

Abstract
We designed a fiber-optic-based optoelectronic fluorometer to measure emitted fluorescence from the auto-fluorescent electron carriers NADH and FAD of the mitochondrial electron transport chain (ETC). The ratio of NADH to FAD is called the redox ratio (RR = NADH/FAD) and is an indicator of the oxidoreductive state of tissue. We evaluated the fluorometer by measuring the fluorescence intensities of NADH and FAD at the surface of isolated, perfused rat lungs. Alterations of lung mitochondrial metabolic state were achieved by the addition of rotenone (complex I inhibitor), potassium cyanide (KCN, complex IV inhibitor) and/or pentachlorophenol (PCP, uncoupler) into the perfusate recirculating through the lung. Rotenone- or KCN-containing perfusate increased RR by 21% and 30%, respectively. In contrast, PCP-containing perfusate decreased RR by 27%. These changes are consistent with the established effects of rotenone, KCN, and PCP on the redox status of the ETC. Addition of blood to perfusate quenched NADH and FAD signal, but had no effect of RR. This study demonstrates the capacity of fluorometry to detect a change in mitochondrial redox state in isolated perfused lungs, and suggests the potential of fluorometry for use in in vivo experiments to extract a sensitive measure of lung tissue’s health in real-time.
Keyterms: Lung surface fluorometry, Nicotinamide Adenine Dinucleotide (NADH), Flavin Adenine Dinucleotide (FADH2), mitochondrial redox
3. Introduction
The mitochondrial metabolic coenzymes NADH (Nicotinamide Adenine Dinucleotide) and FADH2 (Flavin Adenine Dinucleotide) are the primary electron carriers in oxidative phosphorylation. NADH and FADH2 oxidation via the mitochondrial electron transport chain results in the translocation of protons from mitochondrial complexes I, III and IV across the inner mitochondrial membrane into the mitochondrial intermembrane space. The resulting proton gradient along with adenosine diphosphate (ADP) availability control the rate of mitochondrial ATP synthesis, which accounts for ~85% of ATP production in lung tissue [1]. Thus, a change in redox status/concentration of mitochondrial NADH and/or FADH2 is an index of a change in lung tissue mitochondrial bioenergetics, and hence an index of mitochondrial function [2, 3].
Optical fluorescence techniques have the potential to monitor tissue metabolic status in real time in a non-destructive manner in intact organs both in vivo and ex vivo [4–10]. These techniques have been widely used to probe tissue redox state and energy homeostasis in organs such as the heart [8], brain [10], kidney [11], liver [12], skeletal muscle [13], cervix [14], and colon [15], but have not been fully used in lungs [16]. The mitochondrial metabolic coenzymes NADH and FAD (oxidized form of FADH2) are auto-fluorescent and can be monitored using optical techniques [7]. The fluorescence signals of these intrinsic fluorophores have been used as indicators of tissue metabolism in injuries due to hypoxia [17], ischemia, and cell death [4]. We have demonstrated that the ratio of these fluorophores, (NADH/FAD), termed the mitochondrial redox ratio (RR), is a marker of the mitochondrial redox and metabolic state of myocardial tissue in intact hearts and in vivo [4–6, 17]. The redox ratio provides a measure of the mitochondrial redox state that is independent of various factors that affect the measurement of NADH and FAD fluorescence signal, including blood volume and mitochondrial protein concentration [18]. The objective of this study was to demonstrate the utility of these optical fluorescence techniques to evaluate lung tissue mitochondrial redox state (NADH/FAD) in isolated perfused rat lungs with high sensitivity, and quantify the impact of blood on NADH and FAD fluorescence signals and their ratio. Isolated perfused lungs are particularly attractive to study in this regard since perfusate and/or ventilation gas composition can be altered to modify the oxidoreductive state of tissue mitochondria. Our results demonstrate the ability of these techniques to detect a change in lung tissue mitochondrial redox state in isolated perfused lungs, and set the stage for in vivo studies.
4. Materials and Methods
4.1. Materials
Fatty-acid free bovine serum albumin (Standard Powder, BSA) was purchased from Serologicals Corp. (Gaithersburg, MD). All other reagent grade chemicals were purchased from Sigma Chemical Company.
4.2. Fluorometer
The fiber-optic-based fluorometer device designed for this study has undergone several improvements and iterations over our original design [19, 20]. The initial design included a pneumatic filter wheel which was used both for filtering of excitation and emission fluorescence. However, to eliminate the need for compressed air to use the device, the second design replaced the pneumatic filter wheel with two synchronized electric filter wheels. After further investigation, it became apparent that the use of separate detectors for each fluorophore would be beneficial. Therefore, the emission filter wheel was supplanted by the combination of a dichroic mirror and static filters. Finally, to correct for interfering factors such as hemodynamics, a beam splitter was added to detect diffuse reflectance from the tissue. A schematic for the iteration of the fluorometer device used in this study is shown in Figure 1.a. Excitation light is generated from a mercury arc lamp (Intensilight, Nikon, Tokyo, Japan), and coupled to a liquid light guide. The light is then fed into a filter wheel (Lambda-3, Sutter Instrument, CA), where the appropriate excitation wavelength can be selected. On the other side of this filter wheel is one leg of a bifurcated fiber bundle (Newport Instrument, NJ) with a distal end of 3.2 mm inner diameter. This distal tip (Figure 1.b.) is brought into contact with the tissue under investigation to deliver the appropriate excitation light and collect the corresponding fluorescence emission. The emitted light, along with reflection of the excitation is then delivered through the other leg of the bifurcated fiber bundle to the detection optics. After the light exits the fiber bundle, it is collimated and split using a beam splitter (UVBS14-1, Newport Instruments, CA). Half of the light is then incident on an avalanche photodiode (PDA25K, ThorLabs, NJ) for detection of reflected light. The other half of the light then passes through a dichroic mirror (DMLP505R, ThorLabs, NJ) to separate the NADH and FAD channels. In either of the channels, the light is then filtered to select the emitted fluorescence, remove any remaining reflection as background, and is finally incident on a photomultiplier tube (PMT; PMM02, ThorLabs, NJ). Synchronization of excitation and detection is achieved through the use of a custom LabVIEW program. To optimally excite the fluorophores of interest, NADH and FAD, using the mercury arc lamp, the excitation filters used were centered at 370 nm (FF01-370/36, Semrock, NY) and 452 nm (FF01-452/45, Semrock, NY), with bandwidths of 40 nm and 50 nm, respectively. The dichroic mirror used to separate the emission of each fluorophore has its transition wavelength at 505 nm. Finally, the filters used to detect the emitted fluorescence are centered at 460 (D460/40M, Chroma, VT) and 520 nm (D520/40M, Chroma, VT), respectively, each with a bandwidth of 40 nm.
