

Abstract
The conversion of biomass to CH4 (biomethanation) involves an anaerobic microbial food chain composed of at least three metabolic groups of which the first two decompose the complex biomass primarily to acetate, formate, and H2. The thermodynamics of these conversions are unfavorable requiring a symbiosis with the CH4-producing group (methanogens) that metabolize the decomposition products to favorable concentrations. The methanogens produce CH4 by two major pathways, conversion of the methyl group of acetate and reduction of CO2 coupled to the oxidation of formate or H2. This review covers recent advances in the fundamental understanding of both methanogenic pathways with the view of stimulating research towards improving the rate and reliability of the overall biomethanation process.
Introduction
Earth’s biosphere contains diverse oxygen-free (anaerobic) environments where complex organic matter is decomposed to CH4 in a process called biomethanation that is an essential link in the global carbon cycle (Figure 1). In the cycle, atmospheric CO2 is fixed into plant biomass primarily via oxygenic photosynthesis (step 1). Microbes in aerobic environments digest the biomass consuming O2 and returning CO2 to the atmosphere (step 2). A portion of the biomass also enters diverse O2-free environments (step 3) such as the lower intestinal tract of humans, wetlands, rice paddy soils, and the rumen of livestock where anaerobic microbes digest the organic matter producing CO2 and CH4 (steps 4–6) in a ratio of approximately 1:1 (biogas). In freshwater environments, biomethanation involves a minimum of three metabolic groups from the domains Bacteria and Archaea. The fermentative group decomposes biomass primarily to butyrate, propionate, acetate, formate, and H2 plus CO2 (step 4). The obligate proton-reducing acetogenic group (acetogens) converts the butyrate and propionate to acetate, CO2, and H2 or formate (step 5). The thermodynamics of these conversions are unfavorable under standard conditions of equimolar reactants and products (Table 1) requiring a symbiosis with the methane-producing group (methanogens) that metabolizes the products to thermodynamically favorable levels (step 6). The methanogens produce CH4 by two major pathways (Table 1). In the CO2-reduction pathway, formate or H2 is oxidized and CO2 is reduced to CH4. In the aceticlastic pathway, acetate is cleaved with the carbonyl group oxidized to CO2 and the methyl group reduced to CH4. In most freshwater environments, the aceticlastic group is responsible for approximately two-thirds with most of the remaining one-third produced by CO2-reducing methanogens. Smaller, yet significant amounts of CH4, are produced from the methyl groups of methanol, methylamines, and dimethylsulfide. Some of the CH4 is oxidized to CO2 (step 7) by a consortia of anaerobes that reduce either sulfate, nitrate, manganese, or iron [1]. The CH4 also diffuses into aerobic environments (step 8) where O2-requiring methanotrophic microbes oxidize it to CO2 (step 9).
Figure 1.
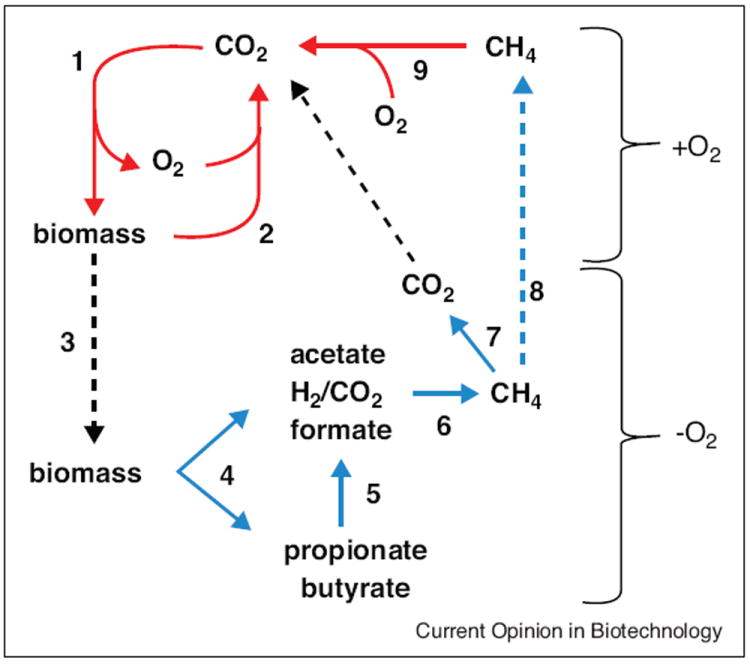
The global carbon cycle. Aerobic O2-requiring conversions are shown in solid red arrows and anaerobic conversions in solid blue arrows. The brackets denote aerobic (+O2) and anaerobic (−O2) habitats. Black dotted arrows symbolize diffusion of substrates and products across the interface of zones.
Table 1.
