear distances estimated among fifteen landmarks selected to represent the braincase (Table II) were included in this analysis. PCA performs a coordinate rotation that aligns transformed axes (or PCs) with the direction of maximum variation for the analyzed groups. PCAs were completed for linear distance data at P0, P2, and combining P0 and P2.
Regression of dissimilarity measures
An alternative means for assessing correlation of brain morphology and skull morphology is through the comparison of dissimilarity measures between pairs of individuals for each tissue type. Dissimilarity measures quantify the resemblance (or lack of resemblance) between two individuals as a single number. In this case we can calculate dissimilarity measures of skull form for each pair of individuals, and a measure for brain form. Calculating a regression of dissimilarity of brain against dissimilarity of skull indicates the nature of the relationship between the brain and skull. Dissimilarity between all possible pairs of mice is expressed as a ( )), where FDM(A,B) is the form difference matrix comparing mouse A and mouse B. This results in a matrix of dissimilarity measures for every pair of mice [Richtsmeier et al., 1998]. If FΩ = 0, then A and B have identical forms. Larger FΩ values indicate greater differences between mice. We calculated four dissimilarity matrices using the 15 brain and 15 skull landmarks as described for the PCA analyses: 1) brain landmark data at P0, 2) skull landmark data at P0, 3) brain landmark data at P2, and 4) skull landmark data at P2. We then performed two regression analyses on brain and skull dissimilarity values, at P0 and at P2. If there is a significant correlation between dissimilarity measures of brain and skull for all possible pairs of mice, then we conclude that there is a strong relationship between brain form and skull form at a given age. If a significant correlation is not evident, then we conclude that brain and skull form are relatively independent of one another at that age.
RESULTS
Overall Size Comparisons
At P0, the Fgfr2+/P253R brain is 1% larger on average and the Fgfr2+/P253R skull is 2% smaller relative to unaffected littermates (Fig 3). At P2, the brain does not differ in size on average between mutant and unaffected mice, but the Fgfr2+/P253R skull is 9% smaller. The results of the ANOVA of brain size finds a significant effect of age (p <0.001), but not of genotype or age*genotype interaction (p=0.082 and 0.437, respectively). The effects of age, genotype, and their interaction are all significant with respect to skull size (p <0.001 for all comparisons). Since overall size is significantly different in several of these analyses, we report analyses of growth and form differences that have been scaled for size differences. Accordingly, each individual was scaled by geometric mean of the appropriate tissue (skull, brain) before all EDMA and PCA analyses were performed. Comparison of the results of analyses employing scaled and unscaled data demonstrated identical patterns of significant differences between Fgfr2+/P253R mice and their unaffected littermates; therefore scaling affects the magnitude of differences only, and not the pattern of differences.
Figure 3.
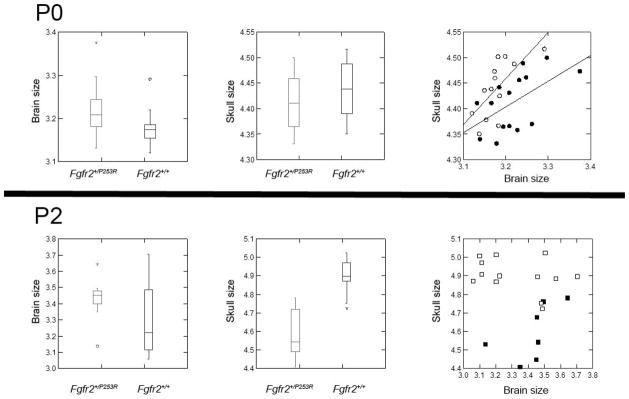
Box-and-whisker plots of brain size (left), skull size (center), and regression of brain and skull size (right) at P0 (above) and P2 (below) for Fgfr2+/P253R (closed symbols) and unaffected littermates (open symbols).
Form analyses
Brain Morphology at P0
Tests for differences in global form for the three regions of the brain at P0 show statistically significant differences of the cerebral surface (p= 0.016) and subcortical structures (p=0.027) of the brain, while the subset describing the cerebellum did not differ significantly in overall form in Fgfr2+/P253R as compared to unaffected littermates (Table III). Examination of individual scaled linear distances determines that 12 of the 105 linear distances differed significantly in the two groups, with the magnitude of these differences ranging from 5 to 12% (Figure 4). Linear distances representing the height of cerebrum are increased in the Fgfr2+/P253R mice compared to unaffected littermates, as are the distances between the genu and splenium of the corpus callosum and between the genu and the anterior commissure. The linear distance describing the length of the corpus callosum is reduced in the Fgfr2+/P253R mice compared to their unaffected littermates. Linear distances that cross from the cerebellum to caudal cerebrum and to the caudate nuclei are reduced in the Fgfr2+/P253R mice. These significant differences in localized form at P0 demonstrate that the Fgfr2+/P253R mice, in general, display reduced brain length along the rostrocaudal axis and increased height of the brain along the dorsoventral axis relative to unaffected littermates.
Table III.
