

Abstract
Epigenetic mechanisms are involved in the pathophysiology of depressive disorders and are unique potential targets for therapeutic intervention. The acetylating agent L-acetylcarnitine (LAC), a well-tolerated drug, behaves as an antidepressant by the epigenetic regulation of type 2 metabotropic glutamate (mGlu2) receptors. It caused a rapid and long-lasting antidepressant effect in Flinders Sensitive Line rats and in mice exposed to chronic unpredictable stress, which, respectively, model genetic and environmentally induced depression. In both models, LAC increased levels of acetylated H3K27 bound to the Grm2 promoter and also increased acetylation of NF-ĸB-p65 subunit, thereby enhancing the transcription of Grm2 gene encoding for the mGlu2 receptor in hippocampus and prefrontal cortex. Importantly, LAC reduced the immobility time in the forced swim test and increased sucrose preference as early as 3 d of treatment, whereas 14 d of treatment were needed for the antidepressant effect of chlorimipramine. Moreover, there was no tolerance to the action of LAC, and the antidepressant effect was still seen 2 wk after drug withdrawal. Conversely, NF-ĸB inhibition prevented the increase in mGlu2 expression induced by LAC, whereas the use of a histone deacetylase inhibitor supported the epigenetic control of mGlu2 expression. Finally, LAC had no effect on mGlu2 knockout mice exposed to chronic unpredictable stress, and a single injection of the mGlu2/3 receptor antagonist LY341495 partially blocked LAC action. The rapid and long-lasting antidepressant action of LAC strongly suggests a unique approach to examine the epigenetic hypothesis of depressive disorders in humans, paving the way for more efficient antidepressants with faster onset of action.
Keywords: BDNF, histone acetylation, MDD, glutamatergic neurotransmission, chromatin
Major depression disorder (MDD) is a severe debilitating medical and life-threatening disorder that displays a variety of psychopathological symptoms, such as anhedonia, dysphoria, irritability, sleep disturbances, abnormalities in appetite and sleep, psychomotor and thinking disturbances with excessive worrying, guilt, and possibly suicidal ideation that carries both a substantial personal burden as well as a social one. As yet, approximately 40% of patients are considered treatment resistant, and all current antidepressants require at least 2–3 wk to significantly improve mood, leaving severely ill patients at risk for suicide in the early phases of treatment (1) and more likely to develop coronary artery disease, type 2 diabetes, and Alzheimer’s disease (2). This highlights the need for novel drugs with a faster onset of action.
Epigenetics is increasingly being implicated in the pathophysiology of major depression and other psychiatric disorders (3, 4). Stochastic or environmental factor-induced chromatin remodeling, resulting from modifications of histone proteins or DNA, without changes in the DNA sequence itself, may lead to faults of gene expression by altering the accessibility of genes to transcription factors. Hence, a clearer understanding of the above-described “epigenetic hypothesis” of major depression may be a unique potent therapeutic target that overcomes the limitations of common antidepressants, resetting the proper expression of multiple genes that are involved in the pathophysiology of depression.
Previous findings demonstrate that chronic defeat stress in mice, which models the pathogenesis of depressive-like states, leads to a long-lasting down-regulation of BDNF in the hippocampus, which is caused by a suppressive histone H3 methylation (5). In addition, pharmacological inhibition of histone deacetylases (HDACs) corrects the hypermethylation of the glucocorticoid receptor gene and improves responses to stress in rats that had received poor maternal care as pups (6). This suggests that drugs that reset the chromatin assembly may correct the pathological epigenetic programming associated with stress-related disorders, such as depression and anxiety. In particular, HDAC inhibitors are currently marketed for the treatment of cancer, including drug-resistant forms of leukemia and lymphomas, but long-term use of these drugs may be seriously limited by a number of adverse effects, including nausea, fatigue, transient thrombocytopenia, and, in some instances, myelosuppression (7, 8). In contrast, drugs that directly promote processes of acetylation represent a potential alternative to the use of HDAC inhibitors in human disorders.
