

Abstract
By binding growth factors (GFs), the ECM tightly regulates their activity. We recently reported that the heparin-binding domain II of fibronectin acts as a promiscuous high-affinity GF-binding domain. Here we hypothesized that fibrin, the provisional ECM during tissue repair, also could be highly promiscuous in its GF-binding capacity. Using multiple affinity-based assays, we found that fibrin(ogen) and its heparin-binding domain bind several GFs from the PDGF/VEGF and FGF families and some GFs from the TGF-β and neurotrophin families. Overall, we identified 15 unique binding interactions. The GF binding ability of fibrinogen caused prolonged retention of many of the identified GFs within fibrin. Thus, based on the promiscuous and high-affinity interactions in fibrin, GF binding may be one of fibrin’s main physiological functions, and these interactions may potentially play an important and ubiquitous role during tissue repair. To prove this role in a gain-of-function model, we incorporated the heparin-binding domain of fibrin into a synthetic fibrin-mimetic matrix. In vivo, the multifunctional synthetic matrix could fully mimic the effect of fibrin in a diabetic mouse model of impaired wound healing, demonstrating the benefits of generating a hybrid biomaterial consisting of a synthetic polymeric scaffold and recombinant bioactive ECM domains. The reproduction of GF–ECM interactions with a fibrin-mimetic matrix could be clinically useful, and has the significant benefit of a more straightforward regulatory path associated with chemical synthesis rather than human sourcing.
Keywords: regenerative medicine, hydrogel, angiogenesis
An important function of the ECM in tissue morphogenesis and repair is sequestration of growth factors (GFs), which control multiple cellular processes. Interactions of GFs and the ECM modulate the partitioning of GFs from the ECM to the soluble phase and thus control their local concentration, diffusion, and signaling (1, 2). For example, many GFs are able to bind the proteoglycan components of the ECM (2). Moreover, it has recently become more evident that ECM proteins such as fibronectin and vitronectin, which do not contain highly negatively charged sugar chains, bind several GFs (3–8). In addition to sequestration of GF by ECM components, the ECM also can modulate cosignaling between adhesion and GF receptor systems. For example, we recently showed that fibronectin domains mediate synergistic signaling between the receptors for VEGF-A, PDGF-BB, and bone morphogenetic protein (BMP)-2 with the integrin α5β1 (9).
Hemostasis in wound healing is achieved initially by the formation of a fibrin clot through the conversion of circulating fibrinogen into a fibrin matrix during blood coagulation (10). Fibrin serves as a provisional ECM for infiltrating cells, and the matrix has been suggested to act as a reservoir for secreted GFs (11). Fibrin(ogen) is known to have specific interactions with FGF-2, VEGF-A165, and insulin-like growth factor binding protein (IGFBP)-3 (12–14). We recently reported that fibronectin, a key adhesion protein found in the blood and in the interstitial ECM, functions as a promiscuous GF-binding protein (3). Through its second heparin-binding domain (FN III12-14), fibronectin binds GFs from the PDGF/VEGF and FGF families and some GFs from the TGF-β and neurotrophin families. Similarly, vitronectin has been shown to bind several GFs, likely through its heparin-binding domain (5, 7). Because the heparin-binding domains of fibronectin and vitronectin bind a wide range of GFs from different families, we hypothesized that fibrin(ogen) could also bind GFs through its heparin-binding domain.
Results
Binding of GF to Fibrinogen.
We first tested the capacity of fibrinogen to bind GFs from different families. GFs from the PDGF/VEGF, FGF, TGF-β, IGF (including IGFBPs), and neurotrophin families were selected. Binding of fibrinogen to absorbed GFs was detected using an antibody against fibrinogen (Fig. 1A). Setting 1 as the highest signal possible, a signal significantly greater than 0.1 was considered to indicate relevant binding. From the PDGF family, VEGF-B, PDGF-AB, PDGF-BB, PDGF-DD, placenta growth factor (PlGF)-2 and PlGF-3 showed binding to fibrinogen, whereas VEGF-A165, VEGF-A121, VEGF-C, PDGF-AA, PDGF-CC, and PlGF-1 did not show relevant binding. From the FGF family, FGF-2, FGF-5, and FGF-7 showed binding to fibrinogen, whereas FGF-1, FGF-4, FGF-6, FGF-8, FGF-9, FGF-10, and FGF-18 did not show strong binding. From the TGF-β superfamily, TGF-β1, TGF-β2, BMP-2, and BMP-2/7 heterodimer showed binding to fibrinogen, but TGF-β3 and BMP-7 did not. None of the IGFs were able to bind fibrinogen directly, although IGFBP-5 demonstrated binding. The neurotrophins neurotrophin-3 (NT-3) and BDNF showed binding to fibrinogen, whereas nerve growth factor (NGF) did not. In addition, EGF, heparin-binding EGF (HB-EGF), and BSA (as a non-GF control) did not show any binding to fibrinogen.
Fig. 1.