Reactions involved in the syntrophic metabolism of obligate proton-reducing acetogens and methanogens.
| Reactions | ΔG°′ (kJ/mol) |
|---|---|
| Propionate− + 3H2O → acetate− + HCO3− + H+ + 3H2 | +76.1 |
| Butyrate− + 2H2O → 2acetate− + H+ + 2H2 | +48.6 |
| 4H2 + HCO3− + H+ → CH4 + 3H2O | −135.6 |
| 4Formate− + H+ + H2O → 3HCO3− + CH4 | −130.4 |
| Acetate− → HCO3− + H+ + CH4 | −36.0 |
The biomethanation of organic matter is exploited for the disposal of organic waste and production of biogas from renewable plant material as an alternative to fossil fuels. Domestic and industrial wastes are disposed of in large-scale municipal sewage treatment plants, reducing the volume of volatile solids as much as 75% and producing electricity with biogas-powered generators that serve the facility and contribute to the power grid. In rural areas of developing countries, biomethanation is utilized for the small-scale disposal of domestic waste and use of the biogas for home heating and cooking. Large-scale production of biogas from sustainable plant biomass has the potential for contributing to the replacement of fossil fuels. On a weight for weight basis, the energy content of CH4 is approximately 3-fold greater than H2, and CH4 is stored and transported in a more efficient and safer manner than H2. Through catalytic reformation, CH4 is converted into methanol as a chemical feed stock that further reduces the dependence on petroleum. This review of the recent literature introduces the physiology of CH4-producing microbes, key players in the production of biogas from organic matter, providing the reader with a background suitable for stimulating research aimed towards improving the process that will ensure biogas among the competitive alternatives to fossil fuels.
Reactions common to aceticlastic and CO2-reducing pathways
Figure 2 shows a composite of CO2-reducing and aceticlastic pathways, and the overall reactions are shown in Table 1. Both pathways have in common reactions 10–12 (Figure 2) but differ in reactions generating either methyl-tetrahydrosarcinapterin (CH3-H4SPT) in the aceticlastic pathway (reactions 1–4) or methyl-tetrahydromethanopterin (CH3-H4MPT) in the CO2-reduction pathway (reactions 5–9). The H4SPT and H4MPT cofactors are functionally equivalent analogs of tetrahydrofolate. In reaction 10, the methyl group of CH3-H4M(S)PT is transferred to HS-CoM followed by reductive demethylation of CH3-S-CoM to CH4 with electrons donated by HS-CoB (reaction 11). Reaction 10 is catalyzed by CH3-H4M(S)PT:coenzyme M (HS-CoM) methyltransferase (Mtr), a membrane-bound eight-subunit complex that couples the exergonic methyl transfer to the generation of a sodium ion gradient (high outside).
Figure 2.
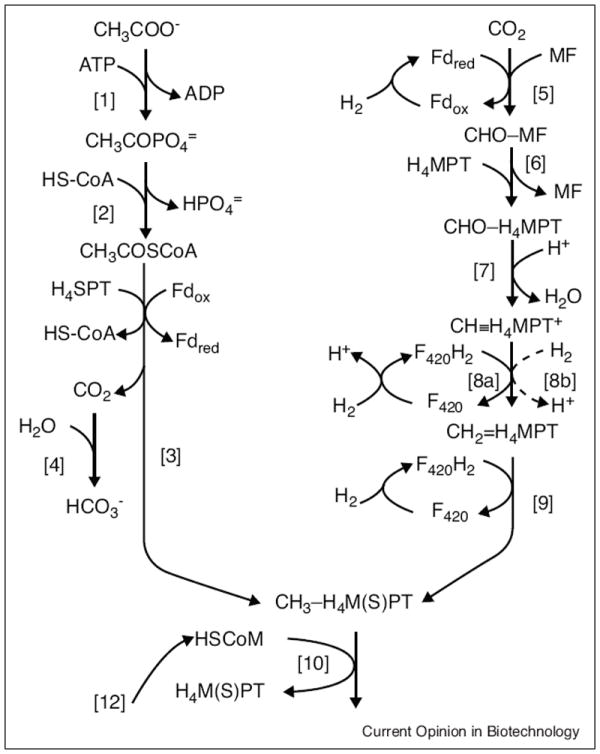
Composite of CO2-reduction and aceticlastic methane-producing pathways. The left arm leading to CH3-H4M(S)PT shows reactions (1–4) unique to the aceticlastic pathway, and the right arm leading to CH3-H4M(S)PT shows reactions (5–9) unique to the CO2-reduction pathway. Both pathways have in common reactions (10–12) leading to the formation of CH4 from the methyl groups of CH3-H4M(S)PT.
Abbreviations: ATP, adenosine triphosphate; H4SPT, tetrahydrosarcinapterin; H4MPT, tetrahydromethanopterin, Fd, ferredoxin; CoA, coenzyme A; CoM, coenzyme M; CoB, coenzyme B; MF, methanofuran; F420, coenzyme F420.
