

Abstract
Nitric oxide (NO) is a potent signaling molecule that needs to be tightly regulated to maintain metabolic and cardiovascular homeostasis. The nitric oxide synthase (NOS)/Dimethylarginine dimethylaminohydrolase (DDAH)/Asymmetric Dimethylarginine (ADMA) pathway is central to this regulation. Specifically, the small molecule ADMA competitively inhibits NOS, thus lowering NO levels. The majority of ADMA is physiologically metabolized by DDAH, thus maintaining NO levels at physiological concentration. However, under pathophysiological conditions, DDAH activity is impaired, in part as a result of its sensitivity to oxidative stress. Therefore, the application of high throughput chemical screening for the discovery of small molecules that could restore or enhance DDAH activity might have significant potential in treating metabolic and vascular diseases characterized by reduced NO levels, including atherosclerosis, hypertension, and insulin resistance. By contrast, excessive generation of NO (primarily driven by iNOS) could play a role in idiopathic pulmonary fibrosis (IPF), sepsis, migraine headaches, and some types of cancer. In these conditions, small molecules that inhibit DDAH activity might be therapeutically useful. Here, we describe optimization and validation of a highly reproducible and robust assay successfully used in a high throughput screen for DDAH modulators.
Keywords: nitric oxide, asymmetric dimethylarginine, diabetes, hypertension, idiopathic pulmonary fibrosis
Introduction
ADMA and monomethyl-L-arginine (L-NMMA) are endogenous competitive inhibitors of nitric oxide synthase (NOS) 1. These methylarginines are generated by the methylation of arginine residues on histones and other proteins by a family of enzymes known as Protein Arginine Methyl Transferases (PRMTs) 2. During the hydrolysis of proteins containing methylarginine residues, free ADMA and L-NMMA are released (Figure-1). The concentration of these methylated arginines is physiologically regulated either by excretion through the renal system or degradation by dimethylarginine dimethylaminohydrolase (DDAH) 3, 4. Of the two arginine analogs, ADMA is present at roughly 10-fold higher levels in plasma and tissue. Plasma ADMA is increased in patients with cardiovascular risk factors, and is predictive of major adverse cardiovascular events, suggesting that it is a pathophysiologically relevant molecule 5.
Figure 1.

The NO/ADMA/DDAH Pathway and the pharmacological Inhibition of DDAH1 leading to higher ADMA levels and subsequent regulation of NO production. DMA = dimethylamine. ADMA = asymmetric Dimethylarginine. SDMA = symmetric Dimethylarginine. L-NMMA = monomethyl arginine. PRMTs = Protein Arginine Methyltransferases. NO = Nitric Oxide. NCE = New Chemical Entity
DDAH is a cysteine hydrolase enzyme that is expressed in all nucleated mammalian cells in one of two isoforms (DDAH1 and DDAH2) 6. About 80% of endogenous ADMA is metabolized by DDAH, principally the DDAH1 isoform 5, 7, 8. Several studies have provided evidence that disruption of DDAH activity is the major cause by which metabolic perturbations (such as hypercholesterolemia or hyperglycemia) elevate levels of ADMA 4, 9. Elevations in plasma ADMA impair vascular function and increase the risk of cardiovascular disease and death 4. Thus, it seems likely that an activator of DDAH, by reducing ADMA levels, might reduce the progression of vascular disease 4, 9.
On the other hand, there are conditions where excessive accumulation of NO (primarily driven by inducible NOS, or iNOS) may be associated with disease. For example, overproduction of NO is associated with vascular collapse in sepsis and is implicated in migraine headache, some types of cancer, and defined autoimmune diseases such as rheumatoid- and spondylo- arthritis 10, 11. In these cases, it seems likely that a DDAH antagonist, by elevating ADMA levels and suppressing NO synthesis, might have therapeutic value. Moreover, a recent study has shown that in an animal model of idiopathic pulmonary fibrosis (IPF) DDAH and iNOS activities are increased. In this model, a small molecule antagonist of DDAH (L-291) reduced fibrosis and improved pulmonary compliance 12. Therefore, interest is growing in the regulation of DDAH activity as a promising therapeutic avenue 10, 13. A growing number of DDAH inhibitors have been reported in the literature 10, 13–21. However, almost all of these compounds, with the exception of the substrate-like competitive inhibitors, are either non-selective or weak antagonists. Furthermore, no allosteric activators of DDAH activity have been reported. Therefore, there is a need for a strategy that will accelerate the search for potent and biologically relevant small molecules that regulate DDAH activity.