GF binding to fibrin(ogen). (A) ELISA plates were coated with GFs or BSA and further incubated with fibrinogen. Bound fibrinogen was detected using an antibody (n ≥ 4; mean ± SEM). A signal significantly greater than 0.1 (gray box) was considered representative of a specific binding. *P < 0.05; **P < 0.01; ***P < 0.001, one-sample Student t test. Binding was highly promiscuous; unique interactions are shown in gray, and previously known interactions are shown in black. (B and C) GF retention in fibrin matrix. Fibrin matrices were produced in the presence of a GF and further incubated in eight volumes of physiological buffer for 7 d. The buffer was changed each day, and released GFs were quantified for each day. (B) Graph showing the cumulative release of GFs over 7 d (n ≥ 3; mean ± SEM). (C) After 7 d, fractions of FGF-2 and PlGF-2 remaining in the matrices were quantified after the gels were digested by plasmin (n = 3; mean ± SEM).
Release of GF from Fibrin Matrix.
We then tested whether a fibrin matrix (clot) could sequester the GFs that showed binding to fibrinogen, considering the GFs VEGF-A121, VEGF-A165, PDGF-BB, PlGF-1, PlGF-2, FGF-2, IGF-I, IGFBP-5, TGF-β1, BMP-2, BDNF, and NGF as examples. Fibrinogen solutions containing GF were polymerized to form fibrin matrix using thrombin and factor XIII. The resulting matrix was then incubated in an excess of physiological buffer that was changed each day, and GF release from the matrix was monitored by ELISA (Fig. 1B and Fig. S1). As expected, the GFs that did not show strong binding to fibrinogen—VEGF-A165, VEGF-A121, PlGF-1, IGF1, and NGF—were quickly released from the fibrin matrix (>85% released after 1 d) (Fig. S1). In contrast, the several GFs that showed binding to fibrinogen—PlGF-2, FGF-2, BMP-2, TGF-β1, IGFBP-5, and BDNF—showed retention within fibrin matrix (Fig. 1B). Only PDGF-BB did not exhibit strong retention (>80% released after 1 d). Interestingly, PlGF-2 and FGF-2 showed very strong retention within the fibrin matrix, with only 12.3% and 32.5% released after 7 d, respectively.
To ensure that the apparent slow release of FGF-2 and PlGF-2 was not related to an issue with their detection, the matrices were digested after 7 d by plasmin to release fibrin-bound GFs. After digestion, 68.9% of FGF-2 and 85.6% of PlGF-2 were still detected (Fig. 1C), confirming mass balance and thus the very strong interaction of these GFs with fibrin.
Binding of GF to the Heparin-Binding Domain of Fibrin(ogen).
Because the heparin-binding domains of fibronectin and vitronectin bind GFs (3, 7), we hypothesized that the heparin-binding domain of fibrin(ogen) could bind GFs as well. We recombinantly produced the heparin-binding domain of fibrin(ogen) (15), located at the N terminus of the fibrin(ogen) β chain (Fg β). The fragment, mimicking the situation in fibrin, starts just after the cleavage site of thrombin (Fg β15–66). Because the heparin-binding domain of fibrin(ogen) is naturally displayed as a dimer (15), the fragment was dimerized at the cysteine residue at position 65 (Fg β15–66(2)) (Fig. 2 A and B). Moreover, for detection purposes, a 6X His tag at the C terminus was added. Binding of the fibrinogen fragment to the absorbed GFs from the different families was detected using an antibody against the tag. The GFs that showed binding to fibrin(ogen)—PlGF-2, PDGF-BB, PDGF-DD, FGF-2, TGF-β1, TGF-β2, BMP-2, IGFBP-5, NT-3, and BDNF—showed binding to Fg β15–66(2) (Fig. 2C). GFs that did not show strong binding to fibrinogen—VEGF-A165, VEGF-A121, PlGF-1, PDGF-CC, FGF-1, FGF-6, FGF-10, TGF-β3, BMP-7, IGF-I, IGF-II, IGFBP-1, IGFBP-3, NGF, and EGF—did not show relevant binding to Fg β15–66(2) (Fig. 2C). Although FGF-7 and TGF-β2 showed relevant binding to fibrinogen, their binding to Fg β15–66(2) was more modest.
Fig. 2.
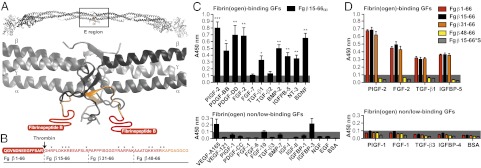
GF binding to the heparin-binding domain of fibrin(ogen). (A) Ribbon diagram representation of human fibrinogen and its central region E (generated using Swiss-PdbViewer). The coiled-coil domains are in gray; the N terminus portions of the Bβ chain are in yellow (Fg β59–66), and the N terminus parts of the Bβ chain missing in the crystal structure are in red (Fg β1–58). Fibrinopeptide B is highlighted in red (Fg β1–14). (B) Amino acid sequences of the fibrin(ogen) fragments (3). The region within Fg β1–66 with unknown tertiary structure is in red, and the known tertiary structure is in orange. The arrow indicates the thrombin cleavage site to remove fibrinopeptide B, and the stars indicate lysine and arginine residues. (C and D) GF binding to fibrinogen fragments. ELISA plates were coated with GFs or BSA and further incubated with fibrin(ogen) fragments. Bound fibrin(ogen) fragments were detected using an antibody against the tag (n ≥ 4; mean ± SEM). A signal significantly greater than 0.1 (gray box) was considered representative of a relevant binding. For D: *P < 0.05; **P < 0.01; ***P < 0.001, one-sample Student t test.