Figure 1.

a) Schematic of the Fluorometer. b) Fluorescence surface measurement in an isolated perfused rat lung.
4.3. Isolated perfused rat lung preparation
The utility of the aforementioned fluorometer for measuring lung surface mitochondrial NADH and FAD signal was evaluated using the isolated perfused lung preparation which has been described previously [21]. This preparation provides a means for manipulating lung tissue mitochondrial redox state without disrupting the multi-cellular environment of the lung, by adding metabolic inhibitor(s) to the recirculating perfusate and/or altering the composition of the ventilation gas. Adult (300–350 g, male) Sprague Dawley rats (Charles Rivers) were anesthetized with pentobarbital sodium (40 mg/kg body wt. i.p.), the trachea was clamped, the chest opened and heparin (0.7 IU/g body wt.) injected into the right ventricle. The pulmonary artery and the trachea were cannulated, and the pulmonary venous outflow was accessed via a cannula in the left atrium. The heart-lung was removed from the chest and attached to a ventilation and perfusion system. The control perfusate contained (in mM) 4.7 KCl, 2.51 CaCl2, 1.19 MgSO4, 2.5 KH2PO4, 118 NaCl, 25 NaHCO3, 5.5 glucose, and 3% bovine serum albumin (BSA) [22]. The perfusion system was primed (Masterflex roller pump) with the control perfusate maintained at 37°C and equilibrated with 15% O2, 6% CO2, balance N2 resulting in perfusate pO2, pCO2 and pH of ~105 Torr, 40 Torr, and 7.4, respectively. Initially, control perfusate was pumped through the lung until it was evenly blanched and venous effluent was clear of blood. The lung was ventilated (40 breaths/min) with end-inspiratory and end-expiratory pressures of ~ 6 and 3 mmHg, respectively, with the above gas mixture. The pulmonary arterial pressure was referenced to atmospheric pressure at the level of the left atrium and monitored continuously during the course of the experiments. The venous effluent pressure was atmospheric pressure. This procedure and the experimental protocols described below were approved by the Institutional Animal Care and Use Committees of the Zablocki Veterans Affairs Medical Center and Marquette University (Milwaukee, WI).
4.4. Experimental protocols
Fluorometer linear response
To evaluate the linearity and sensitivity of the fluorometer to a change in NADH or FAD signal, we measured the NADH and FAD fluorescence signals in cuvettes containing aqueous solution with different NADH and/or FAD concentrations. The range of NADH and FAD concentrations studied encompassed the NADH and FAD concentrations in lung tissue [16, 23].
Lung surface fluorescence measurements
The fluorometer was used in a dark room to minimize stray-light effects [4, 5, 24]. At the beginning of each experiment, NADH and FAD fluorescence standards were measured to account for day-to-day variations in light intensity. Surface fluorescence was then measured by placing the fiber optic probe against the pleural surface of the right lobe (Figure 1.b). For a given lung, NADH and FAD surface fluorescence signals were first acquired under resting conditions (lung perfused with control perfusate and ventilated with 15% O2, 6% CO2 balance N2 ventilation gas), and then following the addition of one or more of the following agents (rotenone, potassium cyanide, and pentachlorophenol) to the recirculation perfusate. Rotenone (20 μM) was used to inhibit mitochondrial complex I activity, which would be expected to increase NADH signal by reducing the chain upstream from complex I, and decrease FAD signal from lipoamide dehydrogenase (LipDH) [25], which is an element of the pyruvate dehydrogenase complex [26]. As a mitochondrial complex IV inhibitor, potassium cyanide (KCN, 2 mM) would be expected to reduce the chain upstream from complex IV and hence increase NADH signal and decrease FAD signal from LipDH, succinate dehydrogenase (complex II), and electron transfer flavoproteins (ETF) which are the main sources of FAD signal [25]. Uncoupled mitochondrial condition was achieved by the addition of pentachlorophenol (PCP, mitochondrial uncoupler; 3 mM) to the recirculating perfusate and by ventilation of the lungs with 95% O2: 5% CO2 to maximize the oxidation of the electron transport chain. As a protonophore, PCP should oxidize the chain and hence decrease NADH signal and increase FAD signal. For some of the lungs, KCN was added after the initial addition of rotenone or PCP. The above concentrations of rotenone, KCN, and PCP were chosen to achieve maximal inhibition or uncoupling and hence maximal changes in NADH and/or FAD signal [16, 21].