To this end we have developed a sensitive, robust and highly reproducible assay that differentially detects the breakdown of the endogenous substrate ADMA. In this biochemical assay, ADMA is metabolized by recombinant human DDAH1 into dimethylamine and L-citrulline. The production of L-citrulline can be quantified colorimetrically using a single derivatization step with a ureido-reactive reagent. This previously reported test-tube method 22 was substantially modified and miniaturized into a 384-well format in order to facilitate the screening of a chemical library of over 130,000 compounds using a robotic arrayer system. In an orthogonal assay, DDAH activity can be monitored fluorimetrically by using an artificial substrate, S-methyl-L-thiocitrulline (SMTC) that produces a methyl mercaptan molecule (methanethiol; CH3- SH) upon metabolism by DDAH. The release of CH3-SH can be quantified upon the addition of a coumarin-containing reagent (CPM).
Materials and Methods
The chemical library of the Stanford High Throughput Bioscience Center (HTBC) contains over 130,000 small molecules selected from diverse sources including Sigma, ChemDiv, MicroSource, ChemBridge, the NIH clinical collection (NIH CC), National Cancer Institute (NCI), natural products and FDA approved drug libraries using stringent criteria to maximize diversity and medicinal drug-like properties (http://htbc.stanford.edu/). The orthogonal assays, using hits derived from the HTBC, were conducted using chemicals purchased from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise. For generation of DDAH protein, we used an E. coli BL21 strain purchased from Invitrogen. Dr Neil McDonald (Birkbeck College, London) kindly provided the plasmid construct pGEX-6P-1-DDAH1. Empty vector control, enzyme purification and cleavage reagents were from GE Healthcare (Piscataway, NJ). Clear and black 384-well plates were from E&K Scientific (Santa Clara, CA). Antibodies directed against DDAH-1 (Abcam; Cambridge, MA) and GST (GE Healthcare) were obtained from commercial purveyors.
Production of recombinant human DDAH1
Human DDAH1 was expressed in E. coli BL21 Star (DE3) strain for protein production. In parallel, cells were also transformed with empty vector. Positive clones were selected by PCR and the clones harboring DDAH were subsequently inoculated into LB broth. Bacteria were grown at 37°C (225 rpm) for 36 hours and preinduction samples were removed prior to inducing the remaining culture by adding isopropyl-beta-D-thiogalactopyranoside (IPTG; 0.1 mM final concentration) at 25°C for 18 hours. The cells were harvested by centrifugation and the supernatant was discarded prior to lysing them with cell disruption buffer (containing 20mM Tris-HCl; pH 8.0; 150 mM NaCl; 2 mM β-mercaptoethanol; 1 mM phenylmethylsulphonyl fluoride (PMSF); 1 mM benzamidine and 10mM DNAse 1) 20 and with 1% triton X-100 and lysozyme to break the peptidoglycan layer. The lysate was centrifuged at 20,000g for 40 min at 4°C and the supernatant was transferred into clean tubes for SDS-PAGE and Western analyses. The protein was purified using Glutathione sepharose 4B column in a batch mode according to the manufacturer’s recommendations. The GST-tag was cleaved off the recombinant protein using Precision Protease. The purified protein was eluted, SDS-PAGE analyzed, and its identity was confirmed by Western and Mass Spectroscopy.
DDAH Activity Assay
The L-citrulline assay was based upon an original test-tube method developed by Prescott and Jones in 1969 22, which we adapted and optimized for a microplate format (see Results section for details). Subsequently, the activity of DDAH was quantified by detecting its conversion of ADMA to citrulline using the optimized protocol. The assay was scaled up to a 384-well format for high throughput chemical screening.