P-values derived from analyses of form and growth for subsets of landmarks. P-values in bold are significantly different between Fgfr2+/P253R mice and their unaffected littermates.
| P0 | P2 | Growth | |
|---|---|---|---|
| Brain | |||
| Cerebral surface | 0.016 | 0.003 | 0.021 |
| Subcortical structures | 0.027 | 0.212 | 0.314 |
| Cerebellum | 0.666 | 0.600 | 1.00 |
|
| |||
| Skull | |||
| Face | 0.001 | 0.000 | 0.001 |
| Neurocranium | 0.001 | 0.001 | 0.010 |
| Cranial base | 0.001 | 0.000 | 0.013 |
Figure 4.

Analyses of brain phenotypes in Fgfr2+/P253R mice relative to unaffected littermates. Results of form difference analysis of the brain at P0 (left) and P2 (middle), where white lines indicate linear distances that were increased in Fgfr2+/P253R mice and black lines indicate linear distances that were decreased in Fgfr2+/P253R mice relative to unaffected littermates. Results of growth difference analysis of the brain (right), where black lines indicate those linear distances that show greater magnitude of change from P0 to P2 in Fgfr2+/P253R and white lines indicate linear distances that show a decreased magnitude of change in the Fgfr2+/P253R mice. Ghosted landmarks and dashed lines indicate landmarks and linear distances that are deep to the surface of the 3D reconstruction. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Brain Morphology at P2
One of the three brain regions was statistically different in Fgfr2+/P253R mice compared to their unaffected littermates at P2 (Table III): the cerebral surface (p=0.030), while the cerebellum (p=0.600) and the subcortical structures (p=0.212) were not significantly different. Relative to the pattern seen at P0, a larger number of linear distances showed local differences that were significantly different at P2 (Fig 4). Twenty-seven linear distances were significantly increased by 5–30% in the two groups of mice, though the majority of these linear distances differed by 5–10%. Distances describing the dorsoventral height of the cerebrum are increased in the Fgfr2+/P253R mice relative to unaffected littermates. All of the remaining linear distances that were increased in the Fgfr2+/P253R mice represent the relationship between the cerebrum and cerebellum. In Fgfr2+/P253R mice, distances originating on the cerebellum and ending on the cerebrum were 5–9% greater than in their unaffected littermates.
Brain growth from P0 to P2
Significant differences in the form of the Fgfr2+/P253R brain relative to that of unaffected littermates were established for both P0 and P2, underscoring differences local to the rostral cerebrum and subcortical structures, such as the corpus callosum. Tests for global differences in growth of the brain from P0 to P2 indicate that the subsets describing the subcortical structures and cerebellum do not show statistically significant differences in relative magnitude of change in the two groups of mice (p=0.314 and 1.00, respectively), while the change in the cerebral surface is statistically different between groups (p=0.021).
Of the 105 possible linear distances, 13 distances demonstrated significantly different relative magnitudes of growth from P0 to P2 between Fgfr2+/P253R mice and unaffected littermates (Fig 4). Distances spanning the mediolateral width of the rostral cerebrum showed significantly decreased magnitudes of growth in the Fgfr2+/P253R mice relative to unaffected mice. The length of the ventral surface of the cerebrum also showed decreased growth in the Fgfr2+/P253R mice. Additionally, distances describing the position of the cerebrum relative to the cerebellum show decreased magnitudes of growth from P0 to P2 in the Fgfr2+/P253R mice.
Five linear distances showed increased magnitude of growth in the brains of the Fgfr2+/P253R mice. These distances spanned the rostrocaudal length of the cerebrum. Specifically, the linear distances from the caudal cerebrum to points within the rostral cerebrum grew more in Fgfr2+/P253R mice compared to unaffected littermates. The length of corpus callosum also shows increased magnitudes of growth in Fgfr2+/P253R mice.
Skull Morphology at P0
Analyses of global form of the skull at P0 indicated that Fgfr2+/P253R mice were significantly different in form for the facial skeleton (p=0.001), neurocranium (p=0.001) and cranial base (p=0.001) (Table III). Of the 253 possible linear distances for skull landmarks, 51 of the distances were significantly different in the two groups (Fig 5). A majority of these distances were reduced in the Fgfr2+/P253R mice relative to unaffected littermates. For example, distances that connect from the palate with points on the rostral neurocranium and face were 5–15% shorter in the Fgfr2+/P253R mice. Conversely, distances describing the width of the caudal neurocranium were significantly increased in the Fgfr2+/P253R mice. These analyses support our previous studies which showed that all regions of the skull are significantly affected by the FGFR2 P253R mutation at P0 but that the greatest differences in the form of the skull are localized to the facial skeleton (Fig 5) [Martínez-Abadias et al., 2010].
Figure 5.