To evaluate the quenching effect of blood on the NADH and FAD signals, surface fluorescence measurements were carried out in a separate group of lungs first perfused with control perfusate, then following the addition of autologous blood to the recirculating perfusate in ~ 0.5 ml increments to achieve perfusate hematocrit (Hct) levels of ~ 0.5%, 1.0%, 1.5%, 2%, 2.5%, 3%, 3.5%, and 4%. This range of perfusate hematocrit was chosen based on the results of a previous study by Chance et. al [18]. At the end of this protocol, KCN was added to the recirculating blood-containing perfusate to evaluate the ability of the system to detect a change in NADH, FAD, and RR = NADH/FAD in the presence of red blood cells (4 % Hct).
4.5. Fluorescent Signal Processing
NADH, FAD, and redox ratio (NADH/FAD)
The signal measured by the fluorometer contains a time shared sequence of NADH and FAD pulses collected from the surface of the lung. The trend of the data was first calculated by extracting the maximum value of each pulse. This trend was then smoothed out using a fourth order median filter followed by a fourth order moving average filter. The resulting NADH and FAD signals were then normalized by dividing each by its baseline value (signal level in the absence of any metabolic inhibitor or uncoupler). The normalized signals were then used to calculate the mitochondrial redox ratio, RR = (normalized NADH)/(normalized FAD), which has a baseline value of 1.
Reflectance signal
For experiments in which blood was added to the perfusate, the NADH and FAD reflectance signals were also measured in both channels. The quenching effect of blood on the surface fluorescence NADH and FAD signals was then corrected for by subtracting the normalized reflectance signals from the normalized fluorescence signals [27]. Thus, the following equation was used for RR in the presence of red blood cells.
Statistical evaluation of data
Data are presented as means ± standard error (SE) unless otherwise stated. Statistical comparisons were carried out in each group using a two-tailed student’s t-test or one way ANOVA followed by Tukey’s HSD (Honestly Significant Difference) post-hoc test, with p < 0.05 as the criterion for statistical significance.
5. Results
The fluorometer demonstrates a linear response to a change in the concentration of NADH (or FAD) in the presence of FAD (or NADH) over a wide range of NADH and FAD concentrations as shown in Figure 2.
Figure 2.
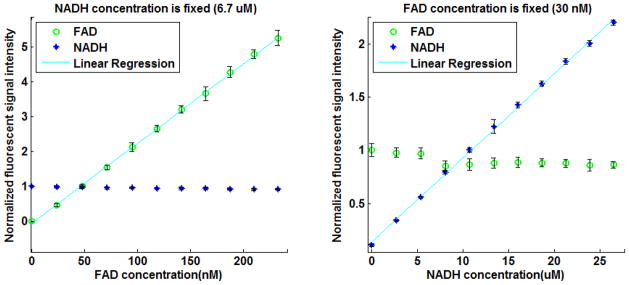
Sensitivity curves of the Fluorometer to changes in a) FAD and b) NADH concentration in an aqueous solution. All intensities are normalized to the signal level obtained at a concentration equal to that in lung tissue. Solid lines are linear regression fits (r2 = 0.999 and 0.998 for NADH and FAD, respectively). The range of NADH and FAD concentrations studied encompassed the NADH and FAD concentrations in lung tissue [23].
Lung ventilation added noise to both NADH and FAD lung surface signals. However, NADH and FAD baseline data (control perfusate) show that this noise was attenuated using a moving average digital filter (Figure 3). The response of both NADH and FAD lung surface signals to lung perfusion with rotenone, KCN, or PCP appeared within a minute of adding the chemical to the perfusate reservoir (Figure 3). An increase in NADH fluorescence signal indicates reduction of the electron transport chain, whereas an increase in the FAD fluorescence signal indicates oxidation of the electron transport chain. Lung perfusion with rotenone (complex I inhibitor) reduced the electron transport chain upstream from complex I resulting in an increase in NADH signal by 21.5 ± 2.5 (SE) %, with no effect on FAD signal, and increased RR by 21.3 ± 2.7 % (Table 1). Lung perfusion with KCN (complex IV inhibitor) reduced the electron transport chain resulting in a 22.0 ± 2.9 % increase in NADH, 6.8 ± 2.0 % decrease in FAD, and as a result a 30.3 ± 2.4 % increase in RR (Table 1). Lung perfusion with PCP, which uncoupled mitochondrial electron transport chain from phosphorylation, decreased NADH signal by 25.4 ± 3.7 % with no effect on FAD signal (p = 0.226), resulting in a 27.2 ± 2.9 % decrease in RR (Table 1). The p-values for the above % changes in NADH, FAD, and RR values are reported in Table 1.
Figure 3.
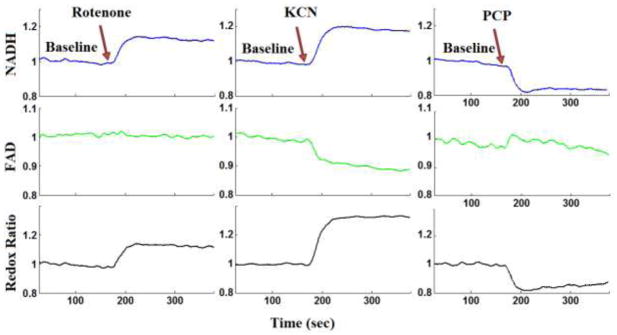
Lung surface NADH (top panel), FAD (middle panel), and mitochondrial redox ratio (bottom panel) signals for (a) baseline followed by lung perfusion with Rotenone, (b) baseline followed by perfusion with potassium cyanide (KCN), (c) baseline followed by lung perfusion with pentachlorophenol (PCP). For each measurement (NADH, FAD and RR) the signal was normalized to the average signal under baseline (control perfusate) conditions.
Table 1.