Methyl-coenzyme M reductase (Mcr) catalyzes reaction 11 wherein the methyl group of CH3-S-CoM is reduced to CH4 with HS-CoB the electron donor and the heterodisulfide CoM-S-S-CoB is an added product. The enzyme is also proposed to activate CH4 for anaerobic oxidation to CO2 [2]. The crystal structure of Mcr from Methanothermobacter marburgensis (f Methanobacterium thermoautotrophicum strain Marburg) shows two active sites, each with a Ni-containing cofactor called F430 [3]. In dispute are two proposed mechanisms, investigated of late, that diverge in how the C–S bond of CH3-S-CoM is cleaved. Mechanism A (also referred to as mechanism II) predicts the nickel of NiIF430 attacking the sulfur atom of CH3-S-CoM producing a •CH3 radical and CoM-S-NiIIF430 [4]. The methyl radical then abstracts a hydrogen from HS-CoB producing •S-CoB and CH4. The •S-CoB radical reacts with CoM-S-NiIIF430 producing the anion radical CoM-S-S•-CoB− that donates the extra electron to NiIIF430 regenerating the active NiIF430 species and yielding CoM-S-S-CoB. Mechanism B (also called mechanism I) proposes a nucleophilic attack of NiI on the methyl of CH3-S-CoM generating CH3-NiIIIF430 and −S-CoM. Transfer of an electron from −S-CoM to CH3-NiIIIF430 produces the •S-CoM radical and CH3-NiIIF430, the protonolysis of which produces CH4. The NiIIF430 is reduced to the active NiIF430 form and CoM-S-S-CoB is produced the same as in mechanism A. Unfortunately a crystal structure of the reduced active enzyme reacted with the natural substrate CH3-S-CoM has not been obtained and the proposed intermediates have not been trapped. Nonetheless, deuterium exchange studies are the basis for a proposal considering both σ-alkane-NiIIIF430 and alkane-NiIF430 complexes as intermediates [5]. X-ray absorption spectroscopy of Mcr reacted with methyl iodide, together with density functional theory, indicates a stable CH3-NiIIIF430 species consistent with mechanism B [6]. Stable synthetic CH3-NiIII complexes are reported [7] that contain Ni–C bond lengths equivalent to the lengths reported for the enzyme-containing CH3-NiIIIF430 species [6]. Further support for mechanism B was obtained by reacting Mcr with the natural substrate CH3-S-CoM and an analog of HS-CoB at reduced rates which allowed transient kinetics to detect an alkyl-NiIII intermediate and an unidentified organic radical that decays at a catalytically competent rate [8]. Finally, it has been suggested that binding of HS-CoB subsequent to binding of CH3-S-CoM induces a conformational change that moves CH3-S-CoM to within a distance of F430 that initiates the reaction. Recent crystal structures of inactive Mcr (NiIIF430) failed to show significant conformational changes when complexed with HS-CoB analogs [9]. On the other hand, crystallographic and spectroscopic approaches investigating Mcr(NiI) complexed with an HS-CoB analog show that binding of HS-CoB induces a major conformational change in the active site with implications for predicting Ni coordination geometries [10]. One approach that has not been employed effectively to resolve the catalytic mechanism is the use of site-directed mutagenesis of the over-expressed enzyme in conjunction with substrate analogs and crystallography to trap and identify reaction intermediates. However, over-expression of an active form of an iron-containing carbonic anhydrase (Cam) from Methanosarcina thermophila in Methanosarcina acetivorans provides precedent for over-expression of other metalloenzymes such as Mcr in an active form [11•].
The CoM-S-S-CoB produced by Mcr is reduced to HS-CoB and HS-CoM (reaction 12, Figure 2) catalyzed by heterodisulfide reductase (Hdr) for which there are two types. A two-subunit (HdrDE) enzyme functions in the aceticlastic pathway of Methanosarcina species [12], and a three-subunit (HdrABC) enzyme functions in obligate CO2-reducers [13••].
Electron transport and ATP synthesis in the aceticlastic and CO2-reducing pathways
All acetotrophic methanogens obtain energy for growth by coupling electron transfer from ferredoxin to CoM-S-S-CoB with generation of a proton gradient that drives ATP synthesis via the proton-translocating “archaeal” A1A0-type ATP synthase [14•,15]. In freshwater Methanosarcina species (Figure 3), ferredoxin donates electrons to a membrane-bound hydrogenase (Ech) that evolves H2 and generates a proton gradient driving ATP synthesis [16,17]. It is proposed that a membrane-bound F420-nonreducing hydrogenase (Vho) reoxidizes H2 and donates electrons to a quinone-like electron carrier methanophenazine that mediates electron transfer to the heterodisulfide reductase while translocating protons that contribute to the proton gradient. On the other hand, the acetotrophic marine isolate M. acetivorans does not encode an Ech hydrogenase, and proteomic analyses indicate that acetate-grown cells synthesize subunits with sequence identity to an Rnf complex first described in Rhodobacter capsulatus from the domain Bacteria [18]. Gene knockouts confirm that the complex is essential for growth on acetate [12]. Thus, it is anticipated that the Rnf complex replaces Ech as an acceptor of electrons from ferredoxin and donor to methanophenazine accompanied by translocation of ions contributing to the gradient driving ATP synthesis.
