

Abstract
As fundamental research advances, it is becoming increasingly clear that a clinically expressed disease implies a mixture of intertwining molecular disturbances. Oxidative stress is one of such pathogenic pathways involved in virtually all central nervous system pathologies, infectious, inflammatory, or degenerative in nature. Since brain homeostasis largely depends on integrity of blood-brain barrier (BBB), many studies focused lately on BBB alteration in a wide spectrum of brain diseases. The proper two-way molecular transfer through BBB depends on several factors, including the functional status of its tight junction (TJ) complexes of proteins sealing neighbour endothelial cells. Although there is abundant experimental work showing that oxidative stress associates BBB permeability alteration, less is known about its implications, at molecular level, in TJ protein expression or TJ-related cell signalling. In this paper, oxidative stress is presented as a common pathway for different brain pathogenic mechanisms which lead to BBB dysregulation. We revise here oxidative-induced molecular mechanisms of BBB disruption and TJ protein expression alteration, in relation to ageing and neurodegeneration.
1. Introduction
It has been extensively proven that a large array of neurological diseases and brain ageing itself are associated with oxidative stress [1–3]. Multiple sclerosis, stroke, brain tumours, and neuroinfections are conditions which associate both reactive oxygen species (ROS) aggression and blood brain barrier (BBB) impairment as well-proven pathogenic mechanisms. Relatively recent data documents BBB disruption not only in vascular or inflammatory brain diseases but in neurodegenerative disorders as well, where oxidative stress plays an important role in the pathogenic scenario [4, 5]. Whether oxidative damage is an important and early event in BBB alteration process, it is not established so far.
BBB is the interface between the periphery of circulatory system and central nervous system. The endothelial cells are the primary components of the BBB, responsible for the controlled environment of the brain. These cells lack fenestrations and have increased mitochondrial content, minimal pinocytotic activity, and a low number of caveolae. A 30–40 nm thin basement membrane is found between endothelial and neighbouring glial cells [6, 7]. At the BBB level, paracellular transport is restricted by tight junctions (TJs), allowing a peculiar “sealing” capacity. However, other cell types—pericytes, astrocytes, and neurons—are required for an accurate organization and function of BBB, not necessarily through direct contact with endothelial cells (Figure 1). Pericytes are the only cell type to intimately connect with endothelial cells, as they lay embedded within the endothelial basement membrane—a fibrillary structure of collagen IV, laminins, and proteoglycans. Pericytes strengthen the barrier integrity and their loss opens the BBB in an age-dependent manner [8]. Recently, pericytes have been added to the classical in vitro 2-cell type model of BBB (coculture of endothelial and astrocytes) [9]. Astrocytes are separated from endothelial cells by the basement membrane around which they extend cell processes called end-feet. Hence, no cell-to-cell junctions are involved in this case, but the molecular flow of information between the two cell types is vital for BBB embryonic development [10] and adult life BBB integrity [11]. In vitro studies indicate astrocytes as regulators of TJ tightness and polarized distribution of transporters at endothelial level [12]. Furthermore, coculture of astrocytes with epithelial (other than brain endothelial) cells leads to induction of BBB properties [13, 14] and this is now a common practice in in vitro BBB models. Neurons are not morphologically involved in BBB formation, but numerous myelinated and nonmyelinated axons are found in close proximity of brain capillaries. The current model of brain homeostasis is based on the neurovascular unit, comprising cellular elements of BBB along with the neurons to which they connect into a functional network [15].
Figure 1.
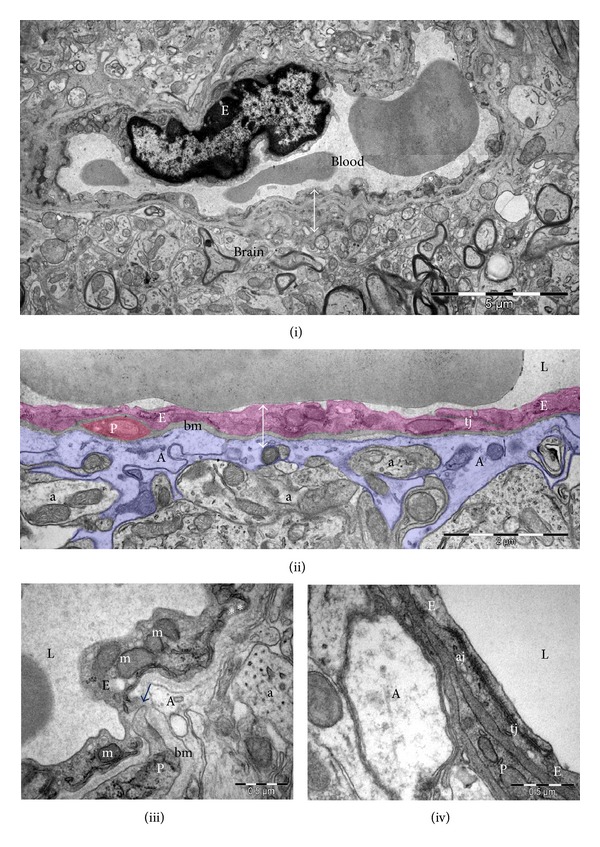
Ultrastructure of blood-brain barrier (↔). (i) Overall electron microscopy image of a cerebral capillary. (ii) Blood-brain barrier components: endothelial cells (E, purple coloured), pericytes (P, brown coloured), basement membrane (bm), and end-feet of astrocytes (A, blue coloured). (iii) Cerebral capillaries have nonfenestrated endothelial cells with numerous mitochondria (m) and rare pinocytotic vesicles (*). Direct membrane-membrane contacts (arrow) often occur between endothelial cells and pericytes. (iv) Tight (tj) and adherens (aj) junctions seal the continuous capillary endothelium. Cerebral capillary lumina (L), axons (a).
The BBB is functionally characterized by highly restrictive transbarrier transport, due to sealing of paracellular pathway by TJs and low transcytotic traffic through caveolae [10]. Transport of virtually all nondiffusible, nonlipidic molecules is controlled through specific carriers present on both sides of endothelial cells, in a time- and concentration-dependent manner. Consequently, quantification of large protein (albumin, dextran) traffic from blood to nervous tissue is an indicator of “tightness” of cell-to-cell endothelial junctions. A barrier “tightness” or “leakiness” is given by expression and molecular organization of different TJ species, which in the case of BBB are unique. In fact, clusters of densely packed molecules form the interendothelial junctions that contain specific components for both adherens and tight junctions.
The TJ is an intricate macromolecular complex [6, 16] formed by:
integral membrane proteins: claudins (claudin-1, 2, 3, 5, 11, 12, 18), MARVEL (the myelin and lymphocyte protein (MAL) and related proteins for vesicle trafficking and membrane link) proteins (occludin, tricellulin/marvelD2 and marvelD3), junctional adhesion molecules (JAMs), endothelial cell selective adhesion molecule (ESAM), and so forth;
cytoplasmic proteins: zonula occludens proteins (ZO-1, 2, 3), afadin (AF-6), calcium/calmodulin dependent serine protein kinase (CASK/LIN-2) from membrane-associated guanylate kinase proteins (MAGUK family), actin binding protein (cingulin), small G-proteins (Rho, Rac, Cdc42), ZO-1-associated nucleic acid binding protein (ZONAB), cyclin-dependent kinase-4 (CDK-4), and so forth;
actin cytoskeleton.