The effect of metabolic inhibitors and uncoupler on the lung tissue surface FAD and NADH fluorescence signals and the redox ratio (RR)
| Treatment | N | Change in FAD % | Change in NADH % | Change in RR % |
|---|---|---|---|---|
| Rotenone | 4 | 0.3 ± 0.3 (0.391) | 21.5 ± 2.5* (0.003) | 21.3 ± 2.7* (0.004) |
| KCN | 6 | −6.8 ± 2.0* (0.019) | 22.0 ± 2.9* (<0.001) | 30.3 ± 2.4* (<0.001) |
| PCP | 5 | 1.4 ± 1.0 (0.226) | −25.4 ± 3.7* (0.002) | −27.2 ± 2.9* (<0.001) |
Values are mean ± SE. N is the number of lungs for each conditions. RR =NADH/FAD.
p values are in parentheses (two-tailed student’s t-test, *p < 0.05).
In the presence of rotenone, KCN resulted in a small increase (4.0 ± 0.86 (SE) %) and decrease (−3.1 ± 0.86 %) in lung surface NADH and FAD signals, respectively, increasing RR by 7.0 ± 0.33 % as compared to RR value in the presence of rotenone only (Figure 4a). The addition of KCN to PCP-treated lungs (Figure 4b) reversed the effect of PCP on the redox status of the electron transport chain, increasing NADH signal by 27.2 ± 2.05 %, decreasing FAD signal by −9.6 ± 1.1 %, and as a result leading to a 38.9 ± 0.75 % increase in RR as compared to values in the presence of PCP only (Table 2). The p-values for the above percentage changes in NADH, FAD, and RR are reported in Table 2.
Figure 4.
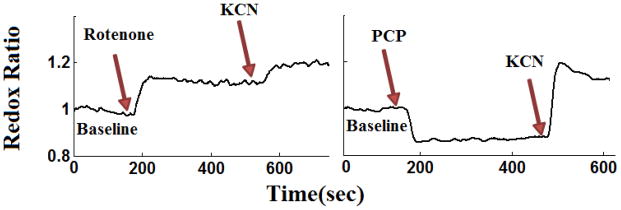
Evaluating the reversibility and additivity in lung surface mitochondrial redox ratio (RR) signals before (baseline) and after lung perfusion with perfusate containing (a) rotenone followed by rotenone + potassium cyanide (KCN) (b) PCP followed by PCP + KCN. The RR values were normalized to the average value under baseline (control perfusate) conditions.
Table 2.
The effect of lung perfusion with potassium cyanide (KCN) on the lung tissue surface fluorescence signals and redox ratio (RR) in the presence of rotenone or pentachlorophenol (PCP).
| Treatment | N | Change in FAD % | Change in NADH % | Change in RR % |
|---|---|---|---|---|
| Rotenone + KCN* | 6 | −3.1 ± 0.86 * (0.049) | 4.0 ± 0.86* (0.033) | 7.0 ± 0.33 * (<0.001) |
| PCP + KCN* | 4 | −9.6 ± 1.1* (<0.001) | 27.2 ± 2.05* (<0.001) | 38.9 ± 0.75* (<0.001) |
Values are mean ± SE. N is the number of lungs for each condition. The percentage change due to additional treatment with KCN is calculated relative to the new baseline in the presence of rotenone or PCP. RR = NADH/FAD
p values are in parentheses (two-tailed student’s t-test, *p < 0.05).
Addition of blood to the control perfusate quenched the lung surface NADH and FAD signals (Figure 5). Increasing perfusate hematocrit (Hct) level from 0% (control perfusate) to 1% decreased NADH and FAD signal by ~40% (Figure 5). Further increases in perfusate Hct had a smaller effect on the signals, with the NADH and FAD signals approaching a steady state value at ~ 4% perfusate hematocrit. This exponential relationship between the degree of quenching of NADH and FAD fluorescence signal and perfusate Hct level is consistent with Beer’s law [28]. The effect of blood on RR was relatively small and was independent of perfusate Hct within the range of Hct studied. This result is consistent with results by Chance et al. [18]. The results in Figure 5 also show that subtracting the normalized reflectance NADH or FAD signal from the corresponding normalized fluorescence signal did not completely eliminate the quenching effect of blood, especially that which occurred between 0% and 1% Hct.
Figure 5.

Lung surface NADH, FAD and mitochondrial redox ratio (RR) signals for a normal lung perfused with sequentially increasing hematocrit levels (0% to 4% Hct, vertical dot lines), before (uncorrected) and after (corrected) subtracting the normalized reflectance signals from the normalized fluorescence signals.
6. Discussion
The results of this study demonstrate the ability of optical fluorescence techniques to detect a change in the redox state of the mitochondrial electron transport chain as measured by lung surface NADH and FAD fluorescent signals. The measured changes in NADH and FAD surface fluorescence following lung treatment with rotenone, KCN, or PCP are qualitatively consistent with the known effects of these metabolic inhibitors on the redox status of the mitochondrial electron transport chain.
Optical fluorescence techniques have been widely used to probe tissue redox state and energy homeostasis in organs such as the heart and liver, but have not been fully used in lungs [16]. The lung tissue presents a greater challenge for quantitative fluorescence studies than metabolically active tissues such as the heart or liver. The reasons include the low mitochondrial density of lung cells compared with metabolically active organs such as the heart [29, 30], the lungs’ air content and high perfusion to metabolic needs ratio, and the lungs’ high collagen content which contributes to high background fluorescence [16]. To the best of our knowledge, the 1976 study by Fisher et al. [16] is the only study in the literature in which lung surface fluorometry was used to probe the redox status of lung tissue NADH. They reported relatively small changes in the NADH signal (Table 3) in response to lung treatment with metabolic inhibitors that are known to oxidize or reduce the mitochondrial electron transport chain. As shown in Table 3, the results of the present study represent a substantial improvement in our ability to detect a change in NADH redox status in response to lung treatment with metabolic inhibitors as compared to what Fisher et al. [16] had reported. This increased sensitivity is mostly due to the use of optical fibers and highly sensitive PMTs. The fiber-optic design injects light into the tissue allowing fluorescence emission to be more heavily weighted from the mitochondrial containing parenchymal tissue than from the surface connective tissue fluorescence as is the case with the lens-based optics approach used by Fisher et al. [16].