Recent evidence suggests that the acetylating agent L-acetylcarnitine (LAC), a drug marketed for the treatment of neuropathic pain (9), causes analgesia increasing type 2 metabotropic glutamate (mGlu2) expression via an epigenetic mechanism shared by HDAC inhibitors (10–12). mGlu2 and its cognate receptor, mGlu3, are coupled to Gi/Go proteins and are preferentially (albeit not exclusively) localized in the preterminal region of axons, where they negatively modulate neurotransmitter release (13). Expression and function of mGlu2/3 receptors is reduced in the hippocampus of spontaneously depressed Flinders Sensitive Line (FSL) rats, and pharmacological activation of mGlu2/3 receptors shortens the time required for the therapeutic efficacy of conventional antidepressants in these rats (14, 15). Together with the observation that there is a clear overlap between the brain regions of the “pain matrix” and the regions involved in the pathophysiology of depression and anxiety, including the prefrontal cortex (16, 17), these studies led us to hypothesize the acetylating agent LAC as a unique epigenetic antidepressant with a fast onset of action.
Results
Rapid and Long-Lasting Antidepressant Effect of LAC in FSL Rats.
We treated FSL and their controls, Flinders Resistant Line (FRL) rats, with LAC, saline, or the reference antidepressant, chlorimipramine (CLO, i.p., daily for 21 d) and assessed the treatment effects using the forced swim test (FST) and the sucrose preference test. Both tests have “pharmacological validity” because they respond to antidepressant drugs (18, 19). Untreated FSL rats, as expected, showed >twofold increase in the immobility time compared with FRL rats (Fig. 1A). Remarkably, LAC displayed a clear-cut antidepressant effect already after 3 and 7 d of daily dosing. No tolerance was developed to the action of LAC. The drug was even more effective after 21 d, and the effect persisted for at least 2 wk after drug withdrawal. In contrast, CLO significantly reduced the immobility time only after 2 wk of treatment, and the effect disappeared after drug withdrawal. Neither LAC nor CLO had any effects in FRL rats (Fig. 1A).
Fig. 1.

Rapid and long-lasting antidepressant activity of LAC in FSL rats.(A) Immobility time in FST in FSL and FRL rats treated i.p. with saline, LAC, or CLO. n = 8. P < 0.05 vs. the respective values at t0 (*), the corresponding values in saline-treated rats (#), or in rats treated with CLO (§). F = 82.1 (time) and 4.7 (treatments). (B) Immobility time in FST in FSL rats treated with LAC for 21 d and with saline or LY341495 on the following day (t22). n = 6. P < 0.05 vs. t0 values (*) or vs. t22 values of FSL rats injected with saline (#). F = 45.5. (C) LAC effect on sucrose preference test in FSL and FRL rats. n = 7. *P < 0.05 vs. the respective values at t0 and the corresponding values of saline-treated rats. F = 46.6 (time) and 8.4 (treatments).
To investigate whether the antidepressant effect of LAC was causally related to mGlu2/3 receptors, we gave a single injection of saline or the brain-permeant mGlu2/3 receptor antagonist LY341495 to subgroups of FSL rats, treated with LAC or saline for 21 d. LY341495 did not affect the immobility time in FSL rats chronically treated with saline but significantly reduced the antidepressant activity of LAC (Fig. 1B).
Moreover, 4 and 20 d of LAC treatment had no effect on locomotor activity in the open field test in either FRL or FSL rats (Table S1).
The antidepressant activity of LAC was confirmed using the sucrose preference test, which models anhedonia associated with major depression. FSL rats treated with saline showed a lower sucrose preference compared with corresponding FRL rats. Sucrose preference in FSL rats was increased already after 3 d of LAC treatment, and the effect persisted for at least 2 wk after drug withdrawal (Fig. 1C), whereas CLO increased sucrose preference in FSL rats only after 14 and 21 d of treatment (Fig. S1).
LAC Corrects Abnormalities in BDNF Levels and Glutamate Release Associated with the Depressive-Like Phenotype of FSL Rats.
To further verify the antidepressant effect of LAC, we measured brain and plasma levels of BDNF. Plasma levels of BDNF are measured in the clinic as a biomarker of efficacy of antidepressant medication. Immunoblot analysis with anti-BDNF antibodies showed a band at 14 kDa corresponding to the mature form of BDNF. FSL rats treated with saline showed reduced BDNF levels in hippocampus and prefrontal cortex compared with corresponding FRL rats. These changes were partially restored by 21 d of LAC treatment, which had no effect in FRL rats (Fig. 2A). Moreover, LAC significantly increased plasma BDNF levels in FSL but not in FRL rats (Fig. 2C).