For a long while, the TJ complexes were considered static structures but new data support a dynamic model of barriers and also suggest that regulation of TJ openings and closings may provide sensitive means to modulate barrier function without changing protein expression [17].
In vivo and in vitro molecular studies of TJ proteins show that alteration of BBB in neurodegeneration usually cooccurs with modified TJ protein expression. As already mentioned, molecular organization of TJs is responsible for the “leakiness” of the BBB, which physiologically is more tight than other epithelial sites, a fact illustrated by a transepithelial resistance 50 times higher than other epithelia [18]. This peculiarity is not only a consequence of protein composition, but also of cellular sensitivity to microenvironment [19].
Although deleterious effects of oxidative stress on neuronal and glial populations in healthy aged and dementia brains are well stated, less is known about its consequences on endothelial cells, BBB, and tight junction protein expression. Based on the functional concept of neurovascular units, the presumption of BBB alteration in a neuronal/glial oxidative stress microenvironment is a plausible theory, possibly involving oxidative stress-related molecules. Furthermore, age is a certain inductor of BBB alteration, as briefly discussed in the following section; therefore, occurrence of oxidative stress in early stages of neurodegeneration might initiate tight junction impairment.
However, in vitro experimental setups used to decipher TJ protein alterations in oxidative environment are very variable in terms of culturing conditions. Most authors acknowledged the need to replicate the results by glial-endothelial cocultures, use of conditioned media, or in vivo conditions [20, 21]. Thus, the results are sometimes conflicting or even contradictory (see Table 1).
Table 1.
Expression of tight junction proteins in various cellular models of oxidative stress.
| BBB in vitro
model |
Type of experiment | Special conditions | Documentation of BBB permeability increase | Tight junction proteins alterations | Reference |
|---|---|---|---|---|---|
| BBMEC monolayers | Hypoxic stress | Glial conditioned-media treatment | Permeability studies with [14]-sucrose | Claudin-1 shows a significant increase following hypoxic stress | [21] |
|
| |||||
| BBMEC monolayers | Hypoxia/reoxygenation | none | TEER measurements and [14]-sucrose transfer across the barrier | Significant increase in expression of occludin, ZO-1, and ZO-2 |
[131] |
|
| |||||
| Rat GP8/3.9 cells | ROS generating environment by a mixture of xanthine oxidase and hypoxanthine | — | TEER FITC-dextran permeability across the barrier |
Decrease of occludin and claudin-5 expression after exposure to oxidative environment | [103] |
|
| |||||
| PBMEC | Hypoxia | Coculture with astrocytes/C6 glioma cells | TEER and passage of [3H]inulin | Decreased ZO-1 immunoreactivity at regions of cell-cell contact |
[43] |
|
| |||||
| BMVECs on a 8.0 μm matrigel-based insert | MMPs aggression | Coculture with leukemic cells | 40 kDa dextran-FITC flux by flow cytometry analysis | Downregulation of ZO-1, claudin-5, and occludin | [132] |
|
| |||||
| hCMEC/D3 (immortalized human BEC line) | Aβ peptides treatments | — | permeability to the paracellular tracer 70 kD FITC-dextran | Decrease in the occludin level, whereas claudin-5 and ZO-1 were unaffected | [85] |
|
| |||||
| Human BMVEC | Exposure to ROS | — | TEER and monocytes migration studies | Decreased occludin and ZO-1 total content, whereas claudin-5 expression depended on the type of stressor used | [91] |
BBMEC: bovine brain microvessel endothelial cells.
TEER: transendothelial electrical resistance.
PBMEC: primary cultures of porcine brain-derived microvascular endothelial cells.
BMVEC: brain microvascular endothelial cells.
ROS: reactive oxygen species.
MMPs: matrix metalloproteinases.
2. Endothelial Ageing and Tight Junctions in Aged Blood-Brain Barrier
Ageing is an independent factor associated with endothelial dysfunction even in the absence of other cardiovascular risk factors [22]. Aged endothelium showed a defective response to certain vasodilators [23], related to reduced NO-mediated dilatation [24], oxidative stress, and vascular inflammation [25]. Brain vasculature in aged animals showed predisposition to increased oxidative stress, activation of NADPH oxidase [26], and of nuclear enzyme poly(ADP ribose) polymerase (PARP) [27]. Ageing is also associated with increased expression of proinflammatory cytokines in vascular endothelial cells from healthy humans [28] which further favours a prooxidative state. Aged brains show increased matrix metalloproteinase- (MMP-) 2 activity and increased MMP-9 expression upon trauma, along with altered BBB repair mechanisms [29].
Molecular studies of BBB impairment in normal ageing explore only superficially the complexity of underling events, usually addressing only few proteins expression and distribution in one experimental paradigm. Results are generated in animal models and convey towards the conclusion that ageing leads to lower tight junction protein expression and a “leaky” BBB status [30–32].
Cumulative damage to mitochondria and mitochondrial DNA caused by ROS accounts for the mitochondrial theory of aging. In Figure 1(iii), we show a typical EM image of rat BBB, where mitochondria are clearly observed in both endothelial and glial cells. EM assessment of BBB in aged laboratory animals might offer a clue about mitochondria content and morphology in different BBB cell types, considering the large number of mitochondria in cerebral endothelium [8]. However, to our knowledge, there are no reports exploring mitochondrial alteration in aged brain endothelia so far.
Age seems to be a BBB frailty-inducing factor, as aged laboratory animals are more prone to brain oedema formation, ischemic injury, neuronal apoptosis following contusion and earlier onset of neuroinflammation than young littermates [33]. In the same manner, BBB dysfunction in old age was shown to be closely related to white matter lesions and lacunar infarctions [34].
There are several studies to address BBB permeability in aged animals (reviewed in [35]), in different experimental models, such as reproductive senescent mouse females [36], or senescence-accelerated mice [37]. They all led to the same conclusion that BBB permeability is altered in aged brain. Nevertheless, how and why this impairment occurs is not clear, and data regarding occludin and claudins expression and distribution in aged brain are scarce.
An overall assessment of BBB integrity can be obtained by immunohistochemistry methods, which show the albumin or immunoglobulin abnormal presence in the brain parenchyma, by elevated CSF albumin to plasma albumin ratio, or by increased perivascular enhancement at brain magnetic resonance imaging (MRI). In human aged brains serum protein immunostaining shows a “leaky” BBB which, interestingly enough, is not associated, at molecular level, with significant changes in endothelial expression of TJ proteins [38], and BBB leakage seems to show a wide individual variation [39].