Table 3.
The comparison of the percentage change in the NADH signal in the presence of metabolic inhibitors and uncoupler in the lung tissue surface fluorescence between our fluorometer and previous results (Fisher et al. [16]).
| Treatment | N | %Change in NADH (Fisher and Chance[16]) | N | %Change in NADH (Present study) |
|---|---|---|---|---|
| Potassium cyanide | 11 | 6.7 ± 0.7 | 6 | 22.0 ± 2.9 |
| Amytal/(Rotenone) | 5 | 5.0 ± 1.2 | 4 | 21.5 ± 2.5 |
| Pentachlorophenol | 3 | −4.3 ± 0.5 | 5 | −25.4 ± 3.7 |
Values are mean ± SE for N experiments.
Our assumption in this study is that changes in the mitochondrial pool of NADH is a key contributor to the measured changes in the NADH fluorescence signal since the metabolic inhibitors target the mitochondrial electron transport chain [31, 32]. However, cytosolic NADH also contributes to the lung surface NADH signal. There are multiple processes that determine cytosolic NADH concentration, including NADH reduction during glycolysis, and NADH oxidation by lactate dehydrogenase. Another important cytosolic process that alters both cytosolic and mitochondrial NADH concentration is the malate-aspartate shuttle which transfers electrons across the mitochondrial membrane from cytosolic NADH, which is oxidized to NAD+, to mitochondrial NAD+, which is reduced to NADH [33]. Lung treatment with rotenone, KCN, or PCP decreases mitochondrial ATP production, stimulates glucoses [34, 35], and hence could alter cytosolic NADH concentration. Data in this report do not permit us to distinguish the contribution of these sources of NADH to the signal used to calculate RR values.
NADPH, which has the same fluorescence characteristics as NADH, may also contribute to the lung surface signal attributed to NADH in this study. The major cytosolic source is the pentose pathway, which requires glucose [25, 36, 37]. Other cytosolic sources include the malic enzyme and NADP+ dependent isocitrate dehydrogenase [25, 36, 37]. These enzymes use citrate acid cycle metabolites to generate NADPH, and hence do not require glucose. Mitochondrial NADPH is generated by energy-dependent mitochondrial nicotinamide nucleotide transhydrogenase, which catalyzes the interconversion of NADH to NADPH. Fisher et al. [37] showed that treatment of isolated perfused rabbit lungs with potassium cyanide, an uncoupler, or antimycin (complex III inhibitor) decreased the rate of the NADPH dependent mixed-function oxidation of p-nitroanisole to p-nitrophenol by 50% to 77% with glucose as substrate. Thus, the increase in lung surface NADH signal in the presence of cyanide (Table 1) cannot be attributed to an increase in the NADPH signal since cyanide decreased the rate of the NADPH-dependent mixed-function oxidation of p-nitroanisole to p-nitrophenol [37]. On the other hand, the decrease in measured lung surface NADH signal in the presence of the uncoupler (Table 1) could be in part due to a decrease in NADPH since lung treatment with the uncoupler decreased the rate of the NADPH dependent reaction. However, the fact that KCN and PCP had opposite effect on the lung surface NADH signal (Table 1), but the same effect on the rate of the NADPH-dependent reaction may suggest that NADPH is not contributing much to the measured NADH signal.
Unlike the NADH signal which has cytosolic and mitochondrial components, the FAD signal derives only from the mitochondria [38]. To the best of our knowledge, this study is the first to measure lung surface FAD fluorescence. Compared to NADH, the change in the FAD signal in response to lung treatment with the various metabolic inhibitors was relatively small (Table 1). One reason could be due to the lower lung tissue concentration of FAD as compared to NADH. In fact, the FAD baseline signal is small relative to the NADH signal as evidenced by the difference in signal amplification for the NADH and FAD channels (103 for NADH compared to ~6 × 105 for FAD).
Sources of redox-sensitive flavoprotein (FAD) include succinate dehydrogenase (complex II), lipoamide dehydrogenase (LipDH), and electron transfer flavoprotein (ETF) [39, 40]. Treatment with rotenone (complex I inhibitor), which raises mitochondrial NADH/NAD+, should decrease LipDH flavoprotein fluorescence signal [25]. In the present study, rotenone had no effect on FAD signal (Table 1), suggesting that lung mitochondrial LipDH does not contribute much to the measured lung surface flavoprotein signal.
The contribution of ETF, a fatty-acid oxidizing flavoprotein, to the measured flavoprotein signal can be evaluated by lung treatment with potassium cyanide (complex IV inhibitor), which reduces ETF, and complex II flavoprotein signal [25]. Thus, the cyanide-dependent decrease in FAD signal measured in the present study (Table 1) could be due to ETF and/or complex II flavoprotein signal. For lung tissue, glucose is the major oxidizable substrate under normal physiological conditions [37]. Since glucose was the only oxidizable substrate in our perfusate, change in FAD signals (Table 1) in the presence of cyanide should be attributed to complex II flavoprotein rather than ETF flavoprotein.
The contributions of complex II, LipDH, and ETF to the measured lung surface flavoprotein signal in the present study are different from those reported by Kunz and Kunz [39]. Whether this difference in the contributions of these flavoprotein enzymes is attributable to a difference between intact lungs and isolated mitochondria, tissue differences [40], or other factors, is not known. Finally, lung treatment with PCP had no effect on the FAD surface fluorescence signal (Table 2). This could be because most of the flavoproteins (FAD + FADH2) are in the oxidized (FAD) form.