Fig. 2.
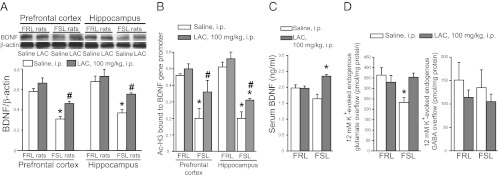
LAC treatment corrects abnormalities in BDNF and glutamate release in FSL rats. (A) BDNF levels in prefrontal cortex and hippocampus of FRL and FSL rats treated with saline or LAC for 21 d. n = 6. P < 0.05 vs. all other values (*) or vs. FRL rats treated with LAC (#). F = 25.4 and 13.6 for hippocampus and prefrontal cortex, respectively. (B) Levels of acetylated H3K27 bound to BDNF promoter gene in prefrontal cortex and hippocampus of FRL and FSL rats treated with saline or LAC for 21 d, n = 4. P < 0.05 vs. all other values (*) or vs. FSL rats treated with saline (#). F = 31.78 and 8.099 for hippocampus and prefrontal cortex, respectively. (C) Serum BDNF levels in FRL and FSL rats treated with saline or LAC for 21 d. n = 6. *P < 0.05 vs. FSL rats treated with saline. F = 8.9. (D) LAC effect on depolarization-evoked glutamate and GABA release in superfused hippocampal synaptosomes, n = 6. *P < 0.05 vs. all other values.
To investigate a potential dysfunction of glutamatergic neurotransmission, we measured glutamate and GABA release in superfused hippocampal synaptosomes from FSL and FRL rats treated with saline or LAC under basal conditions and in response to depolarizing concentrations of potassium ions (12 mM K+). In control experiments, depolarization-evoked release of glutamate or GABA was entirely dependent on extracellular Ca2+. There were no changes in the basal glutamate release regardless of rat strain or treatment (LAC vs. saline). In contrast, depolarization-evoked glutamate release is reduced by >30% in hippocampal synaptosomes from saline-treated FSL rats vs. saline-treated FRL rats. LAC treatment fully reversed the deficit of glutamate release in FSL rats, without affecting glutamate release in FRL rats (Fig. 2D). Basal and depolarization-evoked GABA release did not differ between FSL and FRL rats and was unaffected by LAC (Fig. 2D).
Epigenetic Regulation of mGlu2 Receptors by LAC in FSL Rats.
Searching for an underlying mechanism, we first measured mGlu2 receptor protein levels in FSL and FRL rats treated for 3 or 21 d with saline or LAC. Immunoblots showed a major band at approximately 100 kDa corresponding to mGlu2 receptor monomers. FSL rats treated with saline showed lower mGlu2 receptor levels in hippocampus and prefrontal cortex compared with corresponding FRL rats (Fig. 3A). LAC normalized mGlu2 receptor expression in FSL rats after both 3 and 21 d of treatment and had no effect in FRL rats (Fig. 3A). LAC treatment had no effect on mGlu2 receptor levels in the hypothalamus and nucleus accumbens at least at 21 d (Fig. S2).
Fig. 3.

Epigenetic regulation of mGlu2 receptors by LAC in FSL rats. (A) mGlu2 receptors expression in prefrontal cortex and hippocampus of FRL and FSL rats treated with saline or LAC for 3 or 21 d. n = 4 (3 d) or 6 (21 d). *P < 0.05 vs. all other values. F = 8.54 and 13.9 at 3 d, 12.9 and 12.4 at 21 d, for prefrontal cortex and hippocampus, respectively. (B) mRNA levels of mGlu2 and mGlu3 receptors in prefrontal cortex and hippocampus of FRL and FSL rats treated with saline or LAC. n = 6. P < 0.05 vs. the respective values of FRL rats (*) and vs. FSL rats treated with saline (#). F = 91.6. (C) Increased acetylation of p65/NF-κB in prefrontal cortex and hippocampus of FSL rats, n = 6. *P < 0.05 vs. the respective values of FSL rats treated with saline. (D) mGlu2 receptors expression in prefrontal cortex and hippocampus of FRL and FSL rats treated with sodium salicylate and/or LAC, n = 4. *P < 0.05 vs. the corresponding values obtained in FRL rats. F = 1.58E-002 and 9.22 for prefrontal cortex and hippocampus, respectively. (E) Levels of acetylated H3K27 bound to the Grm2 promoter gene in prefrontal cortex and hippocampus of FRL and FSL rats treated with saline or LAC. n = 6. *P < 0.05 vs. all other values. F = 8.7 and 8.3 for prefrontal cortex and hippocampus, respectively. (F) mGlu2 receptors expression in prefrontal cortex of FRL and FSL rats treated with saline or MS-275 for 21 d. n = 4. *P < 0.05 vs. all other values. F = 10.16.