3. Oxidative Stress Inducers at BBB Level
Although oxidative stress has been extensively studied in central nervous system different injuries, not enough data is available yet about its triggers and effectors on BBB. To some extent, as a result of vicious circles generated at molecular levels, it is difficult to separate or clearly indicate the cause and the effect of oxidative stress on BBB.
3.1. Hypoxia
Hypoxia is probably the best documented pathological process that induces BBB opening. It can be studied in vitro, by exposure of cell cultures to a mixture of hypoxic gas (95% N2/5% CO2; 99% N2/1% O2) or to pure NO2 and in vivo, by exposure of animal models to low oxygen air (6–8% O2) or ligation of cerebral arteries. Permeability may be further assessed by abnormal transport across BBB of large molecules, such as albumin, labelled dextrans, immunoglobulins, or labelled monocyte migration. Proposed mechanisms for altered permeability include increased exposure to free radicals [40] and/or inflammatory cytokines, such as IL-6 and TNF-α [41], activation of MMPs and downregulation of their tissular inhibitors (TIMPs) [42] and induced NOS expression [43], all of them ultimately reflected in the levels of tight junction protein expression.
Opening of BBB in hypoxia/reoxygenation studies is well confirmed in animal models and occurs earlier in aged animals versus young ones [44–46], following a biphasic pattern documented in vivo by MRI studies [47, 48].
Hypoxia is known to change BBB permeability and TJ protein expression in cerebral capillaries [49]. Lipid raft-associated occludin oligomeric assemblies were shown to be internalized during hypoxia [50] and ZO-1 and occludin sub-cellular localization correlated with increased paracellular permeability [51]. Reports of claudins expression during ischemia/reperfusion experiments are, however, contradictory. This can be at least partially explained by different experimental paradigms used in different studies.
3.2. Inflammation, Proinflammatory Cytokines, and Chemokines
Both normal ageing and neurodegenerative disorders are characterized by a degree of neuroinflammation [52]. In the CNS, proinflammatory cytokines are overexpressed as a result of intense/prolonged oxidative stress and are considered marks of neuroinflammation, a well-proven pathogenic mechanism in Alzheimer's disease (AD) and other neurodegenerative conditions. Cytokines, such as IL-1, IL-6, and TNF-α, are increased in plasma and CSF of acute ischemic stroke patients and seem to be associated with increased risk of worsening or recurrence [53–55]. High levels of plasma IL-6, associated with high CRP, seem to be associated with risk of vascular dementia (VaD) [56], and increased levels of IL-6 and TNF-α are also associated with senescence and frailty in old age [57]. Along with other cytokines, growth factors and plasma proteins, IL-1α, IL-8, and TNF-α were proposed as biomarkers able to distinguish AD from controls [58, 59]. A TNF-α inhibitor is reported to improve aphasia in demented patients [60, 61]. Proinflammatory cytokines are important regulators of MMPs and TIMPs expression [41]. In particular, TNF-α-mediated stimulation of MMP expression and synthesis is considered to be an important link between the proinflammatory cytokine network and the local increase of MMP proteolytic activity [41].
Along with TNF-α, IFN-γ has also been repeatedly reported to modify tight junction barrier function in various polarized epithelia [62–64]. Treatment of cell culture with IFN-γ led to decreased protein expression and relocalization of ZO-1 and occludin, occludin and JAM-A [65], in a time and dose-dependent manner. According to Scharl et al., AMP-activated protein kinase (AMPK) in concert with other signals induced by IFN-γ, seems to play a role in mediating reduced epithelial barrier function [66]. Chemokines CCL-2 and CXCL-8 are also reported to be responsible for increased BBB permeability, CCL-2 being produced by both astrocytes and endothelial cells in the late phase of hypoxia/reoxygenation-induced BBB disruption [67].
Some of these cytokines and chemokines appear to exclusively affect the paracellular permeability (e.g., IL-1β and CXCL8), while some others predominantly act to increase transcellular permeability (e.g., TNF-α) [18]. Experimentally induced peripheral inflammation also increases BBB permeability and leads to decreased occludin expression [68] and increased expressions of claudin-3 and 5 [69].
A common experimental animal model used for BBB breakdown in neuroinflammation is the experimental autoimmune encephalomyelitis (EAE), used for the study of multiple sclerosis (MS). An important aspect in the etiopathogeny of MS is loss of immune-privileged environment of the brain and extravasation of leukocytes across the BBB, through chemokine-chemokine receptor interaction. Use of mice with targeted deletions of certain chemokines and their receptors revealed a role for CCL2 and CCR2 in the induction of EAE via effects on infiltrating monocytes [70]. CXCL12 relocation in MS and EAE at the level of the postcapillary venules appears to strongly correlate with the perivascular infiltration of T-cells [71]. CCL19 protein levels in lysates of brain tissue as well as CSF samples were found to be elevated in MS [72]. Regarding the molecular alterations of BBB TJ proteins, in EAE affected mice were noted a coincident loss of both claudin-5 and occludin normal junctional staining patterns [73] and loss of claudin-3 expression that correlated with immune cell infiltration into the CNS and BBB leakiness [74]. Interestingly, although increased expression of claudin-1 in a transgenic EAE mouse model sealed the BBB for paracellular traffic of large molecules, it did not seem to influence immune cell trafficking across the BBB, nor the severity of evolution of the disease [75].
3.3. Beta-Amyloid (Aβ) Peptides and Cerebral Amyloid Angiopathy
AD-related BBB disruption is documented in both animal models [76, 77] and human brains [78]. Aβ peptide, one of AD major pathogenic operators, is considered a strong redox active agent capable of generating peroxide in the presence of metals [79]. Soluble Aβ species have been linked to decreased cytochrome C oxidase activity in the Tg2576 mouse model of AD and were shown to enter the mitochondria and cause a signalling amplification that inactivates SOD-2 and generates additional free radicals [80]. Aβ peptides are known to affect brain small blood vessels by inducement of cerebral amyloid angiopathy (CAA), found in 90% of AD patients and 50% of 90-year-old population [81]. Aβ-loaded capillaries, surrounded by NADPH oxidase-2 (NOX-2)-positive activated microglia are characterized by a dramatic loss of occludin, claudin-5, and ZO-1. Importantly, same brain sections showed abundant vascular expression of the Aβ transporter receptor for advanced glycation end-products (RAGE) [82], that was recently demonstrated to function as a signal transducing cell surface receptor for Aβ1-42, to induce ROS generation from NADPH oxidase [83]. Aβ1-40 perivascular deposition was reported to decrease expression of TJ proteins claudin-1 and claudin-5 and to increase expression of MMP-2 and MMP-9, in both AD brain microvessels and brains of AD transgenic mice [78]. In the neocortex and hippocampus of aged Tg2576 mice, the ratio of occludin to β-actin was reduced by nearly half, when compared to age-matched wild type controls, but also with young transgenic mice [84].