Analyses of skull phenotypes in Fgfr2+/P253R mice relative to unaffected littermates. Results of form difference analysis (left) of the skull landmarks in P2 mice, where white lines indicate linear distances that were increased in Fgfr2+/P253R mice and black lines indicate linear distances that were decreased in Fgfr2+/P253R mice relative to unaffected littermates. Results of the growth difference analyses of skull (right), where black lines indicate those linear distances that show greater magnitudes of change from P0 to P2 in Fgfr2+/P253R mice and white lines indicate linear distances that show decreased magnitudes of change in the Fgfr2+/P253R mice. Ghosted landmarks and dashed lines indicate landmarks and linear distances that are deep to the surface of the 3D reconstruction. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Skull Morphology at P2
Analyses of global form of the skull at P2 indicated that Fgfr2+/P253R mice were significantly different in form for the facial skeleton (p <0.001), neurocranium (p<0.001) and cranial base (p <0.001) (Table III). Of the 253 possible linear distances for skull landmarks, 101 of the distances were significantly different in the two groups (Fig 5). Fifty of these distances were reduced in the Fgfr2+/P253R mice relative to unaffected littermates. For example, distances that connect from the palate with points on the rostral neurocranium and face were 5–15% shorter in the Fgfr2+/P253R mice. Conversely, distances describing the height and width of the neurocranium were significantly increased in the Fgfr2+/P253R mice. These differences, specifically increased height of the neurocranium, correspond with differences in skull form seen in individuals with AS [Marsh et al., 1991].
Skull growth from P0 to P2
Unlike our results for the brain, growth of the skull from P0 to P2 is significantly different for the facial skeleton (p=0.001), neurocranium (p=0.010) and cranial base (p=0.013) in the Fgfr2+/P253R mice relative to unaffected littermates (Table III). Examination of individual linear distances showed that 26 of the possible 253 linear distances showed significantly different magnitudes of growth in the two groups of mice (Fig 5). In general, the Fgfr2+/P253R mice experienced relatively less growth than their unaffected littermates from P0 to P2, with reduced growth especially evident local to the palate and rostral cranial base. Specifically, Fgfr2+/P253R mice demonstrated decreased magnitudes of change along the rostrocaudal axis of the palate relative to unaffected littermates. In contrast, several distances representing the form of the caudal neurocranium showed increased magnitudes of change in the Fgfr2+/P253R mice. The height of the neurocranium and width of the dorsal neurocranium showed increased magnitudes of change in the Fgfr2+/P253R mice relative to unaffected littermates.
Combined brain-skull analyses
Regression of brain and skull size at P0 showed a significant relationship (Fig 3) in all three comparisons: all mice (p=0.014, Pearson correlation 0.439), unaffected mice (p=0.009, Pearson correlation 0.692), and mutant mice (p=0.028, Pearson 0.564). This relationship is not significant at P2, however, in any of the three analyses (all mice p=0.397, Pearson correlation −0.200; unaffected mice p=0.268, Pearson correlation −0.332; mutant mice p=0.166, Pearson correlation 0.587). These results suggest that size of brain and skull are significantly correlated at P0, but not at P2.
Results of the principal components analysis of brain and skull morphology at P0 indicate that mutant and unaffected littermates separate along principal component axis 1 (PC1) (Fig 6). The specific linear distances that contribute up to 30% of the variance associated with PC1 describe rostrocaudal neurocranial length, caudal neurocranial width, and rostral brain width. We performed ANOVA to assess the effects of genotype and litter on PC 1 and PC 2 scores. Results show significant effects of both genotype (p <0.001) and litter (p =0.008) on PC 1, with 70.2% of the variation explained by genotype and 3.2% by litter. Neither litter nor genotype shows significant effects on PC2 (p=0.120 and 0.398, respectively).
Figure 6.
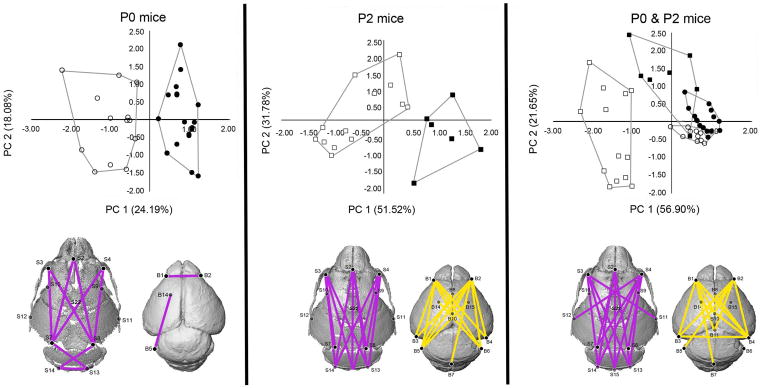
Results of PCA analyses of the linear distances measured in brain and skull for each mouse in the study at P0 (left), P2 (center), and P0 & P2 together (right). Closed symbols indicate Fgfr2+/P253R mice while open symbols indicate unaffected littermates; circles are P0 mice, squares are P2 mice. Linear distances that contribute up to 30% of the variation in PC 1 at P0 and P2 are illustrated in gray. Linear distances that contribute up to 30% of the variation in PC 2 at P2 are illustrated in white. Convex hulls are drawn for Fgfr2+/P253R and unaffected mice. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
PCA of the same measures of brain and neurocranium at P2 reveal a separation between the groups along PC1 (Fig 6). Mutant mice cluster on the positive end of PC1, while unaffected mice tend toward the negative end of the axis. 30% of the variance associated with PC1 is associated with linear distances describing the rostrocaudal length of the neurocranium. Unlike P0, no measures of mediolateral width in either the brain or skull are associated with PC1 or PC2. Tests of the effects of genotype and litter on PC1 show significant effects of genotype (p < 0.001) but not litter (p =0.195), with genotype explaining 64.8% of the variance and litter only 3.6%. There is not a significant effect of genotype on PC2 (p =0.276), but there is an effect of litter (p =0.006), with litter explaining 38.5% of the variance and genotype 9.1%.