The action of LAC was further characterized in rats treated with LAC or saline for 21 d. FSL rats treated with saline showed a significant reduction and a trend to a reduction of mGlu2 mRNA levels in prefrontal cortex and hippocampus, respectively. LAC treatment enhanced mGlu2 mRNA levels in both brain regions of FSL rats but had no effect in FRL rats (Fig. 3B). mGlu3 mRNA levels did not differ between FSL and FRL rats in hippocampus and prefrontal cortex and were not affected by LAC (Fig. 3B). Thus we focused on LAC induction of mGlu2 receptors.
Western blot analysis showed that LAC also increased acetylated p65 levels in hippocampus and prefrontal cortex of FSL but not FRL rats (Fig. 3C). To examine whether p65/NF-κB was involved in the induction of mGlu2 receptors, we combined LAC with sodium salicylate, a nonselective NF-κB inhibitor (20). Sodium salicylate prevented the increase in mGlu2 receptor protein induced by LAC in prefrontal cortex and hippocampus of FSL rats (Fig. 3D).
To assess whether the behavioral effects of LAC and the up-regulation of mGlu2 expression could be due to changes in the chromatin structure, we performed ChIP. Focusing on LAC acetylation proprieties, we analyzed the levels of K27-acetylated H3 histones bound to the Grm2 promoter and, once again, Bdnf promoter. Both were reduced in the hippocampus and prefrontal cortex of FSL rats. LAC treatment reversed these reductions, particularly in prefrontal cortex (Figs. 2B and 3E).
A role for histone acetylation in the control of Grm2 expression was supported by the use of MS-275, an inhibitor of class I HDACs (21). Similarly to LAC, MS-275 enhanced mGlu2 receptor expression in prefrontal cortex of FSL rats (Fig. 3F). In contrast with LAC, however, MS-275 treatment had no effect on mGlu2 receptor levels in hippocampus.
Reduced Endogenous Levels of Acetylcarnitine in the Hippocampus and Prefrontal Cortex of “Spontaneously Depressed” FSL Rats.
Endogenous levels of acetylcarnitine, measured by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), were lower in hippocampus and prefrontal cortex of FSL rats compared with the corresponding regions of FRL rats [acetylcarnitine levels (μg/g wet tissue; means + SEM): FRL hippocampus, 8.02 + 0.42; FSL hippocampus, 3.8 + 0.57 (P < 0.05; t = 8.72; n = 4); FRL prefrontal cortex, 8.8 + 0.43; FSL prefrontal cortex, 5.2 + 0.68 (P < 0.05, t = 3.71, n = 5)].
Rapid and Long-Lasting Antidepressant Activity of LAC Mediated by mGlu2 Receptors in Mice Subjected to Repeated Episodes of Chronic Unpredictable Stress.
To examine whether LAC is also effective in a model of environmentally induced depression, we exposed mice to repeated episodes of unpredictable stress twice daily for 4 wk. These mice showed a depressive phenotype in both FST and sucrose preference test (Fig. 4 A and B). LAC produced an antidepressant effect that was roughly similar to that seen in FSL rats, with the difference that LAC had a longer latency in the FST (Fig. 4 A and B).
Fig. 4.