To further examine the interaction of GFs with the heparin-binding domain of fibrin(ogen), we generated four other monomeric variants (Fig. 2B) and tested them with GFs representative of each family (PlGF-1, PlGF-2, FGF-1, FGF-2, TGF-β1, TGF-β3, IGFBP-4, and IGFBP-5) (Fig. 2D). A fragment mimicking the situation in fibrinogen contained the fibrinopeptide (Fg β1–66); two other fragments were truncations from the N terminus (Fg β31–66 and Fg β48–66). In addition, we generated a variant in which all lysine and arginine residues were substituted with serine (Fg β15–66*S). We found that dimerization of Fg β15–66 improved the binding of GFs (Fig. S2). Fg β1–66 showed similar binding to GFs as Fg β15–66 (Fig. 2D). Moreover, Fg β31–66 displayed a significant GF-binding activity, whereas Fg β48–66 and Fg β15–66*S did not (Fig. 2D).
Specificity of binding was then addressed by competing the binding of GFs to absorbed fibrinogen with soluble Fg β15–66(2). Binding of the GFs (PlGF-2, FGF-2, TGF-β1, and IGFBP-5) to absorbed fibrinogen was detected using an antibody against the GF (Fig. S3). For all of the GFs tested, we could observe a dose-dependent competition. IC50 values of 6.6 nM for PlGF-2, 19.1 nM for FGF-2, 19.6 nM for TGF-β1, and 18.8 nM for IGFBP-5.
Affinity of GF for Fg β15–66(2).
Surface plasmon resonance (SPR) was used to estimate the affinity toward fibrinogen of selected GFs from the foregoing list. SPR chips were functionalized with Fg β15–66(2), and GFs chosen from each family (i.e., PlGF-2, TGF-β1, FGF-2, and IGFBP-5) were analyzed. The curves obtained for the specific binding signal to Fg β15–66(2) were fitted with Langmuir binding kinetics, and dissociation constants (KD), and rate constants (kon and koff) were calculated from the fits (Fig. S4 A and B). KD values were 1.9 nM for PlGF-2, 56.5 nM for TGF-β1, 53.0 nM for FGF-2, and 19.6 nM for IGFBP-5.
Influence of Heparin on Binding of GF to Fg β15–66(2).
Because both Fg β15–66(2) and heparin-binding GFs bind heparin, we explored the potential role of heparin in modulating their interaction. The GFs VEGF-A165, PlGF-2, PDGF-BB, PDGF-DD, FGF-7, FGF-10, TGF-β1, TGF-β2, BMP-2, IGFBP-3, IGFBP-5, NT-3, and BDNF were adsorbed on an ELISA plate, followed by incubation with Fg β15–66(2) with or without heparin in excess (20-fold molar excess relative to Fg β15–66(2)) (Fig. S5A). With heparin in excess, binding to the GF was decreased for VEGF-A165, FGF-2, FGF-7, FGF-10, TGF-β1, TGF-β2, IGFBP-3, IGFBP-5, and NT-3; increased slightly for PDGF-BB, BMP-2, and BDNF; and did not differ for PlGF-2 and PDGF-DD. In addition, a competition assay was performed using a range of heparin concentrations (10- to 10,000-fold molar excess relative to Fg β15–66(2)) with selected GFs from each family: PlGF-2, TGF-β1, FGF-2, and IGFBP-5 (Fig. S5B). IC50 values were 9.4 μM for PlGF-2, 8.4 μM for FGF-2, 0.7 μM for TGF-β1, and 2.0 μM for IGFBP-5.
Influence of Fg β15–66(2) on GF Activity.
We explored the potential modulation of the activity of GFs in the presence of Fg β15–66(2) using a cell proliferation assay (Fig. S6). Proliferation of human endothelial cells was induced with solution of GFs (VEGF-A165, VEGF-A121, PlGF-1, PlGF-2, or FGF-2) containing fibrinogen or Fg β15–66(2) in excess (≥100-fold molar excess relative to GFs). Cells stimulated with GFs were significantly increased in number, but no significant differences were found between cells treated with GFs only and cells treated with GFs plus fibrinogen or Fg β15–66(2), suggesting that the binding of GFs to fibrin(ogen) does not modulate their activity in vitro.
Incorporation of Fg β15–66(2) into a Fibrin-Mimetic Matrix to Treat Chronic Wounds.