Figure 3.
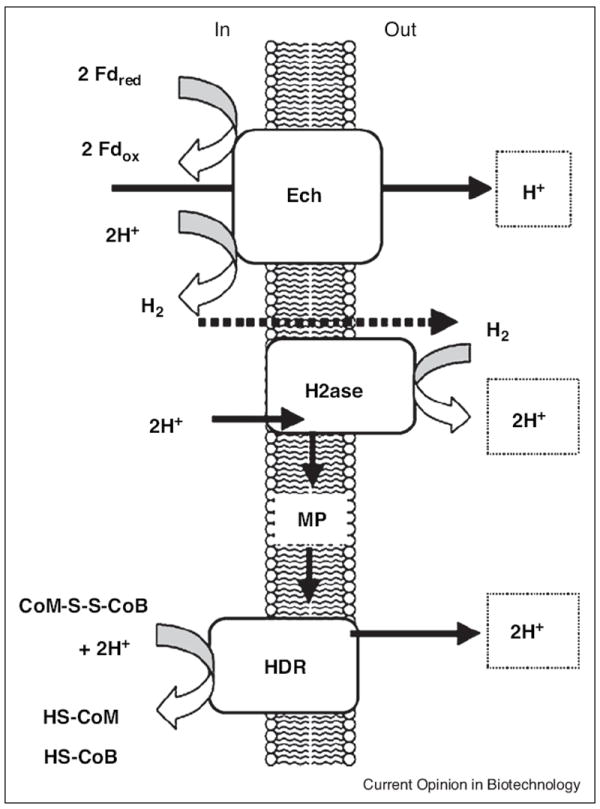
Proposed model of ferredoxin-dependent electron transport chain in the freshwater isolate Methanosarcina mazei. H2ase, F420-nonreducing hydrogenase (Vho); HDR, heterodisulfide reductase; MP, methanophenazine. By permission from Ref. [16].
Electron transfer from H2 to CoM-S-S-CoB (reaction 12, Figure 2) in the CO2-reducing pathways of freshwater Methanosarcina and obligate CO2-reducing species differs significantly. In freshwater Methanosarcina, the H2:CoM-S-S-CoB oxidoreductase system is identical to the proton pumping segment of electron transport in the aceticlastic pathway involving the F420-nonreducing hydrogenase, methanophenazine, and the HdrDE heterodisulfide reductase. However, the membrane-bound electron transport chain of Methanosarcina species is absent in obligate CO2-reducing species with no apparent mechanism for generating an ion gradient. Instead, the H2:CoM-S-S-CoB oxidoreductase system is composed of the cytoplasmic F420-nonreducing hydrogenase (MvhAGD) tightly bound to HdrABC with no experimentally determined mechanism for generating an ion gradient. One possibility for ATP synthesis is the sodium gradient generated by the membrane-bound methyl-H4MPT:coenzyme M methyltransferase complex (Figure 2, reaction 10) driving a sodium translocating ATP synthase [13••].
Electron transport coupled to ATP synthesis in the CO2-reduction pathway of the marine isolate M. acetivorans is distinct from H2-metabolizing Methanosarcina and obligate CO2-reducing methanogens [19•,20]. M. acetivorans does not metabolize H2, and the only electron donor for reduction of CO2 to CH4 is CO. Coenzyme F420 is the electron carrier that donates electrons to a membrane-bound electron transport chain terminating with the heterodisulfide reductase [19•]. Upregulation of the F420H2 dehydrogenase complex (Fpo) in response to growth with acetate suggests it is the entry point to the electron transport chain and that methanophenazine mediates electron transfer from the dehydrogenase to the heterodisulfide reductase generating the proton gradient that drives ATP synthesis.