PCA of the same measures of brain and neurocranium ing data from P0 and P2 mice reveal clusters on PC1 and PC2 (Fig 6). Unaffected P2 mice cluster at the negative end of PC1, while mutant P2 and all P0 mice overlap on this axis. 30% of the variance associated with PC1 is associated with linear distances describing rostrocaudal neurocranial length and width of the rostral neurocranium. Mutant P2 mice cluster separately from the P0 mice on PC2, but overlap with the unaffected P2 mice on this axis. The linear distances that contribute up to 30% of the variance associated with this PC describe rostrocaudal cerebral length and caudal cerebral width. ANOVA testing the effects of age, genotype, and their interaction on PC1 show significant effects of all three (p <0.001), with age explaining 49.7% of the variance, genotype 14.3%, and their interaction only 6.7%. There is a significant effect of all three on PC2 (age p =0.023; genotype p <0.001; age*genotype p =0.015), though the variance is attributed more to genotype on PC 2 (47.9%) than to age (12.7%) or the interaction (14.7%). The P0 mice cluster together along both PC1 and PC2, suggesting constraints in morphology at P0. The within-group variance of the two groups of P2 mice along PC2 is much broader that that for either of the P0 groups along PC2. In general, unaffected and mutant mice have very different growth patterns, with change in unaffected mice defined by PC1, which is primarily associated with skull morphology, while change in mutant mice is defined by PC2, which is more associated with brain morphology.
Results of the regression of dissimilarity measures (FΩ) among all possible pairs of mice at P0 indicate that there is no significant relationship between skull morphology and brain morphology at P0 (p=0.254, Pearson correlation=0.059) (Figure 7). In contrast, there is a highly significant relationship in brain and skull morphology at P2 (p <0.001, Pearson correlation=0.743). These results suggest that in both groups form of the skull and brain are not significantly correlated at P0, but are highly correlated at P2.
Figure 7.
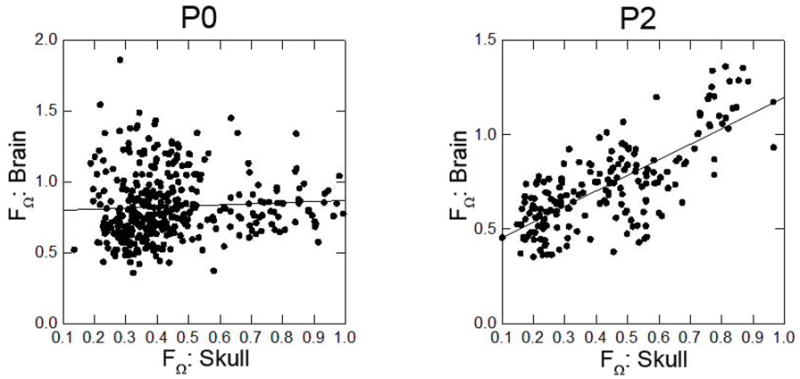
Results of regression of dissimilarity measures of skull and brain for all possible pairs of mice at P0 (left) and P2 (right).
DISCUSSION
The FGFR2 Pro253Arg mutation is associated with approximately 33% of cases of Apert syndrome [von Gernet et al., 2000; Wilkie et al., 1995]. Most studies of both patients and animal models for AS have focused on skeletal and sutural tissues. However, FGFR2 is expressed in many and varied tissues of the developing head, including skull [Hatch 2010; Karaplis 2008; Noden and Trainor 2005] and suture [Chen et al., 2003; Hajihosseini 2008; Merrill et al., 2006], but also in the developing brain [Bansal et al., 2003; Ever et al., 2008; Maric et al., 2007; Yoon et al., 2004].
Our study design excludes two potential sources of variation within the sample, allowing a clearer view of how these tissues interact over time. First, we use an inbred mouse model [Wang et al., 2010] to control for genetic variation that contributes to the phenotypic effects of the Pro253Arg mutation, enabling a more precise characterization of the specific effects of the mutation on CNS and skeletal phenotypes than would be possible using outbred mice. Second, the same mice underwent MRM and μ-CT imaging, such that data collected for the skull and for the brain were obtained from the same individuals. The cost associated with acquiring both sets of imaging data from the mice necessitates smaller sample sizes than a single imaging modality would allow, introducing a potential bias. However, acquisition of both image types from the same mice removes the potential variation introduced by using differing samples of mice for analysis of skull and analysis of brain.