High Throughput Screening of Small Molecules
Over 130,000 small molecules deposited in the Stanford High-throughput Bioscience Center (HTBC) were screened using the enzymatic assay to identify chemicals that regulate DDAH activity. In brief, recombinant human DDAH1 (rhDDAH1) was mixed with ADMA in the presence of screening buffer in 384-well plates using a Staccato multidrop. Small molecules (100nL each) were then added to the wells using a robotic arrayer to yield a final compound screening concentration of up to 50 μM. Plates were incubated at 37°C for 4 hours. Subsequently, color developing reagent (containing 2 volumes of Antipyrin and 1 volume of 2,3-Butanedione oxime reagents) was added using Velocity 11 system and the plates were sealed using an automated plate sealer. Finally, color was developed by incubating the plates at 60°C for 90 min prior to spinning them at 1,500 rpm for 5 min. In this assay absorbance is proportional to the concentration of citrulline generated by DDAH, and was measured using an AnalystGT plate reader at 485nm using a dichroic beamsplitter. The signal-to-noise ratio of separation was calculated using an established formula 23.
Identification of Primary Hits
Inhibitors were defined as compounds that reduce absorbance by at least 30% compared to control wells. The hits were validated using 8-point full dose response study (50 μM to 0.39 μM in serial dilutions). A total of over 150 compounds, about 0.12% of the total compounds, caused a reduction in absorbance of at least 30%. To determine which of these hits were true inhibitors of DDAH activity, we used a modification of a newly validated secondary fluorometric assay 18 as described below. In parallel, compounds were also cross-validated by adding them in reaction mix containing all the components described above with the exception of the enzyme to rule out the possibility that their apparent activity is caused by non-specific reaction quenching and not directly inhibiting DDAH.
Secondary Assay to Validate Potential DDAH inhibitors
For the secondary assay, we adapted a fluorimetric assay that uses SMTC as a substrate. DDAH metabolizes SMTC into L-citrulline and methanethiol (CH3-SH) 18. In brief, DDAH (30 nM final concentration) was mixed with SMTC (100 μM final concentration), CPM (50 μM final concentration) and screening buffer (containing a final concentration of 0.01% Triton-X100 and 1mM EDTA). The reaction mix was added to black 384-well plates to validate primary hits that modulate DDAH activity. The release of CH3-SH was monitored fluorimetrically by adding 7-Diethylamino-3-(4- maleimidophenyl)-4-methylcoumarin (CPM) as described 18.
Application of the L-citrulline assay to cells
The feasibility of the citrulline assay in cell culture was validated by measuring the levels of L-citrulline in cell lysates applying a protocol similar to that described above. First, primary microvascular endothelial cells (HMVECs) were seeded in 75 cm2 cell-culture flasks until confluency. The cells were exposed to 1 mM of L-Arginine for 24 hours in the presence of vehicle control. The cells were washed with PBS and then dissociated with 3 mL of Accutase for 3 min at 37°C/5%CO2. The cells were then pelleted down by centrifugation and lysed by adding lysis buffer (containing 100mM Na2HPO4; 1% NP- 40; 1X protease and phosphatase inhibitors). The suspension was kept on ice for 30 min prior to centrifugation at 13,000 rpm for 30 min at 4°C. Finally, the cell debris was removed and the supernatant was collected for the citrulline assay. The citrulline assay was performed in a microplate assay as described above by transferring equimolar amounts of cell lysate and adding 0.5 volume of color-developing reagent. The mix was incubated at 60°C for 90 min and absorbance was measured as described above. Known concentrations of commercial citrulline were used to construct standard curves and to estimate the concentration of citrulline in the samples.
Results
Human DDAH-1 (858 bp) encoding plasmid was successfully transformed into an E. coli system and the PCR-positive clones were used for the production of rhDDAH1. Western blot analysis using both anti-DDAH1 and anti-GST antibodies confirmed that rhDDAH1 tagged with GST (56.5kDa) was expressed only in the cells transformed with the vector encoding DDAH and induced by IPTG (Figure-2). Sepharose column purification and subsequent SDS-PAGE (Figure-3A) and Western blot analyses (Figure-3B) revealed that the protein can be purified to a single band for use in screening experiments. Despite earlier reports of difficulties producing soluble protein in an E. coli (BL 21) system 24, our protein yield was consistently above 2 mg/L culture, probably due to optimal induction time, RNAse and major protease deficient production system used in our experiments.
Figure 2.

Western blot analysis showing the production of GST-DDAH (56.5kDa). Lanes 1,3: pre-induction; lane-2: post-induction sample of an empty vector; lane 4: IPTG induction of DDAH vector. Lane-M is SeeBlue Plus molecular weight marker.