The measured changes in NADH and FAD fluorescence signals in response to the treatments in the present study are qualitatively and quantitatively similar to those obtained from rat hippocampal slices [41]. Gerich et al. showed that treatment with rotenone (20 μM) increased NADH signal by ~ 10% with no effect on FAD. Potassium cyanide (100 μM or 1 mM) increased NADH by ~ 20% and decreased FAD signal by ~ 9%. Treatment with the uncoupler carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM) decreased NADH by ~ 12% with no effect on FAD. Using isolated rat ventricular trabeculae, Brandes and Bers [33] demonstrated using fluorescence spectroscopy that treatment with KCN increased NADH/NAD+ by 31%, whereas treatment with the uncoupler FCCP decreased the ratio by 49%. These results are again consistent with the results from the present study (Table 1).
Since in vivo estimates of RR are unavoidably acquired in the presence of blood, we determined the effect of blood in perfusate on NADH and FAD lung surface fluorescence signal, and on RR. The results of the study show an exponential relationship of the quenching effect of red blood cells on NADH and FAD fluorescence signals, consistent with Beer’s law [28]. However, the effect of blood on RR is relatively small and appears to be independent of blood hematocrit level. Although the range of Hct studied (0–4%) is much lower than rat blood Hct (44%) [29], the plateau seen in the results at 4% Hct (Figure 5) indicates that higher perfusate Hct will have a minimal additional quenching effect on the NADH and FAD fluorescence signals. These results are consistent with those from a study by Chance et al. in which they showed that perfusion of isolated organs (e.g., liver) with perfusate containing blood (0–4% Hct) decreased NADH and FAD signals but did not change the redox ratio [18]. Thus, we expect that the presence of blood in perfusate will have no significant effect on the percentage changes in RR (Tables 1 and 2). For instance, Figure 6 shows that potassium cyanide increased RR by ~26% in a lung perfused with blood-containing perfusate (4% Hct). This increase in RR is within the 22.0 ± 2.9 % (SE) range measured in lungs perfused with blood-free perfusate (Table 1).
Figure 6.
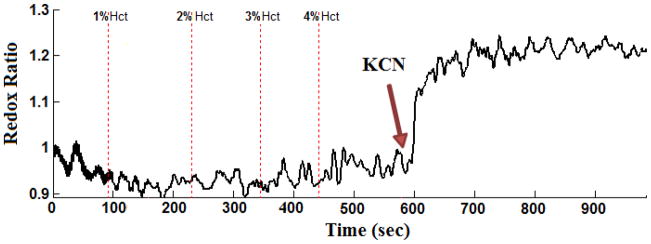
Evaluating the effect of lung perfusion with KCN on surface mitochondrial redox ratio (RR) in the presence of blood (4% Hct = hematocrit).
The lung surface optical imaging data measured in this study does not provide information about the specific types of lung cells contributing to the measured NADH and FAD signals, although endothelial cells would be expected to contribute significantly because of their relatively large surface area and fraction of total lung cells [42]. Although determining the contributions of specific lung cell types to the measured signal is potentially important, the global oxidoreductive state of the lung tissue is a highly valuable piece of information irrespective of the individual cell types contributing to the redox ratio. Another limitation of optical surface fluorescence imaging is that it may not detect deeper than 500 μm, with an initial diameter of 3.2 mm, for a volume of ~ 4 mm3. However, this resolution is more than sufficient for determining the RR of parenchymal tissue which has a thickness (air to plasma) of 1.6 μm [42]. That said, central lung lesions without pleural extension would not be expected to be detected by surface fluorescence measurements such as those used in the present study.
In conclusion, the results of this study demonstrate the utility of optical fluorometry to detect a change in the redox status of lung mitochondrial coenzymes NADH and FAD in isolated perfused lungs. While optical determination of RR in tissues such as brain and myocardium is accepted [4, 43], the applicability of these methodologies in examination of the redox state in lungs, where the density of mitochondria is much lower, has not been established. Using low temperature fluorescence imaging, we have demonstrated that rat exposure to hyperoxia (85% oxygen for 7 days) decreased the NADH signal and increased the FAD signal, resulting in an average decrease in RR of 23% [44]. The potential clinical importance of real time optical imaging of lungs in patients with critical illness, including patients on high O2, or patients with ischemia-reperfusion lung injury secondary to lung transplant, chest contusions, is great. Reliable fluorescence determination of RR could be adopted in the same fashion that near-infrared spectroscopy (NIRS) is gaining favor as a noninvasive measure of tissue oxygenation in critically injured patients [45]. While NIRS is an indirect measure of tissue oxygenation, NADH and FAD data provide information regarding tissue redox and mitochondrial bioenergetics, a truer and more sensitive early measure of organ function. Our studies support the capacity of fluorescence imaging to detect a change in mitochondrial redox state in isolated perfused lungs, and set the stage for in vivo studies.
Acknowledgments
We appreciate the support of University of Wisconsin Milwaukee RGI 6 Grant, Clinical and Translational Science Institute KL2 Grant: NIH 8Kl2TR000056, Wisconsin Applied Research grant (Wi-ARG), NIH Grants HL-24349 (SHA), HL 49294 (ERJ), and the Department of Veterans Affairs that provided resources essential to the completion of these investigations. We acknowledge the technical help from Dr. Steven Haworth in the Clement J. Zablocki VA, Milwaukee, WI.
Contributor Information
Kevin Staniszewski, Email: kevinss2@uwm.edu, Biophotonics Lab, Department of Electrical Engineering, University of Wisconsin Milwaukee, 3200 N Cramer St., Milwaukee, WI 53211.