Antidepressant activity of LAC in mice exposed to CUS. (A) LAC effect on immobility time of CUS mice and unstressed mice. n = 6. P < 0.05 vs. the corresponding t0 values (*) and vs. the respective values of stressed mice treated with saline (#). F = 8.4 and 5.4 for time and treatments, respectively. (B) Sucrose preference test in CUS mice and unstressed mice. n = 6. *P < 0.05 vs. both the corresponding t0 values and the respective values of stressed mice treated with saline. F = 32.2 and 2.28 for time and treatments, respectively. (C) Immobility time in FST in CUS mice treated with LAC for 21 d and with an acute i.p. injection of saline or LY341495 on the following day (t22). n = 6. P < 0.05 vs. t0 values (*) or vs. t22 values of stressed mice injected with saline (#). F = 0.72. (D) Immobility time in wild-type and mGlu2−/− mice treated with saline or LAC for 14 d. n = 6. *P < 0.05 vs. both the corresponding t0 values and the respective values of stressed mice treated with saline. F = 46.7 and 10.8 for time and treatments, respectively.
Again, a single injection of the mGlu2/3 receptor antagonist LY341495 counteracted the antidepressant effects of LAC (Fig. 4C).
Finally, to confirm the relevance of mGlu2 receptors in the antidepressant effects of LAC, we used mice with genetic deletion of mGlu2 receptors. Unstressed mGlu2−/− and wild-type mice did not differ in the FST performed 24 h after a habituation test (Table S2) (22). However, mGlu2−/− mice showed an increased immobility time in response to chronic unpredictable stress (CUS) compared with wild-type mice (Fig. 4D). A 14-d treatment with LAC reduced the immobility time in wild-type mice but had no effects in mGlu2−/− mice (Fig. 4D).
Discussion
Despite intensive research, there have been no major breakthroughs in the treatment of depression in the last decades, although drug targets other than monoamines have been identified (23, 24). Current antidepressant drugs show limited clinical efficacy, and approximately 25–40% of depressed patients are treatment resistant. Moreover, common antidepressants require at least 2–3 wk to significantly improve mood in depressed patients, leaving severely ill patients at risk for suicide in the early phases of treatment. These limitations could be overcome by drugs that target molecular events lying at the core of depressive disorders without interfering with the physiological function of the monoaminergic system.
Our findings provide strong insight into the pathogenesis of major depression that may pave the way to the development of drugs with faster onset of antidepressant effects and good profile of efficacy and tolerability. We found that LAC, a naturally occurring molecule, causes a rapid, robust, and long-lasting antidepressant effect in FSL rat, a genetic model of depression, and in mice exposed to CUS, which model environmentally induced depression. FSL rats exhibit sleep, immune, and neurochemical changes, as well as behaviors similar to those observed in depressed patients (25). Moreover, FSL rats did not show abnormalities in the saccharine preference test unless they had been stressed before (26). Conversely, our FSL rats showed reduced sucrose preference, which might be due to the use of sucrose instead of saccharine as a palatable reward or might be consequent to the stress associated with the change of their housing environment 2 wk before the test (SI Materials and Methods).
LAC is an endogenous compound that acts as a donor of acetyl groups and facilitates the transfer of fatty acids from cytosol to mitochondria during β-oxidation (27). In prefrontal cortex and hippocampus of FRL rats, endogenous LAC levels ranged between 8 and 9 μg/g wet tissue, corresponding to 40–45 nmol/g wet tissue, as previously reported in the mouse brain (28). Interestingly, endogenous LAC levels were reduced by 40–60% in the prefrontal cortex and hippocampus of FSL rat; this may explain why FSL rats but not FRL rats were responsive to LAC treatment in our study.
Here we found that LAC treatment of stressed and genetically vulnerable animals, showing depressive-like behaviors, rapidly reversed those behaviors, an effect that was selectively associated with mGlu2 receptors in brain regions that are critically involved in the pathophysiology of depression, such as the hippocampus and prefrontal cortex. The antidepressant action of LAC was already evident after 3 d and persisted after the end of the treatment, in contrast to CLO, whose antidepressant effects required at least 2 wk of daily dosing and were no longer seen after drug withdrawal. Given the magnitude and efficacy of the effects, as well as the rapidity of LAC responses, the results indicate that LAC and compounds with similar mechanisms of action could represent a truly unique approach to treat depressive disorders.