Figure 3.

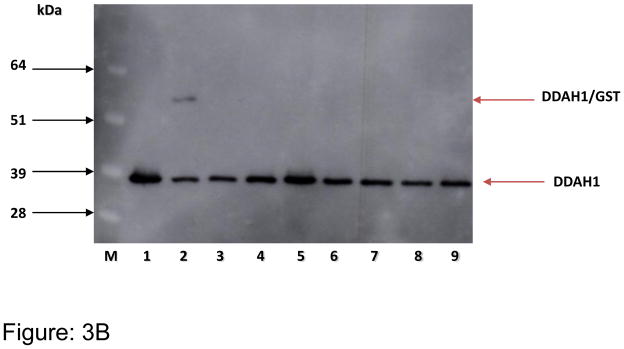
Figure 3A. SDS-PAGE analysis of purified human DDAH1. PI = pre-induction; LY = lysate; FT = flow through; W1 = wash; E = eluent and M = 1kb+ marker
Figure 3B. Western blot showing purified (after GST cleavage) recombinant human DDAH1 (~37 kDa). Purified fractions of DDAH1 (lanes 1–9) were probed using anti-DDAH1 antibody.
To find small molecule modulators of DDAH activity, we first developed an enzymatic assay that would lend itself to high throughput screening. A microplate-based L-citrulline assay to measure DDAH activity has been previously reported 25, but this protocol requires harsh conditions including heating the plates to a temperature of ~100°C. In a high throughput format, these conditions could cause sample evaporation and deformation of the reaction plates and thus this assay is not practical for the purpose of comprehensive screening. Accordingly, we optimized the conditions, including the amount of enzyme, substrate, incubation time and temperature, for a high throughput assay (Figure-4). In brief, various amounts of rhDDAH1 (0.1 to 10 μM) were incubated with various concentrations of ADMA (1 μM to 8 mM) for various time points (30 min to 10 h) at room temperature or 37°C. In the final screening assay we used near catalytic activity (kcat ~ 0.7 min−1) conditions at 0.3 μM of DDAH, 500 μM of ADMA (Km = 180 μM) and 4 hours of incubation time at 37°C. The reactions were stopped and color was developed as described above followed by incubation at 60°C for 90 min. Absorbance, proportional to DDAH activity, was measured at 485nm ± 20nm. In order to screen the entire library, we first optimized the biochemical properties of the assay as described above. We modified the technique so as to reduce the number of steps and facilitate robotic handling and throughput. Subsequently, HTS was performed using 384-well plates. The Stanford HTBC is equipped with a Caliper Life Sciences workstation for sample preparation and analysis, Titertek multidrops, microplate dispenser and automated liquid handler and laboratory robotics for screening of chemical libraries. The Z′-score was consistently found to be between 0.7 and 0.8, indicating the robustness of the assay. Furthermore, the feasibility of the assay was demonstrated in cell culture study by quantifying the amount of L-citrulline in endothelial cells treated with L-arginine or vehicle. As expected, pre-treatment with L-Arginine increased the amount of intracellular L-citrulline significantly (Figure S1; p<0.05).
Figure 4.

Citrulline assay showing that the conversion of ADMA to L-citrulline by DDAH1 is proportional to time, temperature and enzyme concentration. Data is averaged from at least duplicate experiments.