Said H. Audi, Email: said.audi@marquette.edu, Department of Biomedical Engineering, Marquette University, 1515 West Wisconsin Avenue, Milwaukee, WI, 53233.
Reyhaneh Sepehr, Email: rsepehr@uwm.edu, Biophotonics Lab, Department of Electrical Engineering, University of Wisconsin Milwaukee, 3200 N Cramer St., Milwaukee, WI 53211.
Elizabeth R. Jacobs, Email: Elizabeth.Jacobs@va.gov, Associate Chief of Staff, Research and Development, Clement J. Zablocki VA Medical Center, 5000 W. National Avenue Milwaukee, WI 5329 and Associate Dean Research, Medical College of Wisconsin
Mahsa Ranji, Email: ranji@uwm.edu, Biophotonics Lab, Department of Electrical Engineering, University of Wisconsin Milwaukee, 3200 N Cramer St., Milwaukee, WI 53211.
References
- 1.Fisher AB. Intermediary metabolism of the lung. Environ Health Perspect. 1984 Apr;55:149–58. doi: 10.1289/ehp.8455149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramanujam N, et al. Low Temperature Fluorescence Imaging of Freeze-trapped Human Cervical Tissues. Opt Express. 2001 Mar 12;8:335–43. doi: 10.1364/oe.8.000335. [DOI] [PubMed] [Google Scholar]
- 3.Zmijewski JW, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med. 2008 Jul 15;178:168–79. doi: 10.1164/rccm.200710-1602OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ranji M, et al. Fluorescence spectroscopy and imaging of myocardial apoptosis. J Biomed Opt. 2006 Nov-Dec;11:064036. doi: 10.1117/1.2400701. [DOI] [PubMed] [Google Scholar]
- 5.Ranji M, et al. Quantifying acute myocardial injury using ratiometric fluorometry. IEEE Trans Biomed Eng. 2009 May;56:1556–63. doi: 10.1109/TBME.2008.2006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsubara M, et al. In vivo fluorometric assessment of cyclosporine on mitochondrial function during myocardial ischemia and reperfusion. Ann Thorac Surg. May;89:1532–7. doi: 10.1016/j.athoracsur.2010.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chance B, Baltscheffsky H. Respiratory enzymes in oxidative phosphorylation. VII. Binding of intramitochondrial reduced pyridine nucleotide. J Biol Chem. 1958 Sep;233:736–9. [PubMed] [Google Scholar]
- 8.Barlow CH, et al. Fluorescence mapping of mitochondrial redox changes in heart and brain. Crit Care Med. 1979 Sep;7:402–6. doi: 10.1097/00003246-197909000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Balaban RS, Mandel LJ. Coupling of aerobic metabolism to active ion transport in the kidney. J Physiol. 1980 Jul;304:331–48. doi: 10.1113/jphysiol.1980.sp013327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meirovithz E, et al. Effect of hyperbaric oxygenation on brain hemodynamics, hemoglobin oxygenation and mitochondrial NADH. Brain Res Rev. 2007 Jun;54:294–304. doi: 10.1016/j.brainresrev.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Maleki S, et al. Mitochondrial redox studies of oxidative stress in kidneys from diabetic mice. Biomed Opt Express. 2012 Feb 1;3:273–81. doi: 10.1364/BOE.3.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vollmar B, et al. A correlation of intravital microscopically assessed NADH fluorescence, tissue oxygenation, and organ function during shock and resuscitation of the rat liver. Adv Exp Med Biol. 1998;454:95–101. doi: 10.1007/978-1-4615-4863-8_12. [DOI] [PubMed] [Google Scholar]
- 13.Nioka S, et al. Simulation of Mb/Hb in NIRS and oxygen gradient in the human and canine skeletal muscles using H-NMR and NIRS. Adv Exp Med Biol. 2006;578:223–8. doi: 10.1007/0-387-29540-2_36. [DOI] [PubMed] [Google Scholar]
- 14.Ramanujam N, et al. In vivo diagnosis of cervical intraepithelial neoplasia using 337-nm-excited laser-induced fluorescence. Proc Natl Acad Sci U S A. 1994 Oct 11;91:10193–7. doi: 10.1073/pnas.91.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mycek MA, et al. Colonic polyp differentiation using time-resolved autofluorescence spectroscopy. Gastrointest Endosc. 1998 Oct;48:390–4. doi: 10.1016/s0016-5107(98)70009-4. [DOI] [PubMed] [Google Scholar]
- 16.Fisher AB, et al. Evaluation of redox state of isolated perfused rat lung. Am J Physiol. 1976 May;230:1198–1204. doi: 10.1152/ajplegacy.1976.230.5.1198. [DOI] [PubMed] [Google Scholar]
- 17.Sepehr R, et al. Optical imaging of tissue mitochondrial redox state in intact rat lungs in two models of pulmonary oxidative stress. J Biomed Opt. 2012 Apr;17:046010. doi: 10.1117/1.JBO.17.4.046010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chance B, et al. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem. 1979 Jun 10;254:4764–71. [PubMed] [Google Scholar]
- 19.Ranji M, et al. Fluorescence spectroscopy and imaging of myocardial apoptosis. Journal of Biomedical Optics. 2006 Nov-Dec;11 doi: 10.1117/1.2400701. [DOI] [PubMed] [Google Scholar]
- 20.Ranji M, et al. Quantifying Acute Myocardial Injury Using Ratiometric Fluorometry. Ieee Transactions on Biomedical Engineering. 2009 May;56:1556–1563. doi: 10.1109/TBME.2008.2006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Audi SH, et al. Coenzyme Q1 redox metabolism during passage through the rat pulmonary circulation and the effect of hyperoxia. J Appl Physiol. 2008 Oct;105:1114–26. doi: 10.1152/japplphysiol.00177.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audi SH, et al. Duroquinone reduction during passage through the pulmonary circulation. Am J Physiol Lung Cell Mol Physiol. 2003 Nov;285:L1116–31. doi: 10.1152/ajplung.00185.2003. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, et al. Investigation of Synchronous Fluorescence Method in Multicomponent Analysis in Tissue. IEEE J of selected Topics in Quantum Electronics. 2010;16:14. [Google Scholar]