Based on the promiscuous but high-affinity interactions of GFs with fibrin(ogen) and their subsequent retention in fibrin matrices, GF binding may be an important physiological function of fibrin, and these interactions may possibly play an important role during tissue repair. To investigate this function, we incorporated Fg β15–66(2) into a multifunctional poly(ethylene glycol) (PEG) matrix designed to mimic fibrin’s main functions. The fibrin-mimetic matrix backbone comprised two eight-arm PEG peptide conjugates (at a concentration of 1.75%) cross-linkable by the fibrin stabilizing factor, transglutaminase factor XIIIa (16, 17). PEG was conjugated with a peptide containing a factor XIIIa substrate sequence derived from α2-plasmin inhibitor (α2PI1–8, NQEQVSPL) (18) or with a lysine donor peptide containing a substrate sequence for matrix metalloproteases (MMPs) and plasmin (VPMSMRGG) (19), allowing degradation of the matrix in situ. During polymerization by factor XIIIa, the matrix was further functionalized with a peptide containing a promiscuous integrin-binding sequence (α2PI1–8-RGDSPG; 40 μM) that promote cell adhesion and with a cross-linkable version of Fg β15–66(2) (α2PI1–8-Fg β15–66(2); 10 μM), to allow binding of GFs (Fig. 3A). As examples of GFs, we chose FGF-2 and PlGF-2, because these GFs showed the strongest interactions with fibrin(ogen) and Fg β15–66(2), and because they are known to promote wound healing (20, 21). The fibrin matrix (Fig. 1B) and the fibrin-mimetic matrix functionalized with Fg β15–66(2) were both able to sequester FGF-2 and PlGF-2, and after degradation by plasmin, GFs could be completely released (Figs. 1C and 3B).
Fig. 3.
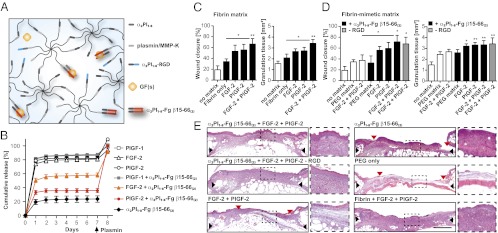
Fibrin-mimetic matrix. (A) The matrix comprises two eight-arm PEG-peptide conjugates (PEG-plasmin/MMP-K and PEG-α2PI1–8), a cell-adhesion peptide (α2PI1–8-RGD), α2PI1–8-Fg β15–66(2), and GF(s). Through the transglutaminase factor XIIIa, peptide bounds are formed between the first glutamine in α2PI1–8 (NQEQVSPL) and the lysine in PEG-plasmin/MMP-K, resulting in simultaneous cross-linking and functionalization of the matrix. (B) Retention of fibrin-binding GFs in fibrin-mimetic matrix functionalized with α2PI1–8-Fg β15–66(2). The graph shows the cumulative release of GFs and α2PI1–8-Fg β15–66(2) over 7 d. Fractions of GFs remaining in the matrices were quantified after the matrix was digested by plasmin (n = 3; mean ± SEM). (C–E) Delivering GFs within the fibrin-mimetic matrix functionalized with α2PI1–8-Fg β15–66(2) enhances skin wound healing in diabetic mice. Full-thickness back skin wounds were treated with FGF-2 and/or PlGF-2 (200 ng of each per wound). Twelve groups were tested: no treatment (no matrix); fibrin only or fibrin containing FGF-2 and/or PlGF-2; PEG matrix (with plasmin/MMP-sensitive and RGD peptides) or PEG matrix containing FGF-2 and PlGF-2; PEG matrix functionalized with α2PI1–8-Fg β15–66(2) or PEG matrix functionalized with α2PI1–8-Fg β15–66(2) containing FGF-2 and/or PlGF-2; and PEG matrix functionalized with α2PI1–8-Fg β15–66(2) containing FGF-2 and PlGF-2 but without RGD (−RGD). (C and D) After 10 d, wound closure and granulation tissue area were evaluated by histological analysis. All points are mean ± SEM (n = 8 per matrix). Statistical comparisons were done using ANOVA with Tukey’s test. *P < 0.05; **P < 0.01. (E) Representative histology (H&E staining). Black arrows indicate wound edges; red arrows indicate tips of epithelium tongue. The granulation tissue, stained in pink-violet, is characterized by a large number of cells (granulocytes) with nuclei that stain in dark violet. Muscle under the wounds is stained in red. Fat tissue appears as transparent bubbles. (Scale bar: 1 mm.) To the right are higher-magnification (5×) views of the granulation tissue.
We then evaluated the synthetic matrix in vivo. We used the db/db mouse, which is a genetic mouse model of diabetes that provides a well-established and clinically relevant experimental system of impaired wound healing (22, 23). We treated full-thickness back skin wounds of these mice with GFs (FGF-2 and PlGF-2, 200 ng of each) delivered by a fibrin matrix or by the fibrin-mimetic matrix. Twelve groups were tested: no treatment (no matrix); fibrin only; fibrin containing FGF-2 and/or PlGF-2; PEG matrix only, PEG matrix containing FGF-2 and PlGF-2, PEG matrix functionalized with α2PI1–8-Fg β15–66(2), PEG matrix functionalized with α2PI1–8-Fg β15–66(2) containing FGF-2 and/or PlGF-2, and PEG matrix functionalized with α2PI1–8-Fg β15–66(2) containing FGF-2 and PlGF-2 without the integrin-binding sequence (RGD). Wounds were analyzed and compared with no treatment after 10 d, considering that wounds are normally fully closed after 15 d when treated only with fibrin matrix (9). The wounds treated with matrices that did not contain GFs or Fg β15–66(2) did not differ significantly from wounds without treatment, in either the amount of granulation tissue or extent of wound closure (the latter indicated by reepithelialization). With the fibrin-mimetic matrix lacking Fg β15–66(2), dual delivery of FGF-2 and PlGF-2 resulted in only slightly improved wound healing (Fig. 3 D and E). In contrast, delivering FGF-2 or PlGF-2 with the fibrin-mimetic matrix functionalized with Fg β15–66(2) or with the fibrin matrix led to faster wound closure and significantly more granulation tissue (Fig. 3 C–E). In addition, the delivery of both FGF-2 and PlGF-2 by fibrin or by the fibrin-mimetic matrix functionalized with Fg β15–66(2) led to significantly faster wound closure and increased development of granulation tissue (Fig. 3 C–E).
