

Abstract
In addition to haem copper oxidases, all higher plants, some algae, yeasts, molds, metazoans, and pathogenic microorganisms such as Trypanosoma brucei contain an additional terminal oxidase, the cyanide-insensitive alternative oxidase (AOX). AOX is a diiron carboxylate protein that catalyzes the four-electron reduction of dioxygen to water by ubiquinol. In T. brucei, a parasite that causes human African sleeping sickness, AOX plays a critical role in the survival of the parasite in its bloodstream form. Because AOX is absent from mammals, this protein represents a unique and promising therapeutic target. Despite its bioenergetic and medical importance, however, structural features of any AOX are yet to be elucidated. Here we report crystal structures of the trypanosomal alternative oxidase in the absence and presence of ascofuranone derivatives. All structures reveal that the oxidase is a homodimer with the nonhaem diiron carboxylate active site buried within a four-helix bundle. Unusually, the active site is ligated solely by four glutamate residues in its oxidized inhibitor-free state; however, inhibitor binding induces the ligation of a histidine residue. A highly conserved Tyr220 is within 4 Å of the active site and is critical for catalytic activity. All structures also reveal that there are two hydrophobic cavities per monomer. Both inhibitors bind to one cavity within 4 Å and 5 Å of the active site and Tyr220, respectively. A second cavity interacts with the inhibitor-binding cavity at the diiron center. We suggest that both cavities bind ubiquinol and along with Tyr220 are required for the catalytic cycle for O2 reduction.
Keywords: diiron protein, neglected tropical diseases, monotopic membrane protein, drug target, ubiquinol oxidase
The alternative oxidase (AOX) is a nonprotonmotive ubiquinol oxidase catalyzing the four-electron reduction of dioxygen to water (1). The gene encoding this protein has been found in all higher plants, some algae, yeast, slime molds, free-living amoebae, eubacteria, nematodes, and some parasites including Trypanosoma brucei (2–5). T. brucei is a parasite that causes human African sleeping sickness and nagana in livestock and is transmitted by the tsetse fly (5). The development of chemotherapy and the continued search for new, unique therapeutic targets for African trypanosomiasis are urgently required, because current treatments, which are poorly targeted, have unacceptable side effects and efficacy (6).
The bloodstream form of T. brucei is equipped with a unique energy metabolism, namely an altered respiratory chain (5) and a modified ATP synthase (7). The parasites live as the bloodstream form in the mammalian host and as the procyclic form in the tsetse fly (5). The procyclic form of T. brucei contains a cytochrome-dependent respiratory chain in addition to an alternative oxidase, whereas within the bloodstream trypanosomes use the glycolytic pathway, localized in the glycosome, as their major source of ATP (5, 8). Once the parasites invade the mammalian host in the bloodstream, both the cytochrome respiratory pathway and oxidative phosphorylation disappear and are replaced by the trypanosomal alternative oxidase (TAO), which functions as the sole terminal oxidase to reoxidize NADH accumulated during glycolysis (5). Because NADH reoxidation is essential for parasite survival and mammalian hosts do not possess this protein, TAO is considered to be a unique target for antitrypanosomal drugs (9). Indeed, we have previously reported that the antibiotic ascofuranone (AF), isolated from the pathogenic fungus Ascochyta viciae, specifically inhibits the quinol oxidase activity of TAO at subnanomolar concentrations and rapidly kills the parasites (10). Furthermore, we have confirmed the chemotherapeutic efficacy of ascofuranone in vivo (11, 12).
Despite universal conservation of the gene encoding the AOX and diversified physiology (2), the molecular features of this protein have yet to be fully characterized. Current structural models predict that the AOX is an integral interfacial membrane protein that interacts with a single leaflet of the lipid bilayer and contains a nonhaem diiron carboxylate active site (1, 13, 14). This model is supported by extensive site-directed mutagenesis and spectroscopic studies (3, 15–20).
There are many proteins that belong to the diiron carboxylate protein family, and in each case they are characterized by the possession of two copies of the diiron binding motifs (21, 22). To date the majority of proteins within this family whose crystal structures have been determined are soluble proteins, and hence determination of a crystal structure of a member of the membrane-bound class is vital, because it would transformationally improve our understanding of the structure–function relationships of this functionally diverse family of proteins. In this paper we report on the crystal structure of the oxidized form of the trypanosomal alternative oxidase at 2.85 Å. In addition to this very important milestone we also describe the structures of the active site of the enzyme in the presence of AF derivatives, AF2779OH and colletochlorin B (CCB), at 2.6 Å and 2.3 Å resolution, respectively. We believe that a detailed knowledge of the active site of the enzyme in the presence of such inhibitors will lead to a greater rational design of further potent and safer antitrypanosomal drugs.
Results and Discussion
Overall Structure of TAO.