In vitro, in cellular barrier models, Aβ treatment increases endothelial permeability, effect documented for both Aβ1-40 [85] and Aβ1-42 [86], while tight junction protein expression is controversial (Table 1). In cultured endothelial cells, Aβ1-42 induced enhanced permeability by disruption of ZO-1 expression in the plasma membrane and increased intracellular calcium and matrix metalloproteinase (MMP) secretion. Neutralizing antibodies against RAGE and inhibitors of calcineurin and MMPs prevented Aβ 1-42-induced changes in ZO-1, suggesting that Aβ-RAGE interactions alter TJ proteins through the Ca2+-calcineurin pathway. Consistent with these in vitro findings, Kook et al. found disrupted microvessels near Aβ plaque-deposition areas, elevated RAGE expression, and enhanced MMP secretion in microvessels of AD mouse brains [86].
3.4. Excessive Alcohol Consumption
Excessive alcohol consumption is a known etiologic factor for cognitive impairment and dementia in humans and long-term treatment of adult laboratory rats with 20% ethanol in drinking water ad libitum resulted in cognitive decline, cholinergic dysfunction, and BBB leakage [87]. Ethanol (EtOH) effects are at least partially mediated by ROS, since, in these mice, superoxide production under basal conditions and in the presence of ADP and NAD(P)H, was increased [88]. In laboratory rats, EtOH consumption has previously been reported to associate increased oxidative stress and cytochrome P450-2E1 activation [89]. EtOH-induced activation of MMP-3/9 led to subsequent degradation of BBB proteins, occludin, claudin-5, and ZO-1 [90].
In vitro, EtOH induces ROS generation and ROS-nitrated protein accumulation in BMVEC [91]. At BBB level, EtOH or its metabolite acetaldehyde increases leakage and TJ protein phosphorylation [92]. Similar effects have been reported in other barriers, such as blood-air barrier [93], or other types of TJ-dependent polarized epithelia [94, 95].
4. Mediators of Oxidative Stress and Their Effects on Tight Junction Proteins
4.1. Reactive Oxygen Species
ROS are the main operators of oxidative stress and are responsible for altering protein structure, DNA denaturation, and lipid peroxidation and may act as messengers in redox-signalling systems [49]. In addition to causing cellular oxidative damage to biomolecules, hydroxyl radicals can also react with Aβ, triggering the formation of dityrosine cross-linking between Aβ peptides which leads to enhanced oligomerization and aggregation [96]. In oxidative-inducing conditions, a number of mechanisms have been proposed to trigger ROS generation, with enzymes such as xanthine oxidase, cyclooxygenase, leukocyte NADPH oxidase, and uncoupled endothelial NOS (eNOS) and mitochondria as putative sources [97]. Increased oxidative stress associated with aging further worsens the outcome of a stroke and favours onset of dementia. Common ROS that are deleterious for the vascular endothelium as well are superoxide, hydroxyl radical and hydrogen peroxide, found in concentrations depending of the balance between oxidases, such as NADPH oxidases (Nox enzymes) and superoxide dismutases (SOD). The impact of ROS on BBB function has been documented on SOD deficient mice, in which ischemia/reperfusion experiments demonstrated increased endothelium permeability to large molecules [98]. The hydrogen peroxide is more stable than superoxide, diffuses easily across cell membrane, can stimulate NADPH oxidase in vascular cells and thus further increase levels of superoxide [99].
In in vitro models, superoxide and other ROS increase permeability of the BBB in a time- and concentration-dependent manner [100, 101]. The reports on TJ proteins expression yielded contrasting results; however, BBB functionality was altered regardless of experimental paradigm. For instance, Lee et al. reported ROS-induced BBB impairment, quantified by transepithelial electrical resistance (TER) measurements, associated with a slight but significant increase in occludin expression [102], whereas Schreibelt et al. provided evidence that short-term oxidative stress-induced redistribution of occludin and claudin-5, with Western blot evidence of loss of these proteins expression [103].
4.2. Nitric Oxide
NO is a signalling molecule and a potent vasodilator, generated at the BBB level by eNOS, from L-arginine, a process that requires 5,6,7,8-tetrahydro-l-biopterin (BH4) as coenzyme. Apart from the constitutive isoform of NOS, endothelial cells also produce inducible NOS (iNOS), activated by interleukins and TNF-α [104]. Activation of iNOS is long-lasting and leads to an increased production of NO, as compared to constitutive isoform. Transgenic iNOS knockout mice develop brain pathology characteristic of AD (amyloid plaques, tau phosphorylation, and neuronal loss) indicating the NO has a protective role [105]. The peroxinitrite resulted from NO during oxidative stress has neurotoxic effects, similar to other ROS, via lipid peroxidation and DNA damage [106], and its presence is documented in astrocytes, neurons as well as blood vessels of AD brains, both in humans and mouse models of AD [80]. Generation of peroxynitrite from NO and superoxide takes place at a faster rate than the dismutation of superoxide by SOD enzymes and results in the loss of normal NO-mediated signalling. Thus, the local concentration of superoxide is a key determinant of the biological half-life of NO [98].
Interestingly, in certain conditions such as reduced levels of BH4, eNOS itself can produce superoxide, a process referred to as “eNOS uncoupling,” in which oxygen becomes terminal electron acceptor instead of L-arginine [107]. NO does not influence the function of BBB during normoxia, but seems to confer protection during ischemia [108].
4.3. Lipid Peroxidation Products
Lipid peroxidation usually designates the oxidative damage of polyunsaturated fatty acids by free radical chain reactions when exposed to O2 in the presence of trace metal ions. Studies of chain reactions in purified chemical systems show that a single initiation event can oxidatively damage 200 to 400 lipid molecules before two radicals react to eliminate the unpaired electrons and terminate the reaction sequence [109]. Lipid peroxidation causes damage at several levels by generation of various reactive aldehydes, such as 4-hydroxynonenal (4-HNE), that can alter the phospholipid asymmetry of the membrane lipid bilayer, and other products of lipid peroxidation, that can react with mitochondrial enzymes and cause disruption of mitochondrial energetics, increase of free radicals release and further oxidative stress [80]. Aβ peptides exert their oxidative effect on membrane lipids as well and there is a strong correlation between lipid peroxides, antioxidant enzymes, amyloid plaques, and neurofibrillary tangles (NFTs) in AD brains [3]. The composition of brain in phospholipids is unique; therefore, specific intermediates are produced upon lipid peroxidation [110]. These intermediates may diffuse into the blood stream and affect red blood cell membrane, as proven by Skoumalova et al. [111]. Oxidized low-density lipoprotein (ox-LDL), which is a hallmark feature of atherosclerosis acts as a stress signal and plays an influential role in BBB permeability [112]. The mechanisms by which lipid peroxidation affects BBB are not elucidated yet, but it has been proven that 4-HNE increases permeability of an in vitro barrier model [113]. In hyperlipemic laboratory mice, lipid peroxidation activates MMP-2/9, which in turn induces RhoA activation, a small GTPase known to phosphorylate TJ proteins and further destabilise BBB [114].