BDNF levels and glutamate release in synaptosomes were measured as surrogate parameters for antidepressant activity. A relationship between BDNF and depressive disorders is suggested by the evidence that (i) expression and activity of BDNF in the hippocampus are reduced in response to several forms of stress and increased by antidepressant treatment; and (ii) BDNF levels are reduced in postmortem hippocampus of depressed patients. BNDF levels are also reduced in the serum of depressed patients and elevated in response to chronic antidepressant treatment (29, 30). Thus the corrective action of LAC on brain and serum BDNF levels is in line with its antidepressant activity in FSL rats, although the precise relationship between BDNF and the depressive-like phenotype of FSL rats is uncertain (31, 32).
Dysfunction of glutamatergic neurotransmission is increasingly considered to be a core feature of stress-related disorders, including MDD (33). Both acute and chronic stress affect glutamate release in hippocampus and prefrontal cortex (34–36), whereas no studies of glutamate release in genetic models of depression have been reported to date. We measured glutamate release from hippocampal synaptosomes using a superfusion technique that allows the specific detection of exocytotic neurotransmitter release (37). Depolarization-evoked glutamate release was reduced in the hippocampus of FSL rats; similar reductions are evident in rats exposed to prenatal stress, which represent an epigenetic model of depression and anxiety. LAC corrected the deficit of glutamate release in FSL rats, which, again, strengthens the antidepressant activity of LAC.
In previous studies, we have shown a selective reduction of mGlu2/3 receptors in hippocampus of FSL rats. Here we show that it is the mGlu2 receptor subtype to be selectively down-regulated in hippocampus and prefrontal cortex of FSL rats, using an antibody that discriminates between mGlu2 and mGlu3 receptors.
Our findings identify at least two acetylation-based mechanisms in the induction of mGlu2 receptors in FSL rats. First, LAC enhances the acetylation of NF-ĸB-p65 subunit, which amplifies the transcriptional activity of NF-κB (38), thereby increasing transcription of the Grm2 gene encoding for the mGlu2 receptor in hippocampus and prefrontal cortex of depressed FSL rats. Importantly, the mGlu2 receptor promoter harbors numerous NF-κB–responsive elements, as opposed to mGlu3 promoter. Moreover, the effect of LAC on the expression of mGlu2 receptors was prevented by a combined treatment with sodium salicylate, a nonselective inhibitor of NF-κB, thereby, supporting the link between mGlu2 induction by LAC and p65/NF-κB.
Second, a notable finding of the present study is the altered histone acetylation state at the Grm2 promoter, which was found to be reduced in FSL rats and enhanced by LAC. In addition, the role of epigenetic mechanisms in the regulation of mGlu2 receptors is supported by the use of MS-275, an HDAC inhibitor that mimicked the action of LAC in enhancing mGlu2 receptor expression, at least in the prefrontal cortex.
The relative contribution of these two mechanisms to the induction of mGlu2 receptors in FSL rats is uncertain. Identification of the epigenetic mechanisms that integrate a better response to antidepressants with pharmacological modulation of mGlu2 function will help in discovering more effective treatment to improve the clinical efficacy of the common antidepressant drugs. The importance of histone acetylation in the regulation of the mGlu2 receptor is also strengthened by the recent finding that chronic treatment with atypical antipsychotics down-regulated the expression of mGlu2 receptors in the mouse and human prefrontal cortex by increasing the binding of HDAC2 to the Grm2 promoter (39).
Other mGlu receptor subtypes might be involved in the pathophysiology of depression, such as mGlu5 or mGlu7 (40). Recent immunohistochemical data show a reduced expression of mGlu5 receptors in several brain regions of FSL rats, including the dorsal hippocampus, cingulated cortex, and parietal cortex (41). Whether acetylating agents such as LAC or HDAC inhibitors up-regulate mGlu5 receptors in the brain of FSL rats is a question that warrants further investigation.