We have recently established protocols to prepare highly purified and stable TAO, which has enabled us to crystallize the enzyme (23, 24). The crystal structure of TAO determined at 2.85 Å resolution (SI Appendix, Table S1) contains four monomers per asymmetric unit that associate to form homodimers (Fig. 1A and SI Appendix, Fig. S1A). Each monomer, which lacks about 30 residues in both N- and C-terminal regions due to faint electron density, consists of a long N-terminal arm, six long α helices (α1–α6), and four short helices (S1–S4). The long helices are arranged in an antiparallel fashion with α2, α3, α5, and α6 forming a four-helix bundle that accommodates a diiron center, as widely observed in other diiron carboxylate proteins (1, 14) (SI Appendix, Fig. S2). Except for the N-terminal arm, each monomer is shaped as a compact cylinder (50 × 35 × 30 Å), and there are no significant structural differences among monomers in the asymmetric unit, as indicated by rms deviations (0.49∼0.68 Å) for superimposed Cα positions of the six helices calculated between a pair of monomers. However, loops connecting adjacent helices show larger differences among monomers, resulting in somewhat larger rms deviations (0.67∼0.88 Å) when calculated using all Cα atoms.
Fig. 1.

Structure of TAO. Long helices are labeled α1 to α6 and short ones S1 to S4. Diiron and hydroxo atoms are shown as magenta spheres. (A) Dimeric structure of TAO viewed roughly perpendicular (Left) and parallel (Right) to the helix axes. Helices are shown as cylinders. Chain A is colored in rainbow from blue (N terminus) to red (C terminus) and chain B in pink. (B) Surface representation of the TAO dimer showing the hydrophobic (Left) and hydrophilic (Right) surfaces. Colors are according to the following hydrophobicity scale: red, high hydrophobicity; white, low hydrophobicity (www.pymolwiki.org/index.php/Color_h). (C) Proposed binding model of the TAO dimer to membranes shown by surface (Left) and cartoon (Right) representations. The hydrophobic region on the molecular surface of the TAO dimer faces the membrane. Conserved basic amino acid residues, which are distributed along a boundary between the hydrophobic and hydrophilic regions of the dimer surface, are colored in blue. Residue names are labeled in black (asterisk denotes in chain B).
In the dimer, two monomers are related by a noncrystallographic twofold axis approximately perpendicular to the bundle (Fig. 1A). Helices α2, α3, and α4 of one monomer and α2*, α3*, and α4* of the other (asterisk denotes helix of a neighboring monomer) build a dimer interface, where six completely conserved residues (H138, L142, R143, R163, L166, and Q187) and 12 highly conserved residues (M131, M135, L139, S141, M145, R147, D148, L156, A159, M167, R180, and I183) are involved in the interaction between monomers (SI Appendix, Fig. S3), suggesting that a dimeric structure is common to all AOXs. In addition, the N-terminal arm (P31∼R62) of one monomer extends into the other monomer (Fig. 1A), suggesting that the arm is important for dimerization. Upon dimerization, about 2,490 Å2 of solvent-accessible surface (35% of the total dimer surface) is buried. A large hydrophobic region is visible on one side of the dimer surface that is formed by α1 and α4 plus the C-terminal region of α2 and the N-terminal region of α5 from both monomers (Fig. 1B Left). Because the opposite side of the dimer surface is relatively hydrophilic (Fig. 1B Right), we propose that the dimer is bound to the mitochondrial inner membrane via this hydrophobic region in an interfacial fashion, as originally suggested by Andersson and Nordlund (13). The membrane penetration depth of TAO, calculated by PPM web server (25), is 8.4 Å, roughly corresponding to the radius of a helix. In addition, basic residues (R106, R143, R180, R203, and R207) are distributed along a boundary between the hydrophobic and hydrophilic regions of the dimer surface (Fig. 1C and SI Appendix, Fig. S4). They are conserved across all amino acid sequences of the membrane-bound AOXs shown in SI Appendix, Fig. S5, and their locations make these residues ideal candidates to interact with the negatively charged phospholipids head groups of membranes.
Structure of Diiron Active Site.