Because angiogenesis is a crucial step in sustaining newly formed granulation tissue within the wound bed (10), we focused on the extent to which angiogenesis differed among the treatments. We detected the standard marker for blood vessels, CD31, which is highly expressed by endothelial cells, although other cells within the granulation tissue, including platelets and some other immune cells, also express CD31 to some extent. In addition, we detected desmin as a specific marker for smooth muscle cells surrounding blood vessels. Immunohistological analysis for CD31 and desmin revealed that angiogenesis within the granulation tissues was much more pronounced when GFs were delivered within fibrin or the fibrin-mimetic matrix functionalized with Fg β15–66(2) (Fig. 4 A–C and Fig. S7). Smooth muscle (i.e., desmin-positive) cells were observed to be generally colocalized with endothelial (i.e., CD31+) cells (Fig. 4 A–C and Fig. S7 A and B), confirming that the structures observed were blood vessels and suggesting that they were mature and stable at day 10.
Fig. 4.
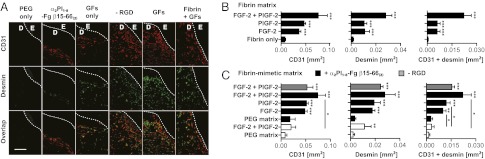
Angiogenesis within the granulation tissue. Angiogenesis was revealed by staining for endothelial (CD31+) cells and smooth muscle (desmin-positive) cells. (A) Representative images are shown. GFs, FGF-2 + PlGF-2. E, epidermis; D, dermis. The hashed line indicates the basement membrane. (Scale bar: 0.2 mm.) (B and C) The graphs show quantification of stained areas for CD31 and desmin as well as the overlay (n ≥ 4; mean ± SEM). *P < 0.05; **P < 0.01; ***P < 0.001, Student t test.
Discussion
During morphogenesis and tissue healing, interactions of GFs with the ECM are critical to regulation of their partitioning, local diffusion, and signaling (1, 2, 9). In tissue healing, the provisional fibrin matrix is supposed to act as a GF reservoir (11), and fibrin(ogen) has been shown to bind to FGF-2 (14, 24), VEGF-A165 (13), and IGFBP-3 (12). Given our recent finding that the second heparin-binding domain of fibronectin acts as a promiscuous GF-binding domain (3), we explored the possibility that fibrin(ogen) could also bind GFs very promiscuously through its heparin-binding domain.
We found that many GFs from the PDGF/VEGF family, FGF family, and TGF-β superfamily, as well as some GFs from the neurotrophin family, bind fibrinogen (Fig. 1A). To graphically illustrate the numerousfindings from this investigation, Fig. 1A shows the interactions reported herein in gray and previously known interactions with fibrinogen in black. Importantly, the lack of signal shown by some GFs likely was not related to poor coating efficiency on the ELISA plate surface; for example, we previously reported no difference in coating efficiency between VEGF-A165 and VEGF-A121 or among the PlGFs (3).
We then examined whether GFs were also able to bind fibrin and, if so, whether the binding was sufficiently strong for the sequestering of GFs into the matrix. As expected, the GFs that did not bind fibrinogen were quickly released from fibrin matrix, whereas the GFs that did show binding were retained (Fig. 1B and Fig. S1). Interestingly, the strength of retention varied among the GFs. FGF-2 and PlGF-2 released very slowly, showing retention of ∼70% and ∼85% after 7 d, respectively. This strong retention correlated with the earlier binding experiment, with FGF-2 and PlGF-2 showing the highest signals for binding to fibrinogen. BDNF, TGF-β1, BMP-2, and IGFBP-5 showed high binding to fibrinogen, as well as retention in fibrin matrix. Interestingly, PDGF-BB showed binding to fibrinogen, but its release from the matrix was relatively rapid (∼80% after 1 d). This difference may be explained by the fact that the ELISA used for the screening of GF binding to fibrinogen was limited to revealing interactions occurring in the solid phase. Thus, it is possible that some interactions were slightly overevaluated or underevaluated compared with those occurring in the physiologically relevant release assay (Fig. 1B).