4.4. Matrix Metalloproteinases
Produced by activated microglia, MMPs are responsible for breaking down of endothelial basal lamina of BBB [57]. The main MMPs studied in relation to BBB alteration are MMP-2 and MMP-9, the first being constitutively expressed in CNS and the latter a marker of neuroinflammation [115]. Expression of MMP-9 within 24 h of an ischemic insult has cellular specificity, being primarily confined to the brain endothelium [116]. As already stated above, MMPs activity is balanced by their endogenous inhibitors, the TIMPs. Direct intracerebral injection of MMP-2 results in opening of the BBB with subsequent haemorrhage, effect that can be prevented by co-administration of TIMP-2 [117]. Blocking MMP-2 activation using either a selective inhibitor or a neutralizing antibody demonstrated that this enzyme is responsible for ischemia-induced occludin degradation. Interestingly, claudin-5 seems to be downregulated by different mechanisms, involving caveolin-1 [118]. On the other hand, Bauer et al. argued that hypoxia-induced oedema formation is mediated by MMP-9-dependent TJ rearrangement by a signalling cascade involving trophic factors, such as VEGF [119].
5. Signalling Pathways Affecting Tight Junction Proteins Phosphorylation Status in Oxidative Environments
Several reports of Saitou et al. showed in different experimental models that absence of occludin expression does not disrupt organization and function of TJs [120–122]. Therefore, TJ proteins emerged as possible signalling molecules. Indeed, there are several phosphorylation sites in the C-terminus sequence of occludin and claudins and phosphate addition in these domains increase protein internalization [65]. As a result, phosphorylation promotes an increase in BBB “leakiness.” These phosphorylation sites are found within consensus sequences for protein kinase C (PKC) and protein kinase A (PKA) [123] and it was further proven that some PKC isoforms are involved in occludin, claudins, and ZO species phosphorylation in normal and hypoxic conditions. Hypoxia-induced BBB changes involved increased paracellular permeability via a PKC activity-dependent mechanism, in both in vitro and in vivo conditions [124].
Cytoplasmic relocation of occludin, claudin-1, and ZO-1 were documented in Ras-transformed Madin-Darby canine kidney epithelial cells (MDCK), effect that was specifically reversed by mitogen-activated protein kinase 1 (MEK-1) inhibition [125]. As demonstrated by Wang et al., an occludin mutant lacking the first extracellular loop rescued cells from Raf-1-mediated transformation [126]. Furthermore, different small GTPases, such as Raf-1 [127], Rho, Rac [128], were shown to influence the expression of occludin and claudin-1 in different epithelial models. Moreover, addition of ROS in cell culture media of immortalized rat endothelial brain cells significantly induced transient PKB phosphorylation and subsequent activation, through RhoA activation [103]. Occludin undergoes phosphorylation at Tyr residues during the disruption of TJs by oxidative stress and acetaldehyde [129]. Occludin and claudin-1 protein expression seems to be influenced by Glycogen Synthase Kinase-3 β (GSK-3β) as well, inhibition of this kinase leading to decreased TJ protein levels [130].
6. Conclusions
Oxidative stress has been involved for a long time and by overwhelming scientific data as a main pathogenic event in brain ageing and neurodegeneration. BBB, as crucial gate of brain-blood molecular exchange, seems to be affected by oxidative stress inducers in early stages of different brain diseases. Further studies are needed to understand which is the relationship between ROS deleterious effects on endothelial cells, BBB impairment, and progress of neurodegeneration, and how specific BBB drug targets can be approached in the future.
Acknowledgments
This paper is supported by the Sectorial Operational Programme Human Resources Development (SOPHRD), financed from the European Social Fund and by the Romanian Government under the Contract no. POSDRU/89/1.5/S/64109 and by the Executive Unit for Financing Higher Education, Research, Development and Innovation, Romania (UEFISCDI) and by PN-II-ID-PCE-2012-4-0566/2013 National Research Council (CNCS) Grant, Romania.
References
- 1.Ceballos-Picot I, Merad-Boudia M, Nicole A, et al. Peripheral antioxidant enzyme activities and selenium in elderly subjects and in dementia of Alzheimer’s type—place of the extracellular glutathione peroxidase. Free Radical Biology and Medicine. 1996;20(4):579–587. doi: 10.1016/0891-5849(95)02058-6. [DOI] [PubMed] [Google Scholar]
- 2.De la Monte SM, Neely TR, Cannon J, Wands JR. Oxidative stress and hypoxia-like injury cause Alzheimer-type molecular abnormalities in central nervous system neurons. Cellular and Molecular Life Sciences. 2000;57(10):1471–1481. doi: 10.1007/PL00000630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adhesion and Migration. 2009;3(1):88–93. doi: 10.4161/cam.3.1.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends in Neurosciences. 2005;28(4):202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Popescu BO, Toescu EC, Popescu LM, et al. Blood-brain barrier alterations in ageing and dementia. Journal of the Neurological Sciences. 2009;283:99–106. doi: 10.1016/j.jns.2009.02.321. [DOI] [PubMed] [Google Scholar]
- 6.Bauer HC, Traweger A, Zweimueller-Mayer J, et al. New aspects of the molecular constituents of tissue barriers. Journal of Neural Transmission. 2011;118:7–21. doi: 10.1007/s00702-010-0484-6. [DOI] [PubMed] [Google Scholar]
- 7.Bednarczyk J, Lukasiuk K. Tight junctions in neurological diseases. Acta Neurobiologiae Experimentalis. 2011;71:393–408. doi: 10.55782/ane-2011-1861. [DOI] [PubMed] [Google Scholar]
- 8.Bell RD, Winkler EA, Sagare AP, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakagawa S, Deli MA, Kawaguchi H, et al. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochemistry International. 2009;54(3-4):253–263. doi: 10.1016/j.neuint.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. International Journal of Developmental Biology. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- 11.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of Disease. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Correale J, Villa A. Cellular elements of the blood-brain barrier. Neurochemical Research. 2009;34(12):2067–2077. doi: 10.1007/s11064-009-0081-y. [DOI] [PubMed] [Google Scholar]
- 13.Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325(6101):253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. [PubMed] [Google Scholar]
- 15.Zlokovic BV. Neurodegeneration and the neurovascular unit. Nature Medicine. 2010;16(12):1370–1371. doi: 10.1038/nm1210-1370. [DOI] [PubMed] [Google Scholar]
- 16.Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Research Reviews. 2003;42(3):221–242. doi: 10.1016/s0165-0173(03)00177-2. [DOI] [PubMed] [Google Scholar]
- 17.Weber CR. Dynamic properties of the tight junction barrier. Annals of the New York Academy of Sciences. 2012;1257:77–84. doi: 10.1111/j.1749-6632.2012.06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Current Neuropharmacology. 2008;6(3):179–192. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasdala I, Wolburg-Buchholz K, Wolburg H, et al. A transmembrane tight junction protein selectively expressed on endothelial cells and platelets. Journal of Biological Chemistry. 2002;277(18):16294–16303. doi: 10.1074/jbc.M111999200. [DOI] [PubMed] [Google Scholar]
- 20.Colgan OC, Collins NT, Ferguson G, et al. Influence of basolateral condition on the regulation of brain microvascular endothelial tight junction properties and barrier function. Brain Research. 2008;1193:84–92. doi: 10.1016/j.brainres.2007.11.072. [DOI] [PubMed] [Google Scholar]