The structure of the diiron active site was refined as an oxidized Fe(III)-Fe(III) form with a single hydroxo-bridge (Fig. 2 and SI Appendix, Fig. S6), as previously predicted from spectroscopic studies (19, 20). The active site, which is located in a hydrophobic environment deep inside the TAO molecule, is composed of the diiron center and four glutamate (E123, E162, E213, and E266) and two histidine residues (H165 and H269), all of which are completely conserved (SI Appendix, Fig. S5). In addition, the conserved hydrophobic residues (L122, A126, L212, A216, Y220, and I262) are within 6 Å of the diiron center (Fig. 2A). The average Fe1–Fe2 distance of the four monomers in the asymmetric unit is 3.3 ± 0.2 Å and, in addition to the hydroxo-bridge, Fe1 and Fe2 are bridged by E162 and E266 and furthermore coordinated in a bidentate fashion by E123 and E213, respectively, thereby resulting in a five-coordinated diiron center possessing a distorted square pyramidal geometry (Fig. 2B and SI Appendix, Fig. S6 and Table S2). The most striking feature of the diiron active site in the oxidized state is that, as predicted from our earlier FTIR studies (26), histidine residues (H165 and H269) are too distant from both Fe1 and Fe2 (H165: 3.3∼4.0 Å, H269: 3.8∼4.4 Å) to coordinate to the diiron center. They do, however, form hydrogen bonds with E123, N161, E162, E213, and D265. N161 and D265 are situated in the center of the hydrogen-bond network and extend the network to W65, Y246, and W247. These residues, apart from W65, are again completely conserved (SI Appendix, Fig. S5), suggesting that the hydrogen bond network is important for the stabilization of the AOX active site. To our knowledge, TAO is the only structure of an oxidized diiron active site that is ligated solely by carboxylate ligands. In contrast, diiron active sites of soluble diiron proteins with known structures, Δ4 ACP desaturase (27) (PDB ID code 2UW1), methane monooxygenase (28) (PDB ID code 1MMO), rubrerythrin (29) (PDB ID code 1LKM), and ribonucleotide reductase R2 subunit (30) (PDB ID code 1RIB), are all coordinated by at least one if not two histidine residues.
Fig. 2.

Diiron structure of TAO. (A) Stereo view of the diiron active site and its environment. Diiron and hydroxo atoms are shown as magenta spheres, four glutamate and two histidine residues important for diiron binding as green sticks, neighboring residues within 6 Å of the diiron in yellow, nitrogen in blue, and oxygen in red. (B) Stereo view of the coordinate bonds (solid lines) and hydrogen bonds (dashed lines) of the diiron active site. Sigma-A weighted electron density map calculated from the refined model of the ligand-free TAO with the diiron centers omitted from the phase calculation is also shown. Contour levels are 1.0 σ (blue) and 3.0 σ (orange). H165 forms hydrogen bonds with E123, E162, and N161 and H269 with E162, E213, and N161. N161, which is situated in the center of the hydrogen network, forms additional hydrogen bonds with Y246 and D265. D265 forms hydrogen bonds with W65 and W247.
Important Tyrosine Residues.
Similar to ribonucleotide reductase, tyrosine residues have also been proposed to play an essential role in the catalytic cycle of AOX (1, 31, 32). Scrutiny of SI Appendix, Fig. S5 reveals that although there seem to be four conserved tyrosine residues (Y198, Y211, Y220, and Y246), only three of which (not Y211) are totally conserved across all amino acid sequences of membrane-bound AOXs, including the plastid AOX (33). Y220 is buried deep within the four-helix bundle and within 4 Å of the diiron center (Fig. 2A), making it the most likely candidate for the amino acid radical involved in the catalytic cycle (32). Indeed, Y220 is absolutely conserved across all AOXs sequenced to date, and mutational analyses have unequivocally demonstrated that this residue is critical for enzymatic activity of all AOXs (1, 33). Y198 has been proposed to be involved in ubiquinol binding, although its mutation does not lead to the complete loss of activity (18, 34). The crystal structure of TAO (SI Appendix, Fig. S7) indicates that Y198, located on the C-terminal portion of helix α4, is separated by more than 15 Å from the diiron center and forms a hydrogen bond with a conserved H206 protruding from the N-terminal portion of helix α5. Such a position suggests it probably stabilizes the structure of TAO rather than being directly involved in ubiquinol binding. Although Y246 on helix S3 is located 10.7 ± 0.2 Å from the diiron center, which is within electron tunneling distance [<14 Å (35)], it is more likely to be involved in the hydrogen-bonding network rather than electron transport, because it is 2.9 ± 0.2 Å from N161 in helix α3 (Fig. 2B and SI Appendix, Fig. S7). This notion is supported by the result that a Y246A mutant retains some activity (1, 34), which would not be the case if it were essential for electron transfer.
Binding Mode of the Potent Inhibitor AF2779OH.