We then examined whether the heparin-binding domain of fibrin(ogen) (Fig. 2 A and B) could act as a GF-binding domain. Among the GFs tested, all GFs that showed relevant binding to fibrin(ogen) also showed binding to Fg β15–66(2) (Fig. 2C). Only FGF-7 and TGF-β2 showed more modest binding to the fibrin fragment. From a structure-function standpoint, we found that mimicking the situation within fibrin(ogen) through the dimerization of Fg β15–66 improves GF binding (Fig. S2). The presence of the fibrinopeptide (i.e., in Fg β1–66) did not change GF binding, suggesting that fibrin and fibrinogen have similar affinity for GFs (Fig. 2D). In addition, because positively charged amino acids within the second heparin-binding domain of fibronectin are known to be involved in the binding of GFs (3), we generated a variant in which all lysine and arginine residues were substituted with serine (Fg β15–66*S). As expected, we found that lysines and arginines within Fg β15–66 are determinant for GF binding, given that their substitution abolished the binding of GFs (Fig. 2D). Moreover, the sequence PAPPPISGGGYRARPAK (Fg β31–47) is critical for GF binding, considering that Fg β31–66 showed significant GF-binding activity, whereas Fg β48–66 did not (Fig. 2D).
We further tested if Fg β15–66(2) could inhibit the binding of GFs to absorbed fibrinogen, testing one GF per family. IC50 values were in the nM range, suggesting that the main GF-binding site within fibrin(ogen) is its heparin-binding domain (Fig. S3). Nevertheless, considering the size of fibrin(ogen) and its complexity, it is possible that other GF-binding sites are present as well.
We estimated the affinity of GFs to Fg β15–66(2) using SPR, measuring the affinity of one GF per family. Remarkably, all GFs tested had equilibrium dissociation constants in the nM range, indicating that the interactions are very strong (Fig. S4). Indeed, the affinities reported here are in the same range as those that have been reported for the binding of FGF-2 to fibrinogen (25). Similar to the binding to fibronectin (3), this broad yet high affinity is somewhat perplexing; very high affinities would be expected to be highly specific, but in this case the high-affinity interaction also appears to be promiscuous. Interestingly, most of the GFs that have been shown to bind fibronectin (3) also bind fibrinogen, suggesting a similar binding mechanism. Only PDGF-AA, FGF-18, and BMP-7 showed specific binding to fibronectin, but not to fibrinogen. Although the affinity values obtained with SPR are on the same order of magnitude, the affinity of GFs for fibronectin seems to be higher, except for PlGF-2, which displays a similar affinity for both ECM proteins (Fig. S4 B and C).
Interestingly, all GFs that were shown to bind fibrin(ogen) through Fg β15–66(2) can bind or are predicted to bind heparin. However, some known heparin-binding GFs, including PDGF-AA, FGF-1, FGF-6, FGF-18, IGF-II, BMP-7, TGF-β3, and NGF (26), did not show binding to fibrinogen. Thus, the presence of a heparin-binding domain within the GFs seems to be necessary, but not sufficient, to provide binding to Fg β15–66(2). We and others have shown enhanced binding of GFs to fibronectin in the presence of heparin, although heparin is not the linker between the two molecules (3, 4, 27). It has been suggested that heparin and heparan sulfate can change the conformation of fibronectin and increase its affinity for GFs (4, 27); however, in the case of GF–Fg β15–66(2) interaction, heparin did not increase GF-binding ability, but rather decreased it (Fig. S5). Thus, the mechanism of GF binding to fibronectin, fibrinogen, or heparan sulfate proteoglycans appears to be complex, and may be resolved by further crystallographic studies of each complex.
The binding of fibrinogen and Fg β15–66(2) to GFs could influence the activity of GF with their respective receptors. For example, synergistic signaling with integrins could occur, as is the case with fibronectin (9), or the binding could inhibit interaction with the receptor and reduce GF activity. To determine whether the binding has an effect on GF signaling, we induced endothelial cell proliferation with fibrin(ogen)-binding GFs with or without Fg β15–66(2) or fibrinogen. Fibrinogen and Fg β15–66(2) slightly increased the proliferative effect of the GFs tested, but did not promote any synergy (Fig. S6), as has been reported for fibronectin with VEGF-A165 (9). Based on these results, we can conclude that fibrin(ogen) binds but does not modulate the activity of these GFs in vitro.
The binding of GF to fibrin(ogen) may be very important for wound healing processes. Platelets and macrophages within the fibrinous matrix provide a continuous source of GFs necessary to stimulate angiogenesis and other healing processes. By binding the newly secreted GFs, fibrin could act as a GF reservoir while also creating biochemical gradients required for appropriate cell infiltration into the lesion. In addition, other ECM proteins present in the fibrin clot, such as fibronectin and vitronectin, bind GFs and act as bridging molecules between cells and fibrin by binding to the integrins on endothelial cells, smooth muscle cells, and other cell types (28). Moreover, GFs critical for angiogenesis during wound healing, such as FGF-2, bind fibrin(ogen) and fibronectin (3).