Studies of overall head shape in individuals with AS have described a flat and elongated forehead, broader bitemporal width, broad and flattened occipital regions, and anteroposteriorly shortened cranial lengths [Marsh et al., 1991] with dramatically increased head height and decreased head length present at birth [Cohen and Kreiborg, 1994]. Studies of postnatal cranial growth in humans with AS, however, have been limited by variation in data types, methods, and small sample size. In a cross-sectional analysis of cephalograms from children with AS, Richtsmeier [1988] found differences in the anterior nasal spine and posterior nasal spine, resulting in a shortened palate [Richtsmeier, 1988]. Another cross-sectional study showed head circumference decreases with age in AS [Cohen and Kreiborg, 1993a]. Analyses of brain growth have been limited to estimates of intracranial volume, which has been found to be larger in infants with AS [Anderson et al., 2004; Gosain et al., 1995] and that this difference increases with age [Gosain et al., 1995].
We may draw some parallels between clinical studies of growth in children with AS and the patterns of early postnatal growth in Fgfr2+/P253R mice discussed here. We found that the overall size of the skull is reduced in Fgfr2+/P253R mice as compared to their unaffected littermates, though this size reduction is not uniformly distributed across the cranium. While the facial skeleton is reduced by 11% in Fgfr2+/P253R mice at P2, the neurocranium is reduced by only 3%. This indicates that head size is disproportionately reduced in the facial skeleton and palate relative to the remainder of the skull. In fact, the greatest differences in growth of the skull of Fgfr2+/P253R mice are localized to the palate, as seen in children with AS [Richtsmeier 1988]. Relative to unaffected littermates, the Fgfr2+/P253R mice show 5–10% reduction of growth of the palate from P0 to P2. Other changes in the Fgfr2+/P253R skull include relatively increased change in the height of the posterior neurocranium with growth (~5%) and increased change in width between the interparietal bones (dorsal neurocranium) with growth, mirroring the increased intracranial volume observed in AS [Anderson et al., 2004; Gosain et al., 1995].
Changes in the brain in Fgfr2+/P253R mice also parallel the findings of clinical studies of brain morphology in children with AS. Dysmorphology of the corpus callosum is a frequent finding in AS [Cohen and Kreiborg 1990; Cohen and Kreiborg, 1991; 1994; de Leon et al., 1987; Posnick et al., 1995], and we find that the corpus callosum is shortened in Fgfr2+/P253R mice at both P0 and P2, in agreement with a previous qualitative study of these mice [Aldridge et al., 2010]. Intracranial volume is often used as a proxy for brain size, and intracranial volume has been observed to be larger in children with AS [Anderson et al., 2004; Gosain et al., 1995], though head circumference decreases with age [Cohen and Kreiborg, 1993b]. The present study did not find statistically significant differences in brain size in mice carrying the Pro253Arg mutation, though we cannot exclude the possibility that these results are affected by small sample size. However, differences in the form of the brain at P0 and P2 were observed. Specifically, Fgfr2+/P253R mice show increased mediolateral growth of the cerebrum, and decreased rostrocaudal cerebral growth, contributing to a brain phenotype that is proportionately shorter rostrocaudally and taller superoinferiorly than in unaffected littermates.
Using images of the brain and skull from the same mice allowed for investigation of the relationship between the two tissues. Our results demonstrate that size of brain and skull are significantly correlated at birth in both Fgfr2+/P253R mutants and unaffected littermates, but not at P2. In contrast, brain and skull form are not significantly correlated at P0, but are correlated at P2. Analysis of overall head form (including brain and skull measures) shows that skull morphology distinguishes Fgfr2+/P253R from unaffected mice at P0 and at P2. However, when P0 and P2 mice are combined, skull morphology distinguishes unaffected P2 mice from the other three groups, while brain morphology distinguishes mutant P2 mice from both groups of P0 mice.
Taken together, these results suggest that size and form show different patterns of growth in both mutant and unaffected mice. Further, though phenotypic differences are observed in both the skull and the brain at P0, the morphology of the skull is most influential in distinguishing groups. However, it is the combination of skull and brain growth that underlies the differentiation of the growth patterns of the two groups. Understanding how these two tissues of the vertebrate head covary is key to understanding both the development and evolution of this complex structure [Jamniczky and Hallgrímsson, 2011].
Our study represents concurrent analyses of both brain and skull in a mouse model for AS. Our results demonstrate that brain and skull development in the Fgfr2+/P253R mouse model mirrors that of infants with AS. Further, we have identified localized regions of the brain and skull that are different in the Fgfr2+/P253R mice as compared to their unaffected littermates during this critical period of early postnatal development. Our findings of differences in growth patterns of skull and brain in Fgfr2+/P253R mice and their unaffected littermates, and the correspondence between them, point to their continued interaction through development. Our demonstration of altered patterns of development in both brain and skull in the Fgfr2+/P253R mice suggest that both tissues display divergent postnatal growth patterns as compared to their unaffected littermates, rather than one tissue directing development of the other.