Until recently little structural information was available on the mode of AF binding to TAO, even given its specificity. Inhibitor kinetic studies indicated that AF showed a mixed-type inhibition against ubiquinol (23), suggesting that the ring moiety and the geranyl portions of AF are important for the interaction of the inhibitor with TAO. To investigate whether this was the case, an AF derivative lacking the furanone ring was synthesized (AF2779OH: 5-chloro-3-[(2E,6E)-8-hydroxy-3,7-dimethylnona-2,6-dienyl]-2,4-dihydroxy-6-methylbenzaldehyde; Fig. 3A). AF2779OH possesses similar inhibitory properties (IC50 = 0.48 nM for TAO; minimum inhibitory concentration = 30 nM for T. brucei brucei) to AF, indicating that the furanone ring is indeed not critical for inhibitory activity, thereby rendering it useful to determine the location of AF binding to TAO. A crystal of the TAO–AF2779OH complex was prepared by soaking in the cryoprotectant solution supplemented with the inhibitor and the structure determined at 2.6 Å by molecular replacement using the inhibitor-free TAO structure as a template (SI Appendix, Table S1 and Fig. S1B). In addition, the crystal structure of TAO complexed with CCB, another AF derivative (5-chloro-3-[(2E)-3,7-dimethylocta-2,6-dienyl]-2,4-dihydroxy-6-methylbenzaldehyde), was also determined at 2.3 Å resolution (SI Appendix, Table S1 and Fig. S1C). SI Appendix, Figs. S8 and S9 show that CCB is bound to the enzyme in a manner similar to AF2779OH. CCB also strongly inhibits TAO (IC50 = 0.20 nM for TAO); however, unlike AF and AF2779OH, it is toxic to mice. Given the toxicity of CCB we will therefore focus further discussion on the structure of TAO complexed with AF2779OH, because it is a safer drug candidate for trypanosomiasis.
Fig. 3.
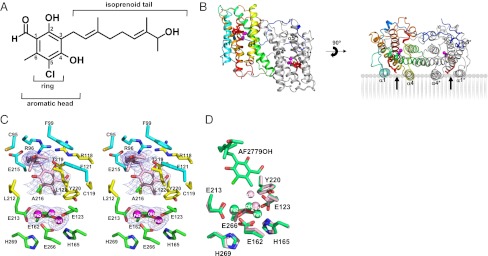
Structure of the TAO–AF2779OH complex. (A) The chemical structure of AF2779OH. (B) Overall structure of the TAO–AF2779OH complex. AF2779OH is shown as a red stick. Chains A and B are shown as rainbow (colored blue to red from N to C terminus) and gray, respectively. The AF2779OH-binding cavity is shown by an arrow. (C) Stereo view of the AF2779OH binding region of chain A. The residues that interact with AF2779OH (pink stick) -ring and -tail are shown as yellow and cyan sticks, respectively. N, O, and Cl atoms are colored in blue, red, and green, respectively. Sigma-A weighted electron density map calculated from the refined model of the TAO–AF2779OH complex with the diiron centers and AF2779OH molecules omitted from the phase calculation is also shown. Contour levels are 1.0 σ (blue) and 3.0 σ (orange). (D) Superimposed diiron active sites of AF2779OH-free (light pink) and -bound (green) forms of TAO. The binding of AF2779OH causes the formation of a coordinate bond between H165 and Fe1.
Fig. 3 B and C show the dimeric structure of the TAO–AF2779OH complex and residues around the bound AF2779OH, respectively. The binding cavity of AF2779OH is located near the membrane surface between helices α1 and α4 and is lined by 16 highly conserved residues (V92, R96, F99, R118, C119, F121, L122, E123, V125, M190, L212, E213, E215, A216, T219, and Y220) plus C95 (Fig. 3C and SI Appendix, Figs. S5 and S8 and Table S3). It is also apparent from Fig. 3C and SI Appendix, Fig. S8 and Table S3 that the aromatic head of AF2779OH is located close to the diiron active site and the C2–OH forms hydrogen bonds with R118 and T219. In addition, the aldehyde oxygen at the C1 position interacts with E123 through a hydrogen bond network, C1–CH = O···C119–SH···Y220–OH···E123–COO−, in the B and D subunit, whereas the aldehyde oxygens of the A and C subunits form an intrasubunit hydrogen bond with C2–OH. These hydrogen bonds are also observed in the TAO–CCB complex and seem to be important for the potent inhibitory activities of both inhibitors. Indeed, IC50 values of AF derivatives lacking this aldehyde group (K2-9 and K4-9 in SI Appendix, Fig. S10) increase substantially (36). It is also likely that van der Waals contacts formed between AF2779OH and TAO (SI Appendix, Table S3) contribute to the potent effect of these inhibitors (36). In the inhibited enzymes the distances between H165 and Fe1 (2.3 ± 0.1 and 2.4 ± 0.1 Å for AF2779OH and CCB, respectively) are shorter than that observed in the inhibitor-free structure (3.5 ± 0.3 Å), and hence H165 can now coordinate with Fe1, unlike H269, which is still separated by 4.3 ± 0.2 Å from Fe2 in both cases (Fig. 3D and SI Appendix, Fig. S12 and Table S2).
Mutational Analysis of Functionally Relevant Residues.
SI Appendix, Table S4 summarizes the catalytic activities of the mutated recombinant proteins that were measured in isolated membrane fractions from each Escherichia coli culture. It is apparent from SI Appendix, Table S4 that all mutated residues that interact either with the diiron (E213A) or the inhibitor (R118A, R118Q, L122A, L122N, E215A, A216L, A216N, T219V, and Y220F; Fig. 4) resulted in almost complete loss of ubiquinol oxidizing activity. Furthermore, the Y246A mutant, which participates in the hydrogen bond network (Fig. 2), also resulted in significant inhibition of catalytic activity. We believe that these residues are important for the correct conformation of the diiron center and interaction with AF2779OH and are consistent with the crystal structure.