To evaluate the GF-binding function of fibrin, we implanted Fg β15–66(2) into a synthetic PEG matrix mimicking fibrin’s main characteristics (Fig. 3A). As such, the PEG matrix was designed to contain a promiscuous integrin-binding sequence to provide adhesion to multiple cell types, an optimized MMP and plasmin degradation-sensitive sequence (19) to allow rapid degradation of the matrix, and Fg β15–66(2) as a GF-binding domain. Furthermore, the biosynthetic matrix can be polymerized in situ using the fibrin-stabilizing factor, factor XIIIa. As expected, similar to fibrin matrix, the fibrin-mimetic matrix functionalized with α2PI1–8-Fg β15–66(2) was able to sequester fibrin(ogen)-binding GFs, such as FGF-2 and PlGF-2 (Fig. 3B). Then, because preclinical evaluations of GFs for chronic skin wound healing are generally performed in rodents, most commonly in db/db diabetic mice (9), we used that model to investigate wound healing by FGF-2 and PlGF-2 delivered by fibrin or by the fibrin-mimetic matrix. We chose FGF-2 and PlGF-2 because they strongly bind Fg β15–66(2), and because both of these GFs are known to improve wound healing (20, 21). Using this gain-of-function model, we sought to explore the extent to which inclusion of Fg β15–66(2) into the matrix could recapitulate the function of fibrin in vivo. We found that effective sequestration and presentation of FGF-2 and/or PlGF-2 by α2PI1–8-Fg β15–66(2) could improve neovessel formation that sustains the newly formed granulation tissue (Fig. 4 A–C and Fig. S7). Granulation tissue morphogenesis translated to improved morphogenesis at the level of the dermal epithelium, as reflected by faster wound reepithelialization and closure (Fig. 3 C–E). As such, the fibrin-mimetic matrix could fully recapitulate fibrin’s efficacy in promoting wound healing by binding FGF-2 and PlGF-2.
To determine whether the RGD sequence is critical, and to simplify the fibrin-mimetic matrix, we removed the integrin-binding peptide in the matrix that demonstrated the best healing. Surprisingly, the delivery of FGF-2 and PlGF-2 with or without RGD resulted in very similar healing (Fig. 3 D and E), with only slightly lower angiogenesis without RGD (Fig. 4C), although the difference was not statistically significant. Thus, the RGD sequence could be removed when designing fibrin-mimetic matrices for skin wound healing. Nevertheless, this finding does not mean that ligation of the appropriate intergins cannot enhance skin repair. Indeed, although promiscuous, RGD cannot engage critical integrins for wound healing, such as integrin α5β1 (9). Additional functionalization of the fibrin-mimetic matrix with integrin-specific fibronectin fragments possibly could further enhance wound healing (9).
Clinically available fibrin is widely used as a sealant and has the potential for use as an efficient GF-delivery system for multiple purposes (29). Of special interest, we show here that an impressive number of GFs strongly bind this material. The substitution of fibrin with a synthetic matrix would have numerous benefits, however. The tailorability of synthetic materials allows greater control of cell–matrix interactions, such as the matrix degradation rate and matrix stiffness, both of which are critical in driving tissue repair or regeneration (29). Thus, this flexibility can fill unmet needs in regenerative medicine by providing a specific and more controlled environment. Moreover, unlike fibrin, which is purified from human plasma, a synthetic fibrin-mimetic matrix benefits strongly from a more straightforward regulatory path associated with chemical synthesis rather than human sourcing.
In conclusion, we found that fibrin(ogen) promiscuously binds GFs from the PDGF, FGF, TGF-β, and neurotrophin families through its heparin-binding domain. Overall, 15 unique binding interactions were established, and all of the GFs evaluated displayed KD values in the nM range. Therefore, fibrin(ogen) could play a broad role in GF binding in the fibrin clot. Finally, by engineering a fibrin-mimetic matrix displaying the GF-binding property of fibrin, we have demonstrated that the binding of GFs to fibrin is critical for wound healing. Furthermore, we have shown that reproduction of the GF–ECM interaction within nonhuman-derived biomaterials could be clinically useful. The synthetic fibrin-mimetic matrix could substitute fibrin for regenerative medicine applications, and, moreover, Fg β15–66(2) could be incorporated into other biomaterials to support GF presentation.
Materials and Methods
Detailed information is provided in SI Materials and Methods. GF binding to fibrin(ogen) and Fg β15–66(2) by ELISA and SPR. Synthesis of the fibrin-mimetic matrix. Wound healing model and analyses of wound tissue sections.
Supplementary Material
Acknowledgments
We thank the staff at the Proteomics and Histology Core Facilities of Ecole Polytechnique Fédérale de Lausanne, as well as Miriella Pasquier, Xavier Quaglia, Céline Dessibourg, and Matteo T. Degiacomi for technical assistance and Jeffrey J. Rice for fruitful discussions and helpful comments. This work was funded in part by the European Community’s Seventh Framework Programme (Project Angioscaff NMP-LA-2008-214402) and the Swiss National Science Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221602110/-/DCSupplemental.