Validation Assay
We first optimized the concentration of SMTC as a substrate in the colorimetric citrulline assay described above. Subsequently, we used this substrate in the fluorimetric assay near its kcat (kcat ~1 min−1) at 100 μM substrate concentration (Km = 1 μM) 18, detecting the metabolite methanethiol (CH3-SH) using the CPM reagent. This study, in conjunction with the citrulline assay, narrowed down the number of hits by reducing potential false positives, such as pan-assay interference compounds (PAINS) 26. About 70% of the selected hits validated in the primary assay (about 35 compounds were retested in the secondary assay) were also inhibitors in the secondary fluorimetric assay. In addition, the validation assay also confirmed known DDAH inhibitors such as chloromercuribenzoate 7 and ebselen 14 and identified several new, potent inhibitors of human DDAH1. Not surprisingly these include other mercury-containing compounds such as phenylmercuric acetate (IC50 < 0.78 μM) and quinone-type compounds such as ChemDiv 2548-0707 (IC50 = 5.5 μM) and 2548-0703 (IC50 = 9.0 μM). However, it also includes more structurally novel compounds such as 4-mercapto-5-methoxy-2-phenyl- 3(2H)-pyridazinone (4-MMP, IC50 = 12.7 μM) and SCH-202676 (IC50 = 13.3 μM) (Figure-5). In addition, our time-dependence kinetics study with two of the compounds (phenylmercuric acetate and 4-chloromercuribenzoic acid) indicates progressive inhibition suggesting that their mode of inhibition might be irreversible (Figure-6). Furthermore, our follow-up study on selected inhibitors using the citrulline assay also confirmed the activity of these inhibitors against human DDAH1. Given their structure, it is possible that many of the compounds inhibit DDAH by reacting with the active site cysteine 20. Additional inhibitors of human DDAH1 and their potencies are shown in the supplemental table (Table-I).
Figure 5.




Curve fit data showing inhibition of human DDAH-1 activity by selected small molecules using the CPM assay: A) ChemDiv Compound 2548-0707 B) ChemDiv Compound 2548–0703 C) 4-MMP and D) SCH-202676. The inhibitory concentration at 50% (IC50) was calculated using Assay Explorer software.
Figure 6.
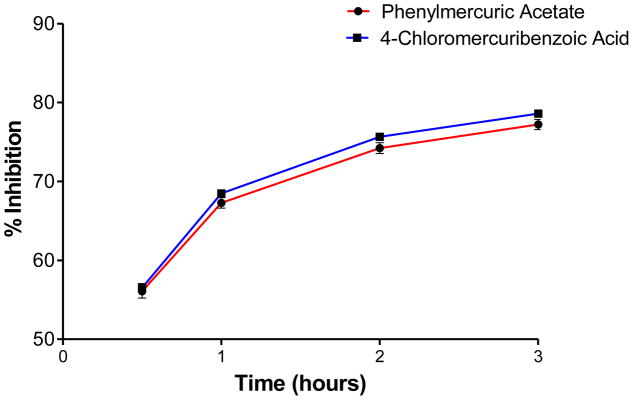
Time-dependent inhibition study of two small molecules, phenylmercuric acetate and 4-chloromercuribenzoic acid (each at 50 μM final concentration), using the fluorometric CPM assay (30 nM final enzyme concentration). Values are mean +/− SEM. Experiments were performed in triplicates.
Discussion
Small molecules that regulate the production of NO would be good molecular probes as well as potential therapeutics for cardiovascular and metabolic diseases. One possible mechanism to regulate NO is by controlling the activity of DDAH using small molecules (Figure-1). Toward this end, we have developed a high throughput assay, and adapted a sensitive orthogonal assay, to detect small molecules that modulate the activity of this enzyme. Our high throughput screening was conducted near Km concentration; a condition that ensured appreciable separation between the noise and signal to provide a good Z-score in our hands using the described colorimetric assay. We found that the concentrations used allowed reproducible linearity over the period of time that our screen was conducted. In addition, our primary assay has an added advantage of using an endogenous substrate over the secondary assay that uses an artificial substrate.
Potential applications of DDAH agonists
Drug development programs are not typically directed towards enzyme activators. Nevertheless, DDAH represents a compelling target for allosteric activation. The ADMA/NO/DDAH pathway is involved in a number of pathophysiological conditions characterized by elevated ADMA and impaired NO function as a result of direct or indirect impairment of DDAH activity. These conditions include hypercholesterolemia, hyperhomocysteinemia, hypertriglyceridemia, stroke, erectile dysfunction, peripheral and coronary artery diseases, systemic and pulmonary hypertension, atherosclerosis, primary dysmenorrhea, cytomegalovirus infection, preeclampsia, chronic kidney disease (CKD), non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), insulin resistance and diabetes mellitus 5, 9, 27, 28 These pathologies could potentially be ameliorated or reversed using small molecules that activate DDAH.