Synthesis of methyl-H4SPT in the aceticlastic pathway
Methyl-H4SPT is synthesized by reactions 1–4 (Figure 2) in the aceticlastic pathway of Methanosarcina species. Unlike the CO2-reduction pathway, homologs of the enzymes catalyzing these reactions are wide-spread in diverse anaerobes from the domain Bacteria. Homologs of acetate kinase and phosphotransacetylase, catalyzing the reverse of reactions 1 and 2 (Figure 1), are the primary energy-conserving enzymes of the fermentative and obligate proton-reducing acetogenic groups (Figure 1) converting acetyl-CoA to ATP and acetate [21••]. Reaction 3 is central to the aceticlastic pathway catalyzed by the CO dehydrogenase/acetyl-CoA synthase (Cdh) complex that cleaves the C–C and C–S bonds of acetyl-CoA transferring the methyl group to H4SPT and oxidizing the carbonyl group to CO2 with transfer of electrons to ferredoxin. Acetogens from the domains Bacteria and Archaea contain Cdh homologs that function in acetate-producing energy-conserving pathways, and also pathways for generating acetyl-CoA from a methyl group and CO2 for cell biosynthesis [22]. The five-subunit CdhABCDE complex from Methanosarcina species is resolvable into three components. The CdhC component cleaves the C–S bond of acetyl-CoA with transfer of the acetyl group to the “A” cluster composed of a 4Fe–4S center bridged by a cysteine thiolate to a binuclear Ni–Ni site [23] similar to the homolog from Moorella thermoacetica, an acetate-producing species from the domain Bacteria [24]. Cleavage of the acetyl group yields enzyme-bound CO derived from the carbonyl group and a methyl group that is transferred to the corrinoid cofactor of the CdhDE component that in turn donates the methyl group to H4SPT. The CdhAE component accepts CO from CdhC oxidizing it to CO2 and reducing ferredoxin. The crystal structure of CdhAE from Methanosarcina barkeri reveals 4Fe–4S clusters proposed to transfer electrons to ferredoxin from the active site “C” center, a pseudocubane NiFe3S4 cluster bridged to an exogeneous iron atom [25•]. A gas channel in the crystal structure extends from the “C” cluster to the protein surface with potential to interact with the CdhC component. A mechanism is proposed in which transfer of an electron from the “C” cluster of CdhAE to cluster “A” of CdhC maintains the reduced catalytically active Ni(I) redox state [26].
The conversion of acetate to CH4 and CO2 provides only a marginal amount of energy available for growth (ΔG°′ = −36 kJ/CH4). Thus, it is postulated that a carbonic anhydrase (Cam) is located outside the cell membrane where it converts CO2 to membrane-impermeable HCO3− (Figure 2, reaction 4) facilitating removal of CO2 from the cytoplasm that optimizes the thermodynamic efficiency of growth. Cam from M. thermophila is the archetype of the γ class of carbonic anhydrases for which homologs are widely distributed in the domains Bacteria and Archaea [27]. Contrary to all prokaryotic carbonic anhydrases that contain zinc, Cam, and the Cam homolog CamH contain Fe2+ in the active site [11•,28].
Although of lesser importance for the biomethanation of renewable plant material to biogas, Methanosarcina species also utilize the methylotrophic substrates methanol, trimethylamines, and methylsulfide for growth and methanogenesis. Compared to the aceticlastic pathway, more energy is available via the methylotrophic pathway for which the encoding genes are upregulated when cells are exposed to both acetate and methylotrophic substrates. A significant understanding of global regulation and genes specific to methylotrophic pathways has been advanced recently for Methanosarcina species [29,30]. However, with one exception [31], nothing is known of the regulatory proteins and mechanisms specific for genes of the aceticlastic pathway for which research is urgently needed.
Synthesis of methyl-H4MPT in the CO2-reduction pathway
The CO2-reducing methanogens can be further divided according to the diversity of substrates used for growth and CH4 production. Obligate CO2-reducing species only reduce CO2 to CH4 with either H2 or formate whereas CO2-reducing Methanosarcina species of the genus also grow and produce CH4 by converting the methyl groups of acetate, methanol, methylamines, and dimethylsulfide to CH4. Both types reduce CO2 via reactions 5–9 (Figure 2) to a methyl group bound either to tetrahydromethanopterin (H4MPT) in obligate CO2 reducers or to tetrahydrosarcinapterin (H4SPT) in Methanosarcina species. The three electron pairs required for reactions 5, 8, and 9 originate from oxidation of H2, CO, or formate and reduction of ferredoxin or coenzyme F420 serving as electron carriers. In freshwater Methanosarcina species CO is first oxidized to H2 and CO2 whereas in the marine isolate M. acetivorans the oxidation of CO is coupled to reduction of ferredoxin and F420 avoiding H2 as an intermediate [19•]. The utilization of formate is limited to obligate CO2-reducing species. Although formic hydrogenlyase systems have been described that convert formate to H2 and CO2, the role of H2 as an intermediate during growth with formate is still in question [32,33].