Acknowledgments
The authors thank Dr. Tim Ryan at the Center for Quantitative X-Ray Imaging and Dr. Thomas Neuberger at the High Field MRI facility at Pennsylvania State University for their work in obtaining μ-CT and MRM images of the mice. Work was supported in part by funds from the Department of Pathology and Anatomical Sciences at the University of Missouri, and NIH grants R01DE018500; R01DE018500-02S1.
References
- Aldridge K, Hill CA, Austin JR, Percival C, Martínez-Abadias N, Neuberger T, Wang Y, Jabs EW, Richtsmeier JT. Brain phenotypes in two FGFR2 mouse models for Apert syndrome. Dev Dyn. 2010;239(3):987–997. doi: 10.1002/dvdy.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge K, Reeves RH, Olson LE, Richtsmeier JT. Differential effects of trisomy on brain shape and volume in related aneuploid mouse models. Am J Med Genet Part A. 2007;143A:1060–1070. doi: 10.1002/ajmg.a.31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Netherway D, Abbott A, Cox T, Roscioli T, David D. Analysis of intracranial volume in Apert syndrome genotypes. Pediatr Neurosurg. 2004;40:161–164. doi: 10.1159/000081933. [DOI] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfra, Olig2 and Plp/dm20: Impliations for oligodendrocyte development. Dev Neurosci. 2003;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Chen L, Li D, Li C, Engel A, Deng C. A Ser250Trp substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33:169–178. doi: 10.1016/s8756-3282(03)00222-9. [DOI] [PubMed] [Google Scholar]
- Cheverud JM. Phenotypic, genetic, and environmental integration in the cranium. Evolution. 1982;36:499–516. doi: 10.1111/j.1558-5646.1982.tb05070.x. [DOI] [PubMed] [Google Scholar]
- Cheverud JM. A comparative analysis of morphological variation patterns in the papionins. Evolution. 1989;43:1737–1747. doi: 10.1111/j.1558-5646.1989.tb02623.x. [DOI] [PubMed] [Google Scholar]
- Cheverud JM. Morphological integration in the saddle-back tamarin (Saguinus fuscicollis) cranium. Am Nat. 1995;145:63–89. [Google Scholar]
- Cohen MM. Apert syndrome. In: Cohen M, MacLean R, editors. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Oxford University Press; 2000. pp. 316–353. [Google Scholar]
- Cohen MM, Jr, Kreiborg S. Growth pattern in the Apert syndrome. Am J Med Genet. 1993a;47(5):617–623. doi: 10.1002/ajmg.1320470508. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Kreiborg S. Suture formation, premature sutural fusion, and suture default zones in Apert syndrome. Am J Med Genet. 1996;62(4):339–344. doi: 10.1002/(SICI)1096-8628(19960424)62:4<339::AID-AJMG3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S. The central nervous system in the Apert syndrome. Am J Med Genet. 1990;35:36–45. doi: 10.1002/ajmg.1320350108. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S. Agenesis of the corpus callosum. Its associated anomalies and syndromes with special reference to the Apert syndrome. Neurosurg Clin N Am. 1991;2:565–568. [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S. An updated pediatric perspective on the Apert syndrome. AJDC. 1993b;147:989–993. doi: 10.1001/archpedi.1993.02160330079025. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S. Cranial size and configuration in the Apert syndrome. J Craniofac Genet Dev Biol. 1994;14:153–162. [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, Roeper P, Martínez-Frias ML. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–659. doi: 10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- Darroch JN, Mosimann JE. Canonical and principal components of shape. Biometrika. 1985;72:241–252. [Google Scholar]
- de Leon GA, de Leon G, Grover WD, Zaeri N, Alburger PD. Agenesis of the corpus callosum and limbic malformation in Apert syndrome (type I acrocephalosyndactyly) Arch Neurol. 1987;44:979–982. doi: 10.1001/archneur.1987.00520210073023. [DOI] [PubMed] [Google Scholar]
- Du X, Weng T, Sun Q, Su N, Chen Z, Qi H, Jin M, Yin L, He Q, Chen L. Dynamic morphological changes in the skulls of mice mimicking human Apert syndrome resulting from gain-of-function mutation of FGFR2 (P253R) J Anat. 2010;217(2):97–105. doi: 10.1111/j.1469-7580.2010.01248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793. doi: 10.1242/dev.129.16.3783. [DOI] [PubMed] [Google Scholar]
- Ever L, Zhao R, Eswarakumar VP, Gaiano N. Fibroblast growth factor receptor 2 plays an essential role in telencephalic progenitors. Dev Neurosci. 2008;30:306–318. doi: 10.1159/000112521. [DOI] [PubMed] [Google Scholar]
- Falsetti AB, Jungers WL, Cole TMr. Morphometrics of the callitrichid forelimb: a case study in size and shape. Inter J Primat. 1993;14:551–572. [Google Scholar]