Potential applications of DDAH antagonists
In contrast, small molecules that inhibit DDAH activity could potentially be useful in diseases where an overly active DDAH needs to be reduced and ADMA levels need to be elevated as in pulmonary fibrosis 12 and conditions where NO production is principally driven by iNOS as in sepsis 10. Our screening effort using a recombinant protein has revealed a number of small molecules that modulate human DDAH1 activity. Among these, we have observed that DDAH is very sensitive to organomercury compounds such as phenylmercuric acetate (IC50 < 0.78μM), Thimerosal (IC50 = 0.46μM) and Cumertilin (100% inhibition at 25μM). This is consistent with the discovery by Ogawa et al of DDAH in 1987 7, at which time they observed that the enzyme (isolated from rat kidney homogenates), was sensitive to the mercury containing compounds p-chloromercuribenzoate and mercury chloride, leading them to correctly predict the involvement of a thiol-containing motif in the active site of the enzyme. This hypothesis was later confirmed in human DDAH1 (which shares 93% homology with rat DDAH) 9, 20. Furthermore, we suspect that electrophilic compounds such as ChemDiv 2548–0707 and 2548–0703 (Figure-5) could also react with the active-site cysteine.
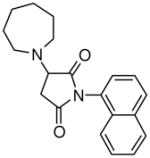
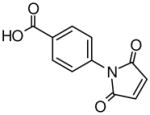
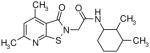
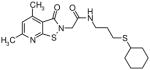
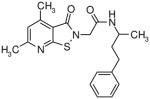
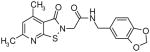
The presence of a cysteine residue (Cys 273) in the catalytic site of DDAH1 appears to have created a vulnerability of DDAH to inhibition by small molecules that contain a sulfhydryl group. Many of the compounds listed in Table-1 contain sulfur and it seems likely that their inhibition of DDAH is by the formation of a disulfide bond with the catalytically active site. Indeed, the isothiazolo[5,4-b]pyridin-3-one core found in many of the molecules bears a striking resemblance to ebselen, which has been previously shown to inhibit DDAH by covalently reacting with the active-site cysteine 14. Similarly, 4-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic acid contains a maleimide moiety, which is known to react with cysteine residues. Although these inhibitors could conceivably be used as probes for in vitro characterization of DDAH, such potentially non-specific inhibitors could have off-target effects via their reaction with other cysteine containing proteins. Nonetheless, several marketed drugs are covalent inhibitors and about 5% of these drugs are in use in cardiovascular medicine 29. For example, aspirin (to reduce the risk of heart attack & stroke) and Plavix (for CAD, CVD and PVD) are examples of widely used covalent inhibitors. However, DDAH inhibitors that possess other functionalities may confound an understanding of their actions in vivo. For example, the seleno-organic compound ebselen strongly inhibits DDAH activity in vitro as shown by others 14 and in our present screening (IC50 = 3.9 μM). However, ebselen is also known to be an antioxidant 30 and thereby could protect DDAH from oxidative stress 9 complicating any simple interpretation of the biological effects of the drug. In addition to these likely covalent inhibitors, some of the molecules, such as 3-[5-(4-methylphenyl)-2-furyl]propanoic acid and 3-(1-azepanyl)-1-(1-naphthyl)-2,5-pyrrolidinedione do not contain obvious electrophilic moieties and could thus be acting non-covalently.
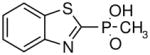
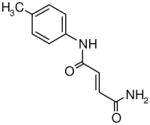
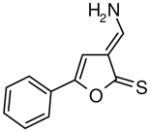
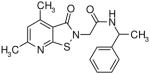
Table I.
Novel Inhibitors of human DDAH-1. The following compounds were validated in full dose-response curves. The inhibitory concentration at 50% (IC50) was calculated using Assay Explorer software.