- 21.Brown RC, Mark KS, Egleton RD, Huber JD, Burroughs AR, Davis TP. Protection against hypoxia-induced increase in blood-brain barrier permeability: role of tight junction proteins and NFκB. Journal of Cell Science. 2003;116(4):693–700. doi: 10.1242/jcs.00264. [DOI] [PubMed] [Google Scholar]
- 22.El Assar M, Angulo J, Vallejo S, Peiro C, Sanchez-Ferrer CF, Rodriguez-Manas L. Mechanisms involved in the aging-induced vascular dysfunction. Frontiers in Physiology. 2012;3, article 132 doi: 10.3389/fphys.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayhan WG, Faraci FM, Baumbach GL, Heistad DD. Effects of aging on responses of cerebral arterioles. American Journal of Physiology. 1990;258(4):H1138–H1143. doi: 10.1152/ajpheart.1990.258.4.H1138. [DOI] [PubMed] [Google Scholar]
- 24.Matz RL, De Sotomayor MA, Schott C, Stoclet JC, Andriantsitohaina R. Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. British Journal of Pharmacology. 2000;131(2):303–311. doi: 10.1038/sj.bjp.0703568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez-Mañas L, El-Assar M, Vallejo S, et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell. 2009;8(3):226–238. doi: 10.1111/j.1474-9726.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 26.Mayhan WG, Arrick DM, Sharpe GM, Sun H. Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation. 2008;15(3):225–236. doi: 10.1080/10739680701641421. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabó C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. British Journal of Pharmacology. 2002;135:1347–1350. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circulation Research. 2007;100(11):1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- 29.Lee P, Kim J, Williams R, et al. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Experimental Neurology. 2012;234(1):50–61. doi: 10.1016/j.expneurol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mooradian AD, Haas MJ, Chehade JM. Age-related changes in rat cerebral occludin and zonula occludens-1 (ZO-1) Mechanisms of Ageing and Development. 2003;124(2):143–146. doi: 10.1016/s0047-6374(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 31.Sandoval KE, Witt KA. Age and 17β-estradiol effects on blood-brain barrier tight junction and estrogen receptor proteins in ovariectomized rats. Microvascular Research. 2011;81(2):198–205. doi: 10.1016/j.mvr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Blau CW, Cowley TR, O’Sullivan J, et al. The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiology of Aging. 2012;33(5):1005.e23–1005.e35. doi: 10.1016/j.neurobiolaging.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 33.Timaru-Kast R, Luh C, Gotthardt P, et al. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0043829.e43829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathologica. 2010;119(3):277–290. doi: 10.1007/s00401-010-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawkins BT, Egleton RD. Pathophysiology of the blood-brain barrier: animal models and methods. Current Topics in Developmental Biology. 2007;80:277–309. doi: 10.1016/S0070-2153(07)80007-X. [DOI] [PubMed] [Google Scholar]
- 36.Bake S, Friedman JA, Sohrabji F. Reproductive age-related changes in the blood brain barrier: expression of IgG and tight junction proteins. Microvascular Research. 2009;78(3):413–424. doi: 10.1016/j.mvr.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueno M, Sakamoto H, Kanenishi K, Onodera M, Akiguchi I, Hosokawa M. Ultrastructural and permeability features of microvessels in the hippocampus, cerebellum and pons of senescence-accelerated mice (SAM) Neurobiology of Aging. 2001;22(3):469–478. doi: 10.1016/s0197-4580(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 38.Simpson JE, Wharton SB, Cooper J, et al. Alterations of the blood-brain barrier in cerebral white matter lesions in the ageing brain. Neuroscience Letters. 2010;486:246–251. doi: 10.1016/j.neulet.2010.09.063. [DOI] [PubMed] [Google Scholar]
- 39.Viggars AP, Wharton SB, Simpson JE, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neuroscience Letters. 2011;505(1):25–30. doi: 10.1016/j.neulet.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 40.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. American Journal of Physiology. 2002;282(2):C227–C241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 41.Krizanac-Bengez L, Hossain M, Fazio V, Mayberg M, Janigro D. Loss of flow induces leukocyte-mediated MMP/TIMP imbalance in dynamic in vitro blood-brain barrier model: role of pro-inflammatory cytokines. American Journal of Physiology. 2006;291(4):C740–C749. doi: 10.1152/ajpcell.00516.2005. [DOI] [PubMed] [Google Scholar]
- 42.Chen W, Hartman R, Ayer R, et al. Matrix metalloproteinases inhibition provides neuroprotection against hypoxia-ischemia in the developing brain. Journal of Neurochemistry. 2009;111(3):726–736. doi: 10.1111/j.1471-4159.2009.06362.x. [DOI] [PubMed] [Google Scholar]
- 43.Fischer S, Clauss M, Wiesnet M, Renz D, Schafer W, Karliczek GF. Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. American Journal of Physiology. 1999;276(4):C812–C820. doi: 10.1152/ajpcell.1999.276.4.C812. [DOI] [PubMed] [Google Scholar]
- 44.Witt KA, Mark KS, Sandoval KE, Davis TP. Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvascular Research. 2008;75(1):91–96. doi: 10.1016/j.mvr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H, Zhang Q, Xue Y, Chen X, Haun RS. Effects of hyperbaric oxygen on the expression of claudins after cerebral ischemia-reperfusion in rats. Experimental Brain Research. 2011;212(1):109–117. doi: 10.1007/s00221-011-2702-3. [DOI] [PubMed] [Google Scholar]
- 46.Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. Journal of Molecular Neuroscience. 2011;44(2):130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 47.Pillai DR, Dittmar MS, Baldaranov D, et al. Cerebral ischemia-reperfusion injury in rats—a 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. Journal of Cerebral Blood Flow and Metabolism. 2009;29(11):1846–1855. doi: 10.1038/jcbfm.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neumann-Haefelin T, Kastrup A, De Crespigny A, et al. Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 2000;31(8):1965–1973. doi: 10.1161/01.str.31.8.1965. [DOI] [PubMed] [Google Scholar]
- 49.Lochhead JJ, McCaffrey G, Quigley CE, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. Journal of Cerebral Blood Flow and Metabolism. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCaffrey G, Willis CL, Staatz WD, et al. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. Journal of Neurochemistry. 2009;110(1):58–71. doi: 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. American Journal of Physiology. 2003;285(6):H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 52.Amor S, Puentes F, Baker D, Van Der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vila N, Castillo J, Dávalos A, Chamorro A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31(10):2325–2329. doi: 10.1161/01.str.31.10.2325. [DOI] [PubMed] [Google Scholar]
- 54.Welsh P, Lowe GDO, Chalmers J, et al. Associations of proinflammatory cytokines with the risk of recurrent stroke. Stroke. 2008;39(8):2226–2230. doi: 10.1161/STROKEAHA.107.504498. [DOI] [PubMed] [Google Scholar]
- 55.Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Current Pharmaceutical Design. 2008;14:3574–3589. doi: 10.2174/138161208786848739. [DOI] [PubMed] [Google Scholar]