- Fragale A, Tartaglia M, Bernardini S, Di Stasi AM, Di Rocco C, Velardi F, Teti A, Battaglia PA, Migliaccio S. Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. Am J Pathol. 1999;154(5):1465–77. doi: 10.1016/S0002-9440(10)65401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosain A, McCarthy J, Glatt P, Staffenberg D, Hoffmann R. A study of intracranial volume in Apert syndrome. Plast Reconstr Surg. 1995;95:284–295. doi: 10.1097/00006534-199502000-00008. [DOI] [PubMed] [Google Scholar]
- Hajihosseini M. Fibroblast growth factor signaling in cranial suture development and pathogenesis. In: Rice D, editor. Craniofacial Sutures Development, Disease and Treatment. Basel: Karger; 2008. pp. 160–177. [DOI] [PubMed] [Google Scholar]
- Hallgrímsson B, Willmore K, Dorval C, Cooper DM. Craniofacial variability and modularity in macaques and mice. J Exp Zool B Mol Dev Evol. 2004;302:207–25. doi: 10.1002/jez.b.21002. [DOI] [PubMed] [Google Scholar]
- Hallgrímsson B, Lieberman DE, Young NM, Parsons TE, Wat S. Evolution of covariance in the mammalian skull. Novartis Found Symp. 2007;284:164–85. doi: 10.1002/9780470319390.ch12. [DOI] [PubMed] [Google Scholar]
- Hatch NE. FGF signaling in craniofacial biological control and pathological craniofacial development. Crit Rev Eukaryot Gene Expr. 2010;20:295–311. doi: 10.1615/critreveukargeneexpr.v20.i4.20. [DOI] [PubMed] [Google Scholar]
- Hill CA, Reeves RH, Richtsmeier JT. Effects of aneuploidy on skull growth in a mouse model of Down syndrome. J Anat. 2007;210(4):394–405. doi: 10.1111/j.1469-7580.2007.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci U S A. 2001;98(13):7182–7. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamniczky HA, Hallgrímsson B. Modularity in the skull and cranial vasculature of laboratory mice: implications for the evolution of complex phenotypes. Evol Dev. 2011;13:28–37. doi: 10.1111/j.1525-142X.2010.00453.x. [DOI] [PubMed] [Google Scholar]
- Jungers WL, Falsetti AB, Wall CE. Shape, relative size, and size-adjustments in morphometrics. Yearbook Phys Anthropol. 1995;38:137–161. [Google Scholar]
- Karaplis AC. Embryonic development of bone and regulation of intramembranous and endochondral bone formation. In: Bilezikian JP, Raisz LG, Martin TJ, editors. Principles of Bone Biology. 3. Elsevier; 2008. pp. 53–84. [Google Scholar]
- Kobayashi T. Brain-to-body ratios and time of maturation of the mouse brain. Am J Physiol. 1963;204:343–6. doi: 10.1152/ajplegacy.1963.204.2.343. [DOI] [PubMed] [Google Scholar]
- Kreiborg S, Cohen MM., Jr Characteristics of the infant Apert skull and its subsequent development. J Craniofac Genet Dev Biol. 1990;10(4):399–410. [PubMed] [Google Scholar]
- Lele S, Richtsmeier JT. An invariant approach to the statistical analysis of shapes. London: Chapman and Hall/CRC Press; 2001. [Google Scholar]
- Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie PJ. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998;101:1310–1317. [PMC free article] [PubMed] [Google Scholar]
- Mao JJ, Wang X, Mooney MP, Kopher RA, Nudera JA. Strain induced osteogenesis of the craniofacial suture upon controlled delivery of low-frequency cyclic forces. Front Biosci. 2003;8:a10–7. doi: 10.2741/917. [DOI] [PubMed] [Google Scholar]
- Marcucio RS, Young NM, Hu D, Hallgrimsson B. Mechanisms that underlie co-variation of the brain and face. genesis. 2011;49:177–189. doi: 10.1002/dvg.20710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric D, Fiorio Pla A, Chang YH, Barker JL. Self-renewing and differentiating properties of cortical neural stem cells are selectively regulated by basic fibroblast growth factor (FGF) signagling via specific FGF receptors. J Neurosci. 2007;27:1836–1852. doi: 10.1523/JNEUROSCI.5141-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JL, Galic M, Vannier MW. The craniofacial anatomy of Apert syndrome. Clin Plast Surg. 1991;18(2):237–49. [PubMed] [Google Scholar]
- Martínez-Abadias N, Heuzé Y, Wang Y, Jabs EW, Aldridge K, Richtsmeier JT. FGF/FGFR signaling coordinates skull development by modulating magnitude of morphological integration: evidence from Apert syndrome mouse models. PLoS ONE. 2011;6:e26425. doi: 10.1371/journal.pone.0026425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abadias N, Percival C, Aldridge K, Hill CA, Sirivunnabood S, Wang Y, Jabs EW, Richstmeier JT. Variation in skull phenotypes of Fgfr2 mouse models for Apert syndrome. Dev Dyn. 2010;239:3058–3071. doi: 10.1002/dvdy.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AE, Bochukova EG, Brugger SM, Ishii M, Pilz DT, Wall SA, Lyons KM, Wilkie AO, Maxson RE., Jr Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum Mol Genet. 2006;15:1319–1328. doi: 10.1093/hmg/ddl052. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219(4):472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Park W-J, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, Cohen MM, Jabs EW. Analysis of phenotypic features of FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–328. [PMC free article] [PubMed] [Google Scholar]