Reaction 5 (Figure 2) catalyzed by formyl-methanofuran (MF) dehydrogenase is endergonic in the environment where partial pressures of H2 are held in the range of 1–10 Pa and therefore requires energy [13••]. The Ech hydrogenase of freshwater Methanosarcina species reduces ferredoxin driven by a proton gradient (high outside) generated by the membrane-bound electron transport chain originating with oxidation of H2 and ending with reduction of CoM-S-S-CoB (reaction 12, Figure 2) [14•]. Less understood is the mechanism that drives reaction 5 in obligate CO2-reducers that do not synthesize a membrane-bound electron transport chain. It is hypothesized that the exergonic H2-dependent reduction of CoM-S-S-CoB is mechanistically coupled to the endergonic reduction of ferredoxin driving reaction 5 [13••]. Supporting this hypothesis is the recent report of a protein complex from Methanococcus maripaludis that contains heterodisulfide reductase, formyl-MF dehydrogenase, and a hydrogenase [34••].
In the next steps (reactions 6–9) the formyl group of formyl-MF is transferred to H4M(S)PT followed by two reduction steps culminating with CH3-H4M(S)PT. The two-electron donor F420 is reduced with a nickel-containing hydrogenase [35]. Under Ni-limiting conditions, involvement of the F420-reducing hydrogenase is bypassed by a novel iron-only hydrogenase that directly oxidizes H2 and reduces CH≡H4MPT+ (reaction 8b) [35].
Biotechnological applications
The biomethanation of easily degradable organic waste and renewable plant material has been abundantly applied in past decades. More recently there has been interest in understanding the biomethanation of more recalcitrant substrates such as peat and higher rank coals to improve yields of coal bed methane with some success [36-39]. Finally, an engineered methanogenic pathway has been described derived from the domains Bacteria and Archaea that utilizes the methyl esters of acetate and propionate for growth and methanogenesis [40••]. The fragile interactions of multispecies food chains converting complex biomass to methane are easily disrupted, a major impediment to efficient and reliable conversion of renewable biomass as an alternative to fossil fuels. The engineered pathway expands the exceptionally narrow range of substrates utilized by methanogens, exemplifying the simplification of food chains leading to the more-efficient conversion of complex biomass to methane.
Conclusions and perspectives
Methanogens are key players in the biomethanation of complex biomass serving as terminal organisms of the food chain and maintaining H2, formate, and acetate concentrations at levels that are thermodynamically favorable for fermentative and acetogenic members at the front of the food chain. Thus, a fundamental understanding of aceticlastic and CO2-reducing methanogenic pathways is necessary to identify factors that optimize the rate and reliability for the biomethanation process to be economically competitive with fossil fuels. Although considerable progress has been made in understanding pathway enzymes, additional research is necessary to understand other factors such as the stress response and regulatory mechanisms, particularly of aceticlastic methanogens. Finally, a fundamental understanding of pathways combined with recent advances in genetics provides a platform for a synthetic genomic approach to engineer methanogens with properties superior to native species that further enhance the biomethanation process.
Acknowledgments
Research in the laboratory of J.G.F. has been supported by the NIH, DOE, NSF, and NASA.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
- 1.Thauer RK. Functionalization of methane in anaerobic microorganisms. Angew Chem Int Ed Engl. 2010;49:6712–6713. doi: 10.1002/anie.201002967. [DOI] [PubMed] [Google Scholar]
- 2.Scheller S, Goenrich M, Boecher R, Thauer RK, Jaun B. The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature. 2010;465:606–608. doi: 10.1038/nature09015. [DOI] [PubMed] [Google Scholar]
- 3.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science. 1997;278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- 4.Dey M, Li X, Zhou Y, Ragsdale SW. Evidence for organometallic intermediates in bacterial methane formation involving the nickel coenzyme f(430) Met Ions Life Sci. 2010;7:71–110. doi: 10.1039/BK9781847551771-00071. [DOI] [PubMed] [Google Scholar]
- 5.Scheller S, Goenrich M, Mayr S, Thauer RK, Jaun B. Intermediates in the catalytic cycle of methyl coenzyme M reductase: isotope exchange is consistent with formation of a σ-alkane-nickel complex. Angew Chem Int Ed Engl. 2010;49:8112–8115. doi: 10.1002/anie.201003214. [DOI] [PubMed] [Google Scholar]