- 56.Ravaglia G, Forti P, Maioli F, et al. Blood inflammatory markers and risk of dementia. The Conselice Study of Brain Aging. Neurobiology of Aging. 2007;28(12):1810–1820. doi: 10.1016/j.neurobiolaging.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 57.Di Napoli M, Shah IM. Neuroinflammation and cerebrovascular disease in old age: a translational medicine perspective. Journal of Aging Research. 2011;2011:18 pages. doi: 10.4061/2011/857484.857484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nature Medicine. 2007;13:1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 59.Ravetti MG, Moscato P. Identification of a 5-protein biomarker molecular signature for predicting Alzheimer’s disease. PLoS ONE. 2008;3(9) doi: 10.1371/journal.pone.0003111.e3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tobinick EL, Gross H. Rapid improvement in verbal fluency and aphasia following perispinal etanercept in Alzheimer’s disease. BMC Neurology. 2008;8, article 27 doi: 10.1186/1471-2377-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tobinick E. Perispinal etanercept produces rapid improvement in primary progressive aphasia: identification of a novel, rapidly reversible TNF-mediated pathophysiologic mechanism. Medscape General Medicine. 2008;10(6, article 135) [PMC free article] [PubMed] [Google Scholar]
- 62.Patrick DM, Leone AK, Shellenberger JJ, Dudowicz KA, King JM. Proinflammatory cytokines tumor necrosis factor-α and interferon-γ modulate epithelial barrier function in Madin-Darby canine kidney cells through mitogen activated protein kinase signaling. BMC Physiology. 2006;6, article 2 doi: 10.1186/1472-6793-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amasheh M, Grotjohann I, Amasheh S, et al. Regulation of mucosal structure and barrier function in rat colon exposed to tumor necrosis factor alpha and interferon gamma in vitro: a novel model for studying the pathomechanisms of inflammatory bowel disease cytokines. Scandinavian Journal of Gastroenterology. 2009;44(10):1226–1235. doi: 10.1080/00365520903131973. [DOI] [PubMed] [Google Scholar]
- 64.Ewert P, Aguilera S, Alliende C, et al. Disruption of tight junction structure in salivary glands from Sjogren's syndrome patients is linked to proinflammatory cytokine exposure. Arthritis and Rheumatism. 2010;62(5):1280–1289. doi: 10.1002/art.27362. [DOI] [PubMed] [Google Scholar]
- 65.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochimica et Biophysica Acta. 2009;1788(4):864–871. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scharl M, Paul G, Barrett KE, McCole DF. AMP-activated protein kinase mediates the interferon-γ-induced decrease in intestinal epithelial barrier function. Journal of Biological Chemistry. 2009;284(41):27952–27963. doi: 10.1074/jbc.M109.046292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. Journal of Cerebral Blood Flow and Metabolism. 2006;26(6):797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 68.Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. American Journal of Physiology. 2001;280(3):H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- 69.Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. American Journal of Physiology. 2005;289(2):H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. Journal of Experimental Medicine. 2000;192(7):1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812:220–230. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krumbholz M, Theil D, Steinmeyer F, et al. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. Journal of Neuroimmunology. 2007;190(1-2):72–79. doi: 10.1016/j.jneuroim.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 73.Errede M, Girolamo F, Ferrara G, et al. Blood-brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. Journal of Neuropathology and Experimental Neurology. 2012;71:840–854. doi: 10.1097/NEN.0b013e31826ac110. [DOI] [PubMed] [Google Scholar]
- 74.Wolburg H, Wolburg-Buchholz K, Kraus J, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathologica. 2003;105(6):586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 75.Pfeiffer F, Schäfer J, Lyck R, et al. Claudin-1 induced sealing of blood-brain barrier tight junctions ameliorates chronic experimental autoimmune encephalomyelitis. Acta Neuropathologica. 2011;122(5):601–614. doi: 10.1007/s00401-011-0883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathologica. 2011;122(3):293–311. doi: 10.1007/s00401-011-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hawkes CA, Härtig W, Kacza J, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathologica. 2011;121(4):431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 78.Hartz AMS, Bauer B, Soldner ELB, et al. Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012;43(2):514–523. doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marlatt MW, Lucassen PJ, Perry G, Smith MA, Zhu X. Alzheimer’s disease: cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. Journal of Alzheimer’s Disease. 2008;15(2):199–210. doi: 10.3233/jad-2008-15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Massaad CA. Neuronal and vascular oxidative stress in Alzheimer’s disease. Current Neuropharmacology. 2011;9(4):662–673. doi: 10.2174/157015911798376244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18(2):311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 82.Carrano A, Hoozemans JJM, Van Der Vies SM, Rozemuller AJM, Van Horssen J, De Vries HE. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxidants and Redox Signaling. 2011;15(5):1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 83.Askarova S, Yang X, Sheng W, Sun GY, Lee JC. Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A2 activation in astrocytes and cerebral endothelial cells. Neuroscience. 2011;199:375–385. doi: 10.1016/j.neuroscience.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in alzheimer’s disease. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0023789.e23789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. Journal of Cellular and Molecular Medicine. 2010;14(5):1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kook SY, Hong HS, Moon M, Ha CM, Chang S. Aβ 1−42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. Journal of Neuroscience. 2012;32:8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ehrlich D, Pirchl M, Humpel C. Effects of long-term moderate ethanol and cholesterol on cognition, cholinergic neurons, inflammation, and vascular impairment in rats. Neuroscience. 2012;205:154–166. doi: 10.1016/j.neuroscience.2011.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sun H, Zheng H, Molacek E, Fang Q, Patel KP, Mayhan WG. Role of NAD(P)H oxidase in alcohol-induced impairment of endothelial nitric oxide synthase-dependent dilation of cerebral arterioles. Stroke. 2006;37(2):495–500. doi: 10.1161/01.STR.0000199033.06678.c3. [DOI] [PubMed] [Google Scholar]
- 89.Sun AY, Sun GY. Ethanol and oxidative mechanisms in the brain. Journal of Biomedical Science. 2001;8(1):37–43. doi: 10.1007/BF02255969. [DOI] [PubMed] [Google Scholar]
- 90.Muneer PMA, Alikunju S, Szlachetka AM, Haorah J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(5):1167–1177. doi: 10.1161/ATVBAHA.112.247668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. Journal of Leukocyte Biology. 2005;78(6):1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- 92.Haorah J, Heilman D, Knipe B, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcoholism. 2005;29(6):999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Y, Li Q, Guo W, Huang Y, Yang J. Effects of chronic ethanol ingestion on tight junction proteins and barrier function of alveolar epithelium in the rat. Shock. 2007;28(2):245–252. doi: 10.1097/SHK.0b013e31803404a9. [DOI] [PubMed] [Google Scholar]
- 94.Rotoli BM, Orlandini G, Guizzardi S, et al. Ethanol increases the paracellular permeability of monolayers of CAPAN-1 pancreatic duct cells. Journal of Molecular Histology. 2004;35(4):355–362. doi: 10.1023/b:hijo.0000039838.56131.02. [DOI] [PubMed] [Google Scholar]