- 24.Ranji M. Fluorescent images of mitochondrial redox states in in situ mouse hypoxic ischemic intestines. Journal of Innovative Optical Health Sciences (JIOHS) 2009;2:365–374. doi: 10.1142/S1793545809000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rocheleau JV, et al. Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response. J Biol Chem. 2004 Jul 23;279:31780–7. doi: 10.1074/jbc.M314005200. [DOI] [PubMed] [Google Scholar]
- 26.Xia L, et al. Reduction of ubiquinone by lipoamide dehydrogenase. An antioxidant regenerating pathway. Eur J Biochem. 2001 Mar;268:1486–90. doi: 10.1046/j.1432-1327.2001.02013.x. [DOI] [PubMed] [Google Scholar]
- 27.Boldt M, et al. A sensitive dual wavelength microspectrophotometer for the measurement of tissue fluorescence and reflectance. Pflugers Arch. 1980 May;385:167–73. doi: 10.1007/BF00588698. [DOI] [PubMed] [Google Scholar]
- 28.Commoner B, Lipkin D. The Application of the Beer-Lambert Law to Optically Anisotropic Systems. Science. 1949 Jul 8;110:41–3. doi: 10.1126/science.110.2845.41-a. [DOI] [PubMed] [Google Scholar]
- 29.Gan Z, et al. Quantifying mitochondrial and plasma membrane potentials in intact pulmonary arterial endothelial cells based on extracellular disposition of rhodamine dyes. Am J Physiol Lung Cell Mol Physiol. 2011 May;300:L762–72. doi: 10.1152/ajplung.00334.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Else PL, Hulbert AJ. Mammals: an allometric study of metabolism at tissue and mitochondrial level. Am J Physiol. 1985 Apr;248:R415–21. doi: 10.1152/ajpregu.1985.248.4.R415. [DOI] [PubMed] [Google Scholar]
- 31.Eng J, et al. Nicotinamide adenine dinucleotide fluorescence spectroscopy and imaging of isolated cardiac myocytes. Biophys J. 1989 Apr;55:621–30. doi: 10.1016/S0006-3495(89)82859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nuutinen EM. Subcellular origin of the surface fluorescence of reduced nicotinamide nucleotides in the isolated perfused rat heart. Basic Res Cardiol. 1984 Jan-Feb;79:49–58. doi: 10.1007/BF01935806. [DOI] [PubMed] [Google Scholar]
- 33.Brandes R, Bers DM. Increased work in cardiac trabeculae causes decreased mitochondrial NADH fluorescence followed by slow recovery. Biophys J. 1996 Aug;71:1024–35. doi: 10.1016/S0006-3495(96)79303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen CB, White CW. Glucose modulates cell death due to normobaric hyperoxia by maintaining cellular ATP. Am J Physiol. 1998 Jan;274:L159–64. doi: 10.1152/ajplung.1998.274.1.L159. [DOI] [PubMed] [Google Scholar]
- 35.Owen MR, et al. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000 Jun 15;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 36.Aldrich TK, et al. Paraquat inhibits mixed-function oxidation by rat lung. J Appl Physiol. 1983 Apr;54:1089–93. doi: 10.1152/jappl.1983.54.4.1089. [DOI] [PubMed] [Google Scholar]
- 37.Fisher AB, et al. Pulmonary mixed-function oxidation: stimulation by glucose and the effects of metabolic inhibitors. Biochem Pharmacol. 1981 Feb 15;30:379–83. doi: 10.1016/0006-2952(81)90070-8. [DOI] [PubMed] [Google Scholar]
- 38.Aldakkak M, et al. Modulation of mitochondrial bioenergetics in the isolated Guinea pig beating heart by potassium and lidocaine cardioplegia: implications for cardioprotection. J Cardiovasc Pharmacol. 2009 Oct;54:298–309. doi: 10.1097/FJC.0b013e3181b2b842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunz WS, Kunz W. Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochimica Et Biophysica Acta. 1985 Sep 6;841:237–46. doi: 10.1016/0304-4165(85)90064-9. [DOI] [PubMed] [Google Scholar]
- 40.Kunz WS, Gellerich FN. Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem Med Metab Biol. 1993 Aug;50:103–10. doi: 10.1006/bmmb.1993.1051. [DOI] [PubMed] [Google Scholar]
- 41.Gerich FJ, et al. Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J Neurophysiol. 2006 Jul;96:492–504. doi: 10.1152/jn.01015.2005. [DOI] [PubMed] [Google Scholar]
- 42.Crapo JD, et al. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis. 1980 Jul;122:123–43. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- 43.Mayevsky A. Brain NADH redox state monitored in vivo by fiber optic surface fluorometry. Brain Res. 1984 Mar;319:49–68. doi: 10.1016/0165-0173(84)90029-8. [DOI] [PubMed] [Google Scholar]
- 44.Sepehr R, et al. Optical imaging of tissue mitochondrial redox state in intact rat lungs in two models of pulmonary oxidative stress. Journal of Biomedical Optics. 2012 Apr;17:046010. doi: 10.1117/1.JBO.17.4.046010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Blasi RA, et al. Noninvasive measurement of forearm blood flow and oxygen consumption by near-infrared spectroscopy. J Appl Physiol. 1994 Mar;76:1388–93. doi: 10.1152/jappl.1994.76.3.1388. [DOI] [PubMed] [Google Scholar]