| Molecular Name | IC50 (μM) | Hill Slope |
|---|---|---|
|
13.1 | 1.8 |
|
23.3 | 1.6 |
|
9.9 | 1.7 |
|
21.2 | 1.1 |
|
17.7 | 1.3 |
|
|
45.8 | 1.1 |
|
13.7 | 1.7 |
|
8.6 | 1.7 |
|
12.8 | 1.6 |
|
|
7.1 | 2.3 |
|
14.1 | 1.6 |
|
11 | 1.5 |
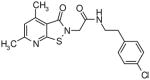
|
13.4 | 1.2 |
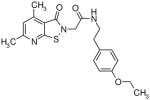
|
10.5 | 1.5 |
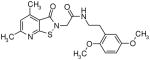
|
9.9 | 1.5 |
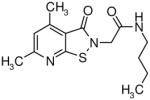
|
5.1 | 1.7 |
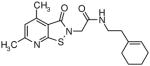
|
11.8 | 2 |
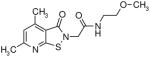
|
3.3 | 1.4 |
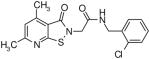
|
39.9 | 0.4 |
In summary, we have developed a robust DDAH assay that uses the natural substrate ADMA and is suitable for high throughput screening to find chemicals that modulate DDAH activity in vitro and in cell-based assays (Figure-S1). This assay is compatible with conventional screening workstations and robotics for rapid throughput. Using this assay, we have screened more than 130,000 compounds and discovered many small molecules that modulate DDAH activity, some of which are reported here. We believe our assay will allow other investigators to screen their libraries or use this assay to validate hits generated using alternative assays 18. As expected, the chemical composition of several of our hits indicates that the active site cysteine might be a preferential target for inhibitors. For example, two previously identified chemicals; homocysteine 9 and ebselen 14 are reported to inhibit DDAH activity by reacting with the active site Cys. Furthermore, fragment-based library screening also revealed that certain aryl halides could inhibit DDAH activity 13. Although we have identified potent DDAH modulators in vitro, their efficacy in cell-based and in vivo studies is still being determined and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the NIH (RC2HL103400, 1U01HL100397, and K12HL087746), AHA (11IRG5180026), Stanford SPARK Program and by the Tobacco-Related Disease Research Program of the University of California (18XT-0098). YTG was a recipient of the Stanford School of Medicine Dean’s fellowship (1049528-149- KAVFB) and is currently supported by the Tobacco-Related Disease Research Program of the University of California (20FT-0090). Keisuke Yamada was a visiting scholar.
The authors are grateful to Prof Neil McDonald (Birkbeck College, London) for providing human DDAH1 plasmid and to Dr David Solow-Cordero and Jason Wu of the Stanford high throughput Center for their technical assistance. We are also grateful to the Stanford PAN facility and Chemistry department for their DNA sequencing and MS services respectively.
Footnotes
Conflict of Interest: None
References
- 1.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 2.Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- 4.Cooke JP. Asymmetrical dimethylarginine: the Uber marker? Circulation. 2004;109:1813–1818. doi: 10.1161/01.CIR.0000126823.07732.D5. [DOI] [PubMed] [Google Scholar]
- 5.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 6.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J. 1999;343(Pt 1):209–214. [PMC free article] [PubMed] [Google Scholar]
- 7.Ogawa T, Kimoto M, Sasaoka K. Occurrence of a new enzyme catalyzing the direct conversion of NG, NG-dimethyl-L-arginine to L-citrulline in rats. Biochem Biophys Res Commun. 1987;148:671–677. doi: 10.1016/0006-291x(87)90929-6. [DOI] [PubMed] [Google Scholar]
- 8.Hu X, Atzler D, Xu X, Zhang P, Guo H, Lu Z, Fassett J, Schwedhelm E, Boger RH, Bache RJ, Chen Y. Global Dimethylarginine Dimethylaminohydrolase-1 (DDAH1) Gene-Deficient Mice Reveal That DDAH1 Is the Critical Enzyme for Degrading the Cardiovascular Risk Factor Asymmetrical Dimethylarginine. Arterioscler Thromb Vasc Biol. 2011;15:1540–1546. doi: 10.1161/ATVBAHA.110.222638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 10.Leiper J, Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat Rev Drug Discov. 2011;10:277–291. doi: 10.1038/nrd3358. [DOI] [PubMed] [Google Scholar]