- 95.Elamin E, Jonkers D, Juuti-Uusitalo K, et al. Effects of ethanol and acetaldehyde on tight junction integrity: in vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0035008.e35008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Atwood CS, Perry G, Zeng H, et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-β . Biochemistry. 2004;43(2):560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 97.Chen C, Lin C, Druhan LJ, Wang T, Chen Y, Zweier JL. Superoxide induces endothelial nitric-oxide synthase protein thiyl radical formation, a novel mechanism regulating eNOS function and coupling. Journal of Biological Chemistry. 2011;286(33):29098–29107. doi: 10.1074/jbc.M111.240127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends in Molecular Medicine. 2008;14(11):495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Faraci FM. Hydrogen peroxide: watery fuel for change in vascular biology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(9):1931–1933. doi: 10.1161/01.ATV.0000238355.56172.b3. [DOI] [PubMed] [Google Scholar]
- 100.Schreibelt G, Musters RJP, Reijerkerk A, et al. Lipoic acid affects cellular migration into the central nervous system and stabilizes blood-brain barrier integrity. Journal of Immunology. 2006;177(4):2630–2637. doi: 10.4049/jimmunol.177.4.2630. [DOI] [PubMed] [Google Scholar]
- 101.Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. Journal of Neurochemistry. 2007;101(2):566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- 102.Lee H, Namkoong K, Kim D, et al. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvascular Research. 2004;68(3):231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 103.Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB Journal. 2007;21(13):3666–3676. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 104.Angeles Muñoz-Fernández M, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-γ and inducible nitric oxide synthase in the development and pathology of the nervous system. Progress in Neurobiology. 1998;56(3):307–340. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 105.Wilcock DM, Lewis MR, Van Nostrand WE, et al. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. Journal of Neuroscience. 2008;28(7):1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eliasson MJL, Huang Z, Ferrante RJ, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. Journal of Neuroscience. 1999;19(14):5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bevers LM, Braam B, Post JA, et al. Tetrahydrobiopterin, but not L-arginine, decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension. 2006;47(1):87–94. doi: 10.1161/01.HYP.0000196735.85398.0e. [DOI] [PubMed] [Google Scholar]
- 108.Utepbergenov DI, Mertsch K, Sporbert A, et al. Nitric oxide protects blood-brain barrier in vitro from hypoxia/reoxygenation-mediated injury. FEBS Letters. 1998;424(3):197–201. doi: 10.1016/s0014-5793(98)00173-2. [DOI] [PubMed] [Google Scholar]
- 109.Jones DP. Radical-free biology of oxidative stress. American Journal of Physiology. 2008;295(4):C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Skoumalova A, Hort J. Blood markers of oxidative stress in Alzheimer's disease. Journal of Cellular and Molecular Medicine. 2012;16:2291–2300. doi: 10.1111/j.1582-4934.2012.01585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Skoumalova A, Madlova P, Topinkova E. End products of lipid peroxidation in erythrocyte membranes in Alzheimer's disease. Cell Biochemistry and Function. 2012;30:205–210. doi: 10.1002/cbf.1836. [DOI] [PubMed] [Google Scholar]
- 112.Wang J, Sun L, Si YF, Li BM. Overexpression of actin-depolymerizing factor blocks oxidized low-density lipoprotein-induced mouse brain microvascular endothelial cell barrier dysfunction. Molecular and Cellular Biochemistry. 2012;371:1–8. doi: 10.1007/s11010-012-1415-7. [DOI] [PubMed] [Google Scholar]
- 113.Mertsch K, Blasig I, Grune T. 4-Hydroxynonenal impairs the permeability of an in vitro rat blood-brain barrier. Neuroscience Letters. 2001;314(3):135–138. doi: 10.1016/s0304-3940(01)02299-6. [DOI] [PubMed] [Google Scholar]
- 114.Elali A, Doeppner TR, Zechariah A, Hermann DM. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and rhoa overactivation. Stroke. 2011;42(11):3238–3244. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- 115.Montaner J, Alvarez-Sabín J, Molina C, et al. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke. 2001;32(8):1759–1766. doi: 10.1161/01.str.32.8.1759. [DOI] [PubMed] [Google Scholar]
- 116.Asahi M, Wang X, Mori T, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. Journal of Neuroscience. 2001;21(19):7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rosenberg GA, Kornfeld M, Estrada E, Kelley RO, Liotta LA, Stetler-Stevenson WG. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Research. 1992;576(2):203–207. doi: 10.1016/0006-8993(92)90681-x. [DOI] [PubMed] [Google Scholar]
- 118.Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. Journal of Neuroscience. 2012;32(9):3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bauer AT, Bürgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. Journal of Cerebral Blood Flow and Metabolism. 2010;30(4):837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saitou M, Furuse M, Sasaki H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Molecular Biology of the Cell. 2000;11(12):4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saitou M, Fujimoto K, Doi Y, et al. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. Journal of Cell Biology. 1998;141(2):397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schulzke JD, Gitter AH, Mankertz J, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochimica et Biophysica Acta. 2005;1669(1):34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 123.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochimica et Biophysica Acta. 2008;1778(3):631–645. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 124.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. American Journal of Physiology. 2005;289(5):H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 125.Chen Y, Lu Q, Schneeberger EE, Goodenough DA. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in Ras-transformed Madin-Darby canine kidney cells. Molecular Biology of the Cell. 2000;11(3):849–862. doi: 10.1091/mbc.11.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Z, Mandell KJ, Parkos CA, Mrsny RJ, Nusrat A. The second loop of occludin is required for suppression of Raf1-induced tumor growth. Oncogene. 2005;24(27):4412–4420. doi: 10.1038/sj.onc.1208634. [DOI] [PubMed] [Google Scholar]
- 127.Li D, Mrsny RJ. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. Journal of Cell Biology. 2000;148(4):791–800. doi: 10.1083/jcb.148.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. Journal of Cell Science. 2003;116(4):725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 129.Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochemical Journal. 2006;393(1):69–77. doi: 10.1042/BJ20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Severson EA, Kwon M, Hilgarth RS, Parkos CA, Nusrat A. Glycogen Synthase Kinase 3 (GSK-3) influences epithelial barrier function by regulating Occludin, Claudin-1 and E-cadherin expression. Biochemical and Biophysical Research Communications. 2010;397(3):592–597. doi: 10.1016/j.bbrc.2010.05.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. American Journal of Physiology. 2002;282(4):H1485–H1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Feng S, Cen J, Huang Y, et al. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0020599.e20599 [DOI] [PMC free article] [PubMed] [Google Scholar]