Fig. 4.

Location of the recombinant TAO mutations within the protein. Diiron and hydroxo atoms are shown as magenta spheres. AF2779OH is shown as a cyan stick. Red sticks show the mutated residues that almost completely abolished activity (specific activity <10%), whereas yellow sticks show the mutated residues that retained some residual activity (specific activity ≥10%).
Ubiquinol Binding Model.
In addition to the inhibitor-binding cavity observed in Figs. 3B and 5 A and C, which is comparable to that observed in other monotopic proteins such as prostaglandin H2 synthase (37), CAVER protein-analysis software (38) predicts that there is another possible hydrophobic cavity near the membrane surface (Fig. 5 A and D). This second cavity connects the diiron active site with the membrane exterior and interacts with the inhibitor-binding cavity at the active site. It is formed by residues from helices α1 (R96 and D100), α2 (R118, L122, E123, and A126), α3 (E162 and H165), α5 (L212, E213, E215, A216, and T219), and α6 (E266), which, similar to that observed in the inhibitor-binding cavity, are also highly conserved (SI Appendix, Fig. S5). It is apparent from Fig. 5 A and D that a part of the aromatic head group of AF2779OH enters this second cavity. Based on the structure of the TAO–AF2779OH complex, a ubiquinol-binding model was built by superposing a ubiquinol molecule onto the bound AF2779OH. The model (Fig. 5B) indicates that the distance between ubiquinol C4–OH and Fe2 is 4.3 Å and C1–OH is connected to the outside of TAO through a hydrogen bond network, C1–OH···R118···D100 (Fig. 5B). On the basis of the structures reported in this study we propose that each hydrophobic cavity binds one ubiquinol close to the active site with their quinol rings located at the bottom of each cavity in a manner similar to AF2779OH. Although the exact route of electron transfer for the four-electron reduction of oxygen to water in any alternative oxidase is unresolved at the present time, we suggest the process involves both ubiquinols and Tyr220 (39). During the sequential electron reduction process we suggest that following the activation of oxygen, free radicals are generated on a tightly bound ubiquinol and Tyr220. The ubisemiquinol is then reoxidized by the tyrosine radical generated during the catalytic cycle and the reduction process is completed following full oxidation of a loosely bound ubiquinol (39).
Fig. 5.

Putative ubiquinol binding cavities in TAO. (A) Two hydrophobic cavities predicted by CAVER protein-analysis software (38). The bound AF2779OH (isoprenoid tail) occupies the green cavity. Putative residues involved in electron transfer are shown as orange sticks. (B) Ubiquinol binding model predicted by the superposition of a ubiquinol molecule (purple stick) onto the bound AF2779OH (translucent white stick) of the TAO–AF2779OH complex. Magenta spheres are diiron (Fe–OH−–Fe), the green stick represents residues coordinating to diiron, and yellow and cyan sticks are residues interacting with the aromatic head and isoprenoid tail, respectively. Hydrogen bonds are depicted as dotted lines. Surface views of the (C) green and (D) orange cavities shown in A. Cyan and pink colors stand for conservation of AOX residues in all eight and over four organisms in SI Appendix, Fig S5, respectively.
Conclusions
The TAO structures reported in this study are a high-resolution view of a membrane bound diiron-carboxylate protein. Although the crystal structures support earlier modeling studies (13, 14, 22) that suggested that the alternative oxidases are monotopic proteins in which the diiron active site is coordinated by carboxylate and histidine residues, they did reveal that in the oxidized state only carboxylate residues act as the coordinating ligands. Such a primary ligation sphere, although unusual for diiron proteins in the oxidized state, is, however, consistent with our earlier reduced minus oxidized IR difference spectra (26). This study clearly demonstrated that upon reduction of purified TAO there was a net protonation of at least one carboxylate residue in addition to alterations in the signals associated with histidine residues consistent with the notion that the oxidation–reduction cycle of the alternative oxidases involves major conformational perturbations and carboxylate shifts. The structure has also revealed that the redox-active Y220, which is totally conserved across all AOXs (1), is within 4 Å of the active site. Such a close-range electron transfer position, comparable to that observed in the R2 subunit of ribonucleotide reductase (31), is further support for the suggestion that radicals play a key role in the AOX catalytic cycle (39).
In addition to providing a structural insight into the active site of this enigmatic protein our structures have also revealed the nature of the inhibitor binding site. The binding site of our AF derivative was within 4 Å of not only the diiron center but also Y220 and resulted in some dramatic conformational changes such that H165 moved within ligating distance of Fe (1). CAVER protein analysis software (38) suggested that the inhibitor-binding cavity connects at the diiron center with an additional cavity, which could also serve as a ubiquinone binding site.