- 11.Chobanyan-Jurgens K, Pham VV, Stichtenoth DO, Tsikas D. Elevated dimethylarginine dimethylaminohydrolase (DDAH) activity in rheumatoid arthritis and spondyloarthritis. Nitric Oxide. 2011;25:436–438. doi: 10.1016/j.niox.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Pullamsetti SS, Savai R, Dumitrascu R, Dahal BK, Wilhelm J, Konigshoff M, Zakrzewicz D, Ghofrani HA, Weissmann N, Eickelberg O, Guenther A, Leiper J, Seeger W, Grimminger F, Schermuly RT. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci Transl Med. 2011;3:87ra53. doi: 10.1126/scitranslmed.3001725. [DOI] [PubMed] [Google Scholar]
- 13.Johnson CM, Monzingo AF, Ke Z, Yoon DW, Linsky TW, Guo H, Robertus JD, Fast W. On the Mechanism of Dimethylarginine Dimethylaminohydrolase Inactivation by 4-Halopyridines. J Am Chem Soc. 2011;133:10951–10959. doi: 10.1021/ja2033684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linsky T, Wang Y, Fast W. Screening for dimethylarginine dimethylaminohydrolase inhibitors reveals ebselen as a bioavailable inactivator. ACS Med Chem Lett. 2011;2:592–596. doi: 10.1021/ml2000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lluis M, Wang Y, Monzingo AF, Fast W, Robertus JD. Characterization of C-alkyl amidines as bioavailable covalent reversible inhibitors of human DDAH-1. ChemMedChem. 2011;6:81–88. doi: 10.1002/cmdc.201000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Monzingo AF, Hu S, Schaller TH, Robertus JD, Fast W. Developing dual and specific inhibitors of dimethylarginine dimethylaminohydrolase-1 and nitric oxide synthase: toward a targeted polypharmacology to control nitric oxide. Biochemistry. 2009;48:8624–8635. doi: 10.1021/bi9007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotthaus J, Schade D, Muschick N, Beitz E, Clement B. Structure-activity relationship of novel and known inhibitors of human dimethylarginine dimethylaminohydrolase-1: alkenyl-amidines as new leads. Bioorg Med Chem. 2008;16:10205–10209. doi: 10.1016/j.bmc.2008.10.058. [DOI] [PubMed] [Google Scholar]
- 18.Linsky T, Fast W. A continuous, fluorescent, high-throughput assay for human dimethylarginine dimethylaminohydrolase-1. J Biomol Screen. 2011;16:1089–1097. doi: 10.1177/1087057111417712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson CM, Linsky TW, Yoon DW, Person MD, Fast W. Discovery of halopyridines as quiescent affinity labels: inactivation of dimethylarginine dimethylaminohydrolase. J Am Chem Soc. 2011;133:1553–1562. doi: 10.1021/ja109207m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 21.Hartzoulakis B, Rossiter S, Gill H, O’Hara B, Steinke E, Gane PJ, Hurtado-Guerrero R, Leiper JM, Vallance P, Rust JM, Selwood DL. Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg Med Chem Lett. 2007;17:3953–3956. doi: 10.1016/j.bmcl.2007.04.095. [DOI] [PubMed] [Google Scholar]
- 22.Prescott LM, Jones ME. Modified methods for the determination of carbamyl aspartate. Anal Biochem. 1969;32:408–419. doi: 10.1016/s0003-2697(69)80008-4. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 24.Knipp M, Charnock JM, Garner CD, Vasak M. Structural and functional characterization of the Zn(II) site in dimethylargininase-1 (DDAH-1) from bovine brain. Zn(II) release activates DDAH-1. J Biol Chem. 2001;276:40449–40456. doi: 10.1074/jbc.M104056200. [DOI] [PubMed] [Google Scholar]
- 25.Knipp M, Vasak M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem. 2000;286:257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 26.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 27.Bloomgarden ZT. Insulin resistance, dyslipidemia, and cardiovascular disease. Diabetes Care. 2007;30:2164–2170. doi: 10.2337/dc07-zb08. [DOI] [PubMed] [Google Scholar]
- 28.Kielstein JT, Donnerstag F, Gasper S, Menne J, Kielstein A, Martens-Lobenhoffer J, Scalera F, Cooke JP, Fliser D, Bode-Boger SM. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke. 2006;37:2024–2029. doi: 10.1161/01.STR.0000231640.32543.11. [DOI] [PubMed] [Google Scholar]
- 29.LG Covalent Drugs Form Long-Lived Ties. Chemical & Engineering News. 2011;89:19–26. [Google Scholar]
- 30.Nakamura Y, Feng Q, Kumagai T, Torikai K, Ohigashi H, Osawa T, Noguchi N, Niki E, Uchida K. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J Biol Chem. 2002;277:2687–2694. doi: 10.1074/jbc.M109641200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.