- 6.Sarangi R, Dey M, Ragsdale SW. Geometric and electronic structures of the Ni(I) and methyl-Ni(III) intermediates of methyl-coenzyme M reductase. Biochemistry. 2009;48:3146–3156. doi: 10.1021/bi900087w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee CM, Chen CH, Liao FX, Hu CH, Lee GH. Mononuclear Ni(III)-alkyl complexes (alkyl = Me and Et): relevance to the acetyl-CoA synthase and methyl-CoM reductase. J Am Chem Soc. 2010;132:9256–9258. doi: 10.1021/ja102430d. [DOI] [PubMed] [Google Scholar]
- 8.Dey M, Li X, Kunz RC, Ragsdale SW. Detection of organometallic and radical intermediates in the catalytic mechanism of methyl-coenzyme M reductase using the natural substrate methyl-coenzyme M and a coenzyme B substrate analogue. Biochemistry. 2010;49:10902–10911. doi: 10.1021/bi101562m. [DOI] [PubMed] [Google Scholar]
- 9.Cedervall PE, Dey M, Pearson AR, Ragsdale SW, Wilmot CM. Structural insight into methyl-coenzyme M reductase chemistry using coenzyme B analogues. Biochemistry. 2010;49:7683–7693. doi: 10.1021/bi100458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebner S, Jaun B, Goenrich M, Thauer RK, Harmer J. Binding of coenzyme B induces a major conformational change in the active site of methyl-coenzyme M reductase. J Am Chem Soc. 2010;132:567–575. doi: 10.1021/ja906367h. [DOI] [PubMed] [Google Scholar]
- 11•.MacAuley SR, Zimmerman SA, Apolinario EE, Evilia C, Hou Y, Ferry JG, Sowers KR. The archetype γ-class carbonic anhydrase (Cam) contains iron when synthesized in vivo. Biochemistry. 2009;48:817–819. doi: 10.1021/bi802246s. Describes the first over-production of a metalloenzyme in a native methanogen host. [DOI] [PubMed] [Google Scholar]
- 12.Buan NR, Metcalf WW. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol Microbiol. 2010;75:843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 13••.Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–591. doi: 10.1038/nrmicro1931. Reviews electron transport in Methanosarcina and obligate CO2-reducing species, and hypothesizes thermodynamic coupling of the exergonic reduction of heterodisulfide with the endergonic first reaction of the pathway. [DOI] [PubMed] [Google Scholar]
- 14•.Deppenmeier U, Muller V. Life close to the thermodynamic limit: how methanogenic archaea conserve energy. Results Probl Cell Differ. 2008;45:123–152. doi: 10.1007/400_2006_026. Overall review of electron transport and ATP synthesis in all methanogenic pathways. [DOI] [PubMed] [Google Scholar]
- 15.Saum R, Schlegel K, Meyer B, Muller V. The F1FO ATP synthase genes in Methanosarcina acetivorans are dispensable for growth and ATP synthesis. FEMS Microbiol Lett. 2009;300:230–236. doi: 10.1111/j.1574-6968.2009.01785.x. [DOI] [PubMed] [Google Scholar]
- 16.Welte C, Kratzer C, Deppenmeier U. Involvement of Ech hydrogenase in energy conservation of Methanosarcina mazei. FEBS J. 2010;277:3396–3403. doi: 10.1111/j.1742-4658.2010.07744.x. [DOI] [PubMed] [Google Scholar]
- 17.Welte C, Kallnik V, Grapp M, Bender G, Ragsdale S, Deppenmeier U. Function of Ech hydrogenase in ferredoxin-dependent, membrane-bound electron transport in Methanosarcina mazei. J Bacteriol. 2010;192:674–678. doi: 10.1128/JB.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Li L, Rejtar T, Lessner DJ, Karger BL, Ferry JG. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J Bacteriol. 2006;188:702–710. doi: 10.1128/JB.188.2.702-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Lessner DJ, Li L, Li Q, Rejtar T, Andreev VP, Reichlen M, Hill K, Moran JJ, Karger BL, Ferry JG. An unconventional pathway for reduction of CO2 to methane in CO-grown Methanosarcina acetivorans revealed by proteomics. Proc Natl Acad Sci U S A. 2006;103:17921–17926. doi: 10.1073/pnas.0608833103. Describes a novel CO2-reduction pathway dependendent on CO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rother M, Oelgeschlager E, Metcalf WM. Genetic and proteomic analyses of CO utilization by Methanosarcina acetivorans. Arch Microbiol. 2007;188:463–472. doi: 10.1007/s00203-007-0266-1. [DOI] [PubMed] [Google Scholar]
- 21••.McInerney MJ, Sieber JR, Gunsalus RP. Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol. 2009;20:623–632. doi: 10.1016/j.copbio.2009.10.001. Reviews syntrophic species dependent on methanogens to lower metabolic products allowing thermodynamically favorable conditions for growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drake HL, Gossner AS, Daniel SL. Old acetogens, new light. Ann N Y Acad Sci. 2008;1125:100–128. doi: 10.1196/annals.1419.016. [DOI] [PubMed] [Google Scholar]