In conclusion, we believe that the structures presented in this report will contribute to a more complete understanding of the function and inhibition of all AOXs. It will not only be beneficial for the control of trypanosomiasis and other human diseases, such as cryptosporidiosis and candidiasis, but also for the control of plant diseases caused by phytopathogenic fungi (1, 40).
Materials and Methods
Crystallization.
The oxidized form of alternative oxidase from Trypanosoma brucei brucei was expressed, purified, and crystallized essentially according to the method described previously (23, 24) using 28–34% (wt/vol) PEG 400, 100 mM imidazole buffer (pH 7.4), and 500 mM potassium formate as the reservoir solution. Detailed information is presented in SI Appendix, SI Materials and Methods.
Data Collection and Phasing.
For phasing by the single-wavelength anomalous dispersion (SAD) method, anomalous scattering effects caused by Fe were measured to 3.2 Å resolution. The dataset was processed and scaled with HKL2000 (41). The program SOLVE (42) was used to locate and refine four “diiron sites” (figure of merit = 0.195). The RESOLVE (43) program was used for solvent flattening (figure of merit = 0.645). The resulting electron density map was clear enough to trace the TAO molecules. Initial models were built using RESOLVE (43) and BUCANER (43). Detailed analysis of diffraction data showed that the crystal used for the data collection of Fe-SAD was pseudohemihedral twinning. Amplitude-based twin-refinement using REFMAC5 (45) decreased Rwork/Rfree drastically from 0.307/0.363 to 0.250/0.310. X-ray diffraction data of ligand-free TAO and AF derivatives complex crystals were collected to 2.85, 2.6, and 2.3 Å resolution, respectively. All datasets were processed and scaled with HKL2000 (42). Detail information is presented in SI Appendix, SI Materials and Methods.
Refinement.
The initial model of inhibitor-free TAO was determined by molecular replacement (MR) using the model obtained by SAD (3.2 Å resolution) as a search model. The program Phaser (46) in CCP4i was used for MR. The models of ligand-free TAO and TAO-AF2779OH complex were rebuilt with reference to the well-refined model of the TAO–CCB complex at 2.3 Å resolution. Manual rebuilding and crystallographic refinement of all structures were performed using COOT (47) and REFMAC5 (45). All structures were refined by amplitude-based twin-refinement in REFMAC5 (45) to final Rwork/Rfree values of 0.192/0.247 (twin fraction of 0.476), 0.214/0.256 (twin fraction of 0.552), and 0.185/0.227 (twin fraction of 0.527) for ligand-free TAO, TAO–AF2779OH, and TAO–CCB, respectively. The omit electron density maps of ligand-free TAO, TAO–AF2779OH, and TAO–CCB around helix 5 are shown in SI Appendix, Fig. S14. On average, about 30 residues of N and C termini of TAO were missing as a result of flexibility. Data collection and structural refinement statistics are summarized in SI Appendix, Table S1. Figures showing protein structures were prepared with the graphics program PyMol (www.pymol.org). Detailed information is presented in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank all staff members of beamlines BL44XU and BL41XU at SPring-8, BL17A at the High Energy Accelerator Research Organization Photon Factory for their help with X-ray diffraction data collection. This work was supported in part by Grant-in-Aid for Young Scientists (B) 21790402 (to Y.K.); Grant-in-Aid for Scientific Research (C) 23570131 (to T.S.); Creative Scientific Research Grant 18GS0314 (to K.K.); Grant-in-Aid for Scientific Research on Priority Areas 18073004 (to K.K.) from the Japanese Society for the Promotion of Science and by a grant from the Targeted Proteins Research Program (to T.N., T.A., T.H., A.T., M.I., S.M., S.H., and K.K.) from the Japanese Ministry of Education, Science, Culture, Sports and Technology; a grant-in-aid for research on emerging and reemerging infectious diseases from the Japanese Ministry of Health and Welfare (to K.K.); and by the Programme for Promotion of Basic and Applied Researches for Innovations in Bio-Oriented Industry (S.H. and K.K.). A.L.M. gratefully acknowledges the Biotechnology and Biological Sciences Research Council for financial support. A.L.M. and K.K. acknowledge support from the Prime Minister’s Initiative for International Education fund for collaborative twinning.
Footnotes
The authors declare no conflict of interest.
†This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3VV9 [trypanosomal alternative oxidase (TAO)], 3VVA [TAO-AF2779OH complex], and 3W54 [TAO-colletochlorin B complex]).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218386110/-/DCSupplemental.