- Parsons TE, Schmidt EJ, Boughner JC, Jamniczky HA, Marcucio RS, Hallgrimsson B. Epigenetic integration of the developing brain and face. Dev Dyn. 2011;240:2233–2244. doi: 10.1002/dvdy.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooh R, Nakagawa Y, Pooh K, Nakagawa Y, Nagamachi N. Fetal craniofacial structure and intracranial morphology in a case of Apert syndrome. Ultrasound Obstet Gynecol. 1999;13:274–280. doi: 10.1046/j.1469-0705.1999.13040274.x. [DOI] [PubMed] [Google Scholar]
- Posnick J, Armstrong D, Bite U. Crouzon and Apert syndromes: intracranial volume measurements before and after cranio-orbital reshaping in childhood. Plast Reconstr Surg. 1995;96:539–548. [PubMed] [Google Scholar]
- Renier D, Lajeunie E, Arnaud E, Marchac D. Management of craniosynostoses. Childs Nerv Syst. 2000;16:645–658. doi: 10.1007/s003810000320. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Cole TM, Krovitz G, Valeri CJ, Lele S. Preoperative morphology and development in sagittal synostosis. J Craniofac Genet Dev Biol. 1998;18:64–78. [PubMed] [Google Scholar]
- Richtsmeier JT. Craniofacial growth in apert syndrome as measured by finite-element scaling analysis. Acta Anat (Basel) 1988;133(1):50–6. doi: 10.1159/000146614. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Aldridge K, DeLeon VB, Panchal J, Kane AA, Marsh JL, Yan P, Cole TM. Phenotypic integration of neurocranium and brain. J Exp Zool B Mol Dev Evol. 2006;306:360–78. doi: 10.1002/jez.b.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtsmeier JT, Corner BD, Grausz HM, Cheverud JM, Danahey SM. The role of postnatal growth pattern in the production of facial morphology. Syst Biol. 1993;42:307–330. [Google Scholar]
- Richtsmeier JT, DeLeon VB, Lele SR. The promise of geometric morphometrics. Am J Phys Anthropol Suppl. 2002;35:63–91. doi: 10.1002/ajpa.10174. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Lele S. A coordinate-free approach to the analysis of growth patterns: models and theoretical considerations. Biol Rev Camb Philos Soc. 1993;68:381–411. doi: 10.1111/j.1469-185x.1993.tb00737.x. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Paik C, Elfert P, Cole TM, Dahlman H. Precision, repeatability and validation of the localization of cranial landmarks using computed tomography scans. Cleft Palate and Craniofacial Journal. 1995;32:217–27. doi: 10.1597/1545-1569_1995_032_0217_pravot_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Tokumaru A, Barkovich A, Ciricillo S, Edwards M. Skull base and calvarial deformities: Association with intracranial changes in craniofacial syndromes. AJNR Am J Neuroradiol. 1996;17:619–630. [PMC free article] [PubMed] [Google Scholar]
- von Gernet S, Golla A, Ehrensfels Y, Schuffenhauer S, Fairley JD. Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin Genet. 2000;57:137–139. doi: 10.1034/j.1399-0004.2000.570208.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sun M, Uhlhorn V, Peter I, Hill CA, Percival CJ, Richtsmeier JT, Huso DL, Jabs EW. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2+/P253R mice. BMC Dev Biol. 2010;10:22. doi: 10.1186/1471-213X-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F, Karim BO, Iocovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, Yu K, Ornitz DM, Elisseeff J, Huso DL, Jabs EW. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development. 2005;132:3537–3548. doi: 10.1242/dev.01914. [DOI] [PubMed] [Google Scholar]
- Wilke TA, Gubbels S, Schwartz J, Richman JM. Expression of fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3) in the developing head and face. Dev Dyn. 1997;210:41–52. doi: 10.1002/(SICI)1097-0177(199709)210:1<41::AID-AJA5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9(2):165–72. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Yacubian-Fernandes A, Palhares A, Giglio A, Gabarra R, Zanini S, et al. Apert syndrome: analysis of associated brain malformations and conformational changes determined by surgical treatment. J Neuroradiol. 2004;31:116–122. doi: 10.1016/s0150-9861(04)96978-7. [DOI] [PubMed] [Google Scholar]
- Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, Yang J, Jin M, Deng C, Chen L. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631–643. doi: 10.1016/j.bone.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yoon K, Nery S, Rutlin ML, Radtke F, Fishell G, Gaiano N. Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci. 2004;24:9497–9506. doi: 10.1523/JNEUROSCI.0993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad Sci U S A. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. Uncoupling fibroblast growth factor receptor 2 ligand binding specificity leads to Apert syndrome-like phenotypes. Proc Natl Acad Sci U S A. 2001;98:3641–3643. doi: 10.1073/pnas.081082498. [DOI] [PMC free article] [PubMed] [Google Scholar]