A relationship between induction of mGlu2 receptors and antidepressant effect of LAC was demonstrated by the finding that a single injection of the mGlu2/3 receptor antagonist LY341495 (42) was sufficient to significantly attenuate the action of LAC in both FSL rats and CUS mice. In addition, mGlu2−/− mice exposed to CUS showed an increased immobility time compared with wild-type mice and did not respond to LAC. The use of pharmacological agents that do not discriminate between mGlu2 and mGlu3 receptors has raised the question of whether agonists or antagonists should be used in the treatment of depressive disorders. Thus, the mGlu2/3 receptor antagonist MGS0039 exerts a clear-cut antidepressant effect (43, 44), whereas, conversely, the mGlu2/3 receptor agonist LY379268 reinforces the antidepressant activity of CLO or fluoxetine. The lack of subtype-selectivity of MGS0039 and LY379268 may confound the interpretation of these findings. Recent evidence shows that a selective mGlu2 receptor enhancer produces antidepressant effects (45), and here, LAC relieved depressive symptoms by selectively inducing mGlu2 receptors. A potential involvement of mGlu3 receptors in the pathophysiology of depression is suggested by the association between a polymorphic variant of Grm3 (the gene encoding the mGlu3 receptor) and MDD (46). Perhaps mGlu2 and mGlu3 receptors have opposite roles in depression and the reported antidepressant effects of mGlu2/3 receptor antagonists are mediated by mGlu3 receptor blockade.
In a double-blind, randomized, controlled clinical trial, LAC was not inferior to amisulpride in relieving symptoms of dysthymic patients, showing an excellent profile of safety and tolerability (47). LAC has also shown efficacy in a limited cohort of senile patients with depression (48). Our findings strongly indicate that the antidepressant effects of LAC should be translationally investigated in the treatment of MDD by comparison with a classical antidepressant, with particular focus on the effects in the early stages of treatment. LAC is quite popular in the medical practice because it is highly tolerated and does not cause any of the major adverse effects associated with the use of HDAC inhibitors. Thus, LAC can be a useful pharmacological tool to examine the validity of the epigenetic hypothesis of depressive disorders in humans.
Materials and Methods
Animal Models and Drug Treatments.
Experiments were carried out according to the European (86/609/EEC) and Italian (D.Lgs 116/92) guidelines of animal care. All efforts were made to minimize animal suffering. Male FSL and FRL rats (200–300 g body weight) and male CD1 mice (20–25 g; Charles River) were housed five per cage under controlled conditions (12-h light/dark cycle, 22 °C, food and water ad libitum) for 2 wk before the beginning of experiments. Male CD1 mice were exposed to CUS (SI Materials and Methods). Animals were tested in the FST (SI Materials and Methods) and sucrose preference test (SI Materials and Methods) after 3, 7, 14, and 21 d of treatment with saline, LAC (100 mg/kg, i.p.) or CLO (10 mg/kg, i.p.) and 7 and 14 d after drug withdrawal. Additional animal groups received a single injection of either saline or LY341495 (1 mg/kg, i.p.) 24 h after the end of the chronic treatment and were tested in FST at t = 0 and t = 22 1 h after injection of LY341495 or saline. Additional groups of FRL and FSL rats, treated daily with saline or LAC (n = 5/group) were used for measurements of spontaneous motor activity in an open field apparatus (SI Materials and Methods). Motor activity was measured after 4 and 20 d of treatment. In the same animals, immobility time in the FST was measured after 3 and 21 d of treatment. Additional groups of FSL or FRL rats (n = 4 per group) were used exclusively for measurements of mGlu2 receptor expression by immunoblotting and treated as follows: (i) saline or LAC for 3 d; (ii) sodium salicylate (50 mg/kg, i.p.) with saline or LAC for 21 d; and (iii) saline or MS-275 (3 mg/kg, i.p.) for 21 d.
We also subjected mGlu2 receptor knockout mice (mGlu2−/− mice, 20–25 g body weight) and age-matched C57BL/6J wild-type mice to CUS for 28 d. At the end of the stress session, two groups of mGlu2−/− mice and two groups of wild-type mice were treated with saline or LAC for 14 d and evaluated at FST at t = 7 and t = 14. Two independent groups of untreated wild-type and mGlu2−/− mice not subjected to CUS (n = 5 per group) were tested for the immobility time in the FST. Details in SI Materials and Methods.
Measurements of Acetylcarnitine Levels.
Acetylcarnitine levels in prefrontal cortex and hippocampus of FRL and FSL rats were measured by LC-MS/MS (SI Materials and Methods).