References
- 1.Moore AL, Albury MS. Further insights into the structure of the alternative oxidase: From plants to parasites. Biochem Soc Trans. 2008;36(Pt 5):1022–1026. doi: 10.1042/BST0361022. [DOI] [PubMed] [Google Scholar]
- 2.McDonald AE, Vanlerberghe GC. Origins, evolutionary history, and taxonomic distribution of alternative oxidase and plastoquinol terminal oxidase. Comp Biochem Physiol Part D Genomics Proteomics. 2006;1(3):357–364. doi: 10.1016/j.cbd.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Chaudhuri M, Ajayi W, Hill GC. Biochemical and molecular properties of the Trypanosoma brucei alternative oxidase. Mol Biochem Parasitol. 1998;95(1):53–68. doi: 10.1016/s0166-6851(98)00091-7. [DOI] [PubMed] [Google Scholar]
- 4.Roberts CW, et al. Evidence for mitochondrial-derived alternative oxidase in the apicomplexan parasite Cryptosporidium parvum: A potential anti-microbial agent target. Int J Parasitol. 2004;34(3):297–308. doi: 10.1016/j.ijpara.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri M, Ott RD, Hill GC. Trypanosome alternative oxidase: From molecule to function. Trends Parasitol. 2006;22(10):484–491. doi: 10.1016/j.pt.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Phillips MA. Stoking the drug target pipeline for human African trypanosomiasis. Mol Microbiol. 2012;86(1):10–14. doi: 10.1111/mmi.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zíkova A, Schnaufer A, Dalley RA, Panigrahi AK, Stuart KD. The Fo-F1-ATP synthase complex contains novel subunits and is essential for procyclic Trypanosoma brucei. PLoS Pathog. 2009;5:1–15. doi: 10.1371/journal.ppat.1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opperdoes FR, Borst P, Bakker S, Leene W. Localization of glycerol-3-phosphate oxidase in the mitochondrion and particulate NAD+-linked glycerol-3-phosphate dehydrogenase in the microbodies of the bloodstream form to Trypanosoma brucei. Eur J Biochem. 1977;76(1):29–39. doi: 10.1111/j.1432-1033.1977.tb11567.x. [DOI] [PubMed] [Google Scholar]
- 9.Nihei C, Fukai Y, Kita K. Trypanosome alternative oxidase as a target of chemotherapy. Biochim Biophys Acta. 2002;1587(2–3):234–239. doi: 10.1016/s0925-4439(02)00086-8. [DOI] [PubMed] [Google Scholar]
- 10.Minagawa N, et al. An antibiotic, ascofuranone, specifically inhibits respiration and in vitro growth of long slender bloodstream forms of Trypanosoma brucei brucei. Mol Biochem Parasitol. 1997;84(2):271–280. doi: 10.1016/s0166-6851(96)02797-1. [DOI] [PubMed] [Google Scholar]
- 11.Yabu Y, et al. The efficacy of ascofuranone in a consecutive treatment on Trypanosoma brucei brucei in mice. Parasitol Int. 2003;52(2):155–164. doi: 10.1016/s1383-5769(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 12.Yabu Y, et al. Chemotherapeutic efficacy of ascofuranone in Trypanosoma vivax-infected mice without glycerol. Parasitol Int. 2006;55(1):39–43. doi: 10.1016/j.parint.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Andersson ME, Nordlund P. A revised model of the active site of alternative oxidase. FEBS Lett. 1999;449(1):17–22. doi: 10.1016/s0014-5793(99)00376-2. [DOI] [PubMed] [Google Scholar]
- 14.Berthold DA, Andersson ME, Nordlund P. New insight into the structure and function of the alternative oxidase. Biochim Biophys Acta. 2000;1460(2-3):241–254. doi: 10.1016/s0005-2728(00)00149-3. [DOI] [PubMed] [Google Scholar]
- 15.Albury MS, Affourtit C, Moore AL. A highly conserved glutamate residue (Glu-270) is essential for plant alternative oxidase activity. J Biol Chem. 1998;273(46):30301–30305. doi: 10.1074/jbc.273.46.30301. [DOI] [PubMed] [Google Scholar]
- 16.Ajayi WU, Chaudhuri M, Hill GC. Site-directed mutagenesis reveals the essentiality of the conserved residues in the putative diiron active site of the trypanosome alternative oxidase. J Biol Chem. 2002;277(10):8187–8193. doi: 10.1074/jbc.M111477200. [DOI] [PubMed] [Google Scholar]
- 17.Albury MS, Affourtit C, Crichton PG, Moore AL. Structure of the plant alternative oxidase. Site-directed mutagenesis provides new information on the active site and membrane topology. J Biol Chem. 2002;277(2):1190–1194. doi: 10.1074/jbc.M109853200. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura K, et al. Mutational analysis of the Trypanosoma vivax alternative oxidase: The E(X)6Y motif is conserved in both mitochondrial alternative oxidase and plastid terminal oxidase and is indispensable for enzyme activity. Biochem Biophys Res Commun. 2005;334(2):593–600. doi: 10.1016/j.bbrc.2005.06.131. [DOI] [PubMed] [Google Scholar]