References
- 1.Schultz GS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17(2):153–162. doi: 10.1111/j.1524-475X.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 2.Macri L, Silverstein D, Clark RA. Growth factor binding to the pericellular matrix and its importance in tissue engineering. Adv Drug Deliv Rev. 2007;59(13):1366–1381. doi: 10.1016/j.addr.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Martino MM, Hubbell JA. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010;24(12):4711–4721. doi: 10.1096/fj.09-151282. [DOI] [PubMed] [Google Scholar]
- 4.Smith EM, Mitsi M, Nugent MA, Symes K. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. Proc Natl Acad Sci USA. 2009;106(51):21683–21688. doi: 10.1073/pnas.0902510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kricker JA, Towne CL, Firth SM, Herington AC, Upton Z. Structural and functional evidence for the interaction of insulin-like growth factors (IGFs) and IGF binding proteins with vitronectin. Endocrinology. 2003;144(7):2807–2815. doi: 10.1210/en.2002-221086. [DOI] [PubMed] [Google Scholar]
- 6.Wijelath ES, et al. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91(1):25–31. doi: 10.1161/01.res.0000026420.22406.79. [DOI] [PubMed] [Google Scholar]
- 7.Schoppet M, Chavakis T, Al-Fakhri N, Kanse SM, Preissner KT. Molecular interactions and functional interference between vitronectin and transforming growth factor-beta. Lab Invest. 2002;82(1):37–46. doi: 10.1038/labinvest.3780393. [DOI] [PubMed] [Google Scholar]
- 8.Gui YT, Murphy LJ. Insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) binds to fibronectin (FN): Demonstration of IGF-I/IGFBP-3/fn ternary complexes in human plasma. J Clin Endocrinol Metab. 2001;86(5):2104–2110. doi: 10.1210/jcem.86.5.7472. [DOI] [PubMed] [Google Scholar]
- 9.Martino MM, et al. Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Sci Transl Med. 2011;3(100):100ra189. doi: 10.1126/scitranslmed.3002614. [DOI] [PubMed] [Google Scholar]
- 10.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 11.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3(8):1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 12.Campbell PG, Durham SK, Hayes JD, Suwanichkul A, Powell DR. Insulin-like growth factor-binding protein-3 binds fibrinogen and fibrin. J Biol Chem. 1999;274(42):30215–30221. doi: 10.1074/jbc.274.42.30215. [DOI] [PubMed] [Google Scholar]
- 13.Sahni A, Francis CW. Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood. 2000;96(12):3772–3778. [PubMed] [Google Scholar]
- 14.Sahni A, Odrljin T, Francis CW. Binding of basic fibroblast growth factor to fibrinogen and fibrin. J Biol Chem. 1998;273(13):7554–7559. doi: 10.1074/jbc.273.13.7554. [DOI] [PubMed] [Google Scholar]
- 15.Yakovlev S, Gorlatov S, Ingham K, Medved L. Interaction of fibrin(ogen) with heparin: Further characterization and localization of the heparin-binding site. Biochemistry. 2003;42(25):7709–7716. doi: 10.1021/bi0344073. [DOI] [PubMed] [Google Scholar]
- 16.Ehrbar M, et al. Enzymatic formation of modular cell-instructive fibrin analogs for tissue engineering. Biomaterials. 2007;28(26):3856–3866. doi: 10.1016/j.biomaterials.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 17.Ehrbar M, et al. Biomolecular hydrogels formed and degraded via site-specific enzymatic reactions. Biomacromolecules. 2007;8(10):3000–3007. doi: 10.1021/bm070228f. [DOI] [PubMed] [Google Scholar]
- 18.Schense JC, Hubbell JA. Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjug Chem. 1999;10(1):75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]
- 19.Patterson J, Hubbell JA. SPARC-derived protease substrates to enhance the plasmin sensitivity of molecularly engineered PEG hydrogels. Biomaterials. 2011;32(5):1301–1310. doi: 10.1016/j.biomaterials.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 20.Cianfarani F, et al. Placenta growth factor in diabetic wound healing: Altered expression and therapeutic potential. Am J Pathol. 2006;169(4):1167–1182. doi: 10.2353/ajpath.2006.051314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83(3):835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan SR, et al. Validation of a model for the study of multiple wounds in the diabetic mouse (db/db) Plast Reconstr Surg. 2004;113(3):953–960. doi: 10.1097/01.prs.0000105044.03230.f4. [DOI] [PubMed] [Google Scholar]
- 23.Davidson JM. Animal models for wound repair. Arch Dermatol Res. 1998;290(Suppl):S1–S11. doi: 10.1007/pl00007448. [DOI] [PubMed] [Google Scholar]
- 24.Sahni A, Khorana AA, Baggs RB, Peng H, Francis CW. FGF-2 binding to fibrin(ogen) is required for augmented angiogenesis. Blood. 2006;107(1):126–131. doi: 10.1182/blood-2005-06-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng H, et al. Identification of a binding site on human FGF-2 for fibrinogen. Blood. 2004;103(6):2114–2120. doi: 10.1182/blood-2003-08-2638. [DOI] [PubMed] [Google Scholar]
- 26.Bernfield M, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 27.Mitsi M, Forsten-Williams K, Gopalakrishnan M, Nugent MA. A catalytic role of heparin within the extracellular matrix. J Biol Chem. 2008;283(50):34796–34807. doi: 10.1074/jbc.M806692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. doi: 10.1111/j.1538-7836.2006.01861.x. [DOI] [PubMed] [Google Scholar]
- 29.Rice JJ, et al. Engineering the regenerative microenvironment with biomaterials. Adv Healthc Mater. 2013;2(1):57–71. doi: 10.1002/adhm.201200197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.