Quantitative PCR Analysis of mGlu2 and mGlu3 Receptors.
DNA synthesis and real-time PCR were carried out as described in SI Materials and Methods. The following primers were used. mGlu2 receptor: forward: 5′-CTACAGTGATGTCTCCATCC-3′, reverse: 5′-AAAGCCTCAATGCCTGTCTC-3′; mGlu3 receptor: forward: 5′-CAAGTGACTACAGAGTGCAG-3′, reverse: 5′-CTGTCACCAATGCTCAGCTC-3′; β-actin: forward: 5′-TGAACCCTAAGGCCAACCGTG-3′, reverse: 5′-GCTCATAGCTCTTCTCCAGGG-3′.
Western Blot Analysis.
The following primary antibodies were used: mouse anti p65/Rel-A (1:200; Santa Cruz Biotechnology), rabbit anti–acetyl-K310-p65Rel-A (2 μg/mL; Abcam), mouse anti-mGlu2 receptor (1 μg/mL; Abcam), mouse anti-BDNF (1:1,000; Santa Cruz Biotechnology), and mouse anti–β-actin (1:5,000; Sigma) (SI Materials and Methods).
Release Experiments in Hippocampal Synaptosomes.
Glutamate and GABA release were measured in hippocampal synaptosome prepared from FRL and FSL rats chronically treated with saline or LAC, as described in SI Materials and Methods.
ChIP Analysis.
ChIP analysis of acetyl-K27-H3 histone bound to the Grm2 and Bdnf promoter genes was performed as detailed in SI Materials and Methods.
Statistics.
Values are means + SEM of different n as reported in SI Materials and Methods. Either ANOVA or Student t test were used, depending on the number of groups in the comparison. Tukey’s test was used for post hoc analysis of ANOVA results. A P value <0.05 was regarded as significant.
Supplementary Material
Acknowledgments
We thank Benedetta Bigio for bioinformatics support. The work was partially supported by Swedish Medical Research Council Grant 10414 and the Karolinska Institute (A.A.M.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216100110/-/DCSupplemental.
See Commentary on page 4441.
References
- 1.Perroud N. Suicidal ideation during antidepressant treatment: Do genetic predictors exist? CNS Drugs. 2011;25:459–471. doi: 10.2165/11589420-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 2.Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: Systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530–538. doi: 10.1001/archpsyc.63.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mill J, Petronis A. Molecular studies of major depressive disorder: The epigenetic perspective. Mol Psychiatry. 2007;12:799–814. doi: 10.1038/sj.mp.4001992. [DOI] [PubMed] [Google Scholar]
- 4.Autry AE, Monteggia LM. Epigenetics in suicide and depression. Biol Psychiatry. 2009;66:812–813. doi: 10.1016/j.biopsych.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 6.Weaver IC, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 7.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15:3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 8.Galli M, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89:185–190. doi: 10.1007/s00277-009-0793-8. [DOI] [PubMed] [Google Scholar]
- 9.Youle M. Acetyl-L-carnitine in HIV-associated antiretroviral toxic neuropathy. CNS Drugs. 2007;21(Suppl 1):25–30. doi: 10.2165/00023210-200721001-00004. [DOI] [PubMed] [Google Scholar]
- 10.Chiechio S, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75:1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- 11.Chiechio S, et al. Transcriptional regulation of metabotropic glutamate receptor 2/3 expression by the NF-kappaB pathway in primary dorsal root ganglia neurons: A possible mechanism for the analgesic effect of L-acetylcarnitine. Mol Pain. 2006;2:20. doi: 10.1186/1744-8069-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiechio S, et al. Transcriptional regulation of type-2 metabotropic glutamate receptors: An epigenetic path to novel treatments for chronic pain. Trends Pharmacol Sci. 2010;31:153–160. doi: 10.1016/j.tips.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Nicoletti F, et al. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matrisciano F, et al. Group-II metabotropic glutamate receptor ligands as adjunctive drugs in the treatment of depression: A new strategy to shorten the latency of antidepressant medication? Mol Psychiatry. 2007;12:704–706. doi: 10.1038/sj.mp.4002005. [DOI] [PubMed] [