- 19.Berthold DA, Voevodskaya N, Stenmark P, Gräslund A, Nordlund P. EPR studies of the mitochondrial alternative oxidase. Evidence for a diiron carboxylate center. J Biol Chem. 2002;277(46):43608–43614. doi: 10.1074/jbc.M206724200. [DOI] [PubMed] [Google Scholar]
- 20.Moore AL, et al. Compelling EPR evidence that the alternative oxidase is a diiron carboxylate protein. Biochim Biophys Acta. 2008;1777(4):327–330. doi: 10.1016/j.bbabio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Nordlund P, Eklund H. Di-iron-carboxylate proteins. Curr Opin Struct Biol. 1995;5(6):758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 22.Berthold DA, Stenmark P. Membrane-bound diiron carboxylate proteins. Annu Rev Plant Biol. 2003;54:497–517. doi: 10.1146/annurev.arplant.54.031902.134915. [DOI] [PubMed] [Google Scholar]
- 23.Kido Y, et al. Purification and kinetic characterization of recombinant alternative oxidase from Trypanosoma brucei brucei. Biochim Biophys Acta. 2010;1797(4):443–450. doi: 10.1016/j.bbabio.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Kido Y, et al. Crystallization and preliminary crystallographic analysis of cyanide-insensitive alternative oxidase from Trypanosoma brucei brucei. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 3):275–278. doi: 10.1107/S1744309109054062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40(Database issue):D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maréchal A, Kido Y, Kita K, Moore AL, Rich PR. Three redox states of Trypanosoma brucei alternative oxidase identified by infrared spectroscopy and electrochemistry. J Biol Chem. 2009;284(46):31827–31833. doi: 10.1074/jbc.M109.059980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guy JE, Whittle E, Kumaran D, Lindqvist Y, Shanklin J. The crystal structure of the ivy Δ4-16:0-ACP desaturase reveals structural details of the oxidized active site and potential determinants of regioselectivity. J Biol Chem. 2007;282(27):19863–19871. doi: 10.1074/jbc.M702520200. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366(6455):537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 29.Jin S, Kurtz DM, Jr, Liu ZJ, Rose J, Wang BC. X-ray crystal structures of reduced rubrerythrin and its azide adduct: A structure-based mechanism for a non-heme diiron peroxidase. J Am Chem Soc. 2002;124(33):9845–9855. doi: 10.1021/ja026587u. [DOI] [PubMed] [Google Scholar]
- 30.Nordlund P, Eklund H. Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J Mol Biol. 1993;232(1):123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt PP, Rova U, Katterle B, Thelander L, Gräslund A. Kinetic evidence that a radical transfer pathway in protein R2 of mouse ribonucleotide reductase is involved in generation of the tyrosyl free radical. J Biol Chem. 1998;273(34):21463–21472. doi: 10.1074/jbc.273.34.21463. [DOI] [PubMed] [Google Scholar]
- 32.Affourtit C, Albury MS, Crichton PG, Moore AL. Exploring the molecular nature of alternative oxidase regulation and catalysis. FEBS Lett. 2002;510(3):121–126. doi: 10.1016/s0014-5793(01)03261-6. [DOI] [PubMed] [Google Scholar]
- 33.McDonald AE. Alternative oxidase: What information can protein sequence comparisons give us? Physiol Plant. 2009;137(4):328–341. doi: 10.1111/j.1399-3054.2009.01242.x. [DOI] [PubMed] [Google Scholar]
- 34.Albury MS, Elliott C, Moore AL. Ubiquinol-binding site in the alternative oxidase: Mutagenesis reveals features important for substrate binding and inhibition. Biochim Biophys Acta. 2010;1797(12):1933–1939. doi: 10.1016/j.bbabio.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402(6757):47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 36.Saimoto H, Kido Y, Haga Y, Sakamoto K, Kita K. Pharmacophore identification of ascofuranone, potent inhibitor of cyanide-insensitive alternative oxidase of Trypanosoma brucei. J Biochem. 2013;153(3):267–273. doi: 10.1093/jb/mvs135. [DOI] [PubMed] [Google Scholar]
- 37.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367(6460):243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 38.Medek P, Benes P, Sochor J. Computation of tunnels in protein molecules using Delaunay triangulation. J WSCG. 2007;15:107–114. [Google Scholar]
- 39.Moore AL, et al. Unraveling the heater – New insights into the structure of the alternative. Annu Rev Plant Biol. 2013 doi: 10.1146/annurev-arplant-042811-105432. in press. [DOI] [PubMed] [Google Scholar]
- 40.Fernández-Ortuño D, Torés JA, de Vicente A, Pérez-García A. Mechanisms of resistance to QoI fungicides in phytopathogenic fungi. Int Microbiol. 2008;11(1):1–9. [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 9):1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.