- 23.Funk T, Gu WW, Friedrich S, Wang HX, Gencic S, Grahame DA, Cramer SP. Chemically distinct Ni sites in the A-cluster in subunit beta of the Acetyl-CoA decarbonylase/synthase complex from Methanosarcina thermophila: Ni L-edge absorption and X-ray magnetic circular dichroism analyses. J Am Chem Soc. 2004;126:88–95. doi: 10.1021/ja0366033. [DOI] [PubMed] [Google Scholar]
- 24.Ragsdale SW. Nickel and the carbon cycle. J Inorg Biochem. 2007;101:1657–1666. doi: 10.1016/j.jinorgbio.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Gong W, Hao B, Wei Z, Ferguson DJ, Jr, Tallant T, Krzycki JA, Chan MK. Structure of the α2ε2 Ni-dependent CO dehydrogenase component of the Methanosarcina barkeri acetyl-CoA decarbonylase/synthase complex. Proc Natl Acad Sci U S A. 2008;105:9558–9563. doi: 10.1073/pnas.0800415105. Describes the first crystal structure for a component of the central enzyme in the aceticlastic pathway of methanogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gencic S, Grahame DA. Two separate one-electron steps in the reductive activation of the A cluster in subunit beta of the ACDS complex in Methanosarcina thermophila. Biochemistry. 2008;47:5544–5555. doi: 10.1021/bi7024035. [DOI] [PubMed] [Google Scholar]
- 27.Smith KS, Jakubzick C, Whittam TC, Ferry JG. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc Natl Acad Sci U S A. 1999;96:15184–15189. doi: 10.1073/pnas.96.26.15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmerman SA, Tomb JF, Ferry JG. Characterization of CamH from Methanosarcina thermophila, founding member of a subclass of the {gamma} class of carbonic anhydrases. J Bacteriol. 2010;192:1353–1360. doi: 10.1128/JB.01164-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Opulencia RB, Bose A, Metcalf WW. Physiology and posttranscriptional regulation of methanol:coenzyme M methyltransferase isozymes in Methanosarcina acetivorans C2A. J Bacteriol. 2009;191:6928–6935. doi: 10.1128/JB.00947-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichlen MJ, Murakami KS, Ferry JG. Functional analysis of the three TBP homologs in Methanosarcina acetivorans. J Bacteriol. 2010;192:1511–1517. doi: 10.1128/JB.01165-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson KL, Apolinario EE, MacAuley SR, Sowers KR. A 5′ leader sequence regulates expression of methanosarcinal CO dehydrogenase/acetyl coenzyme A synthase. J Bacteriol. 2009;191:7123–7128. doi: 10.1128/JB.00731-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupa B, Hendrickson EL, Leigh JA, Whitman WB. Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. Appl Environ Microbiol. 2008;74:6584–6590. doi: 10.1128/AEM.01455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendrickson EL, Leigh JA. Roles of coenzyme F420-reducing hydrogenases and hydrogen- and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. J Bacteriol. 2008;190:4818–4821. doi: 10.1128/JB.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34••.Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci U S A. 2010;107:11050–11055. doi: 10.1073/pnas.1003653107. Describes the first experimental evidence for the hypothesized coupling of heterodisulfide reduction with the first reductive step in the CO2 pathway of obloigate CO2-reducing methanogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thauer RK, Kaster AK, Goenrich M, Schick M, Hiromoto T, Shima S. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem. 2010;79:507–536. doi: 10.1146/annurev.biochem.030508.152103. [DOI] [PubMed] [Google Scholar]
- 36.Midgley DJ, Hendry P, Pinetown KL, Fuentes D, Gong S, Mitchell DL, Faiz M. Characterisation of a microbial community associated with a deep, coal seam methane reservoir in the Gippsland Basin, Australia. Int J Coal Geol. 2010;82:232–239. [Google Scholar]
- 37.Jones EJ, Voytek MA, Corum MD, Orem WH. Stimulation of methane generation from a non-productive coal by addition of nutrients or a microbial consortium. Appl Environ Microbiol. 2010 doi: 10.1128/AEM.00728-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doerfert SN, Reichlen M, Iyer P, Wang M, Ferry JG. Methanolobus zinderi sp. nov., a methylotrophic methanogen isolated from a deep subsurface coal seam. Int J Syst Evol Microbiol. 2009;59:1064–1069. doi: 10.1099/ijs.0.003772-0. [DOI] [PubMed] [Google Scholar]
- 39.Brauer S, Cadillo-Quiroz H, Ward RJ, Yavitt J, Zinder S. Methanoregula boonei gen. nov., sp nov., an acidiphilic methanogen isolated from an acidic peat bog. Int J Syst Evol Microbiol. 2010;61:45–52. doi: 10.1099/ijs.0.021782-0. [DOI] [PubMed] [Google Scholar]
- 40••.Lessner DJ, Lhu L, Wahal CS, Ferry JG. An engineered methanogenic pathway derived from the domains Bacteria and Archaea. mBio. 2010;1 doi: 10.1128/mBio.00243-10. Describes the first engineered methanogenic pathway expanding the substrate range of methanogens.e00243–10 [DOI] [PMC free article] [PubMed] [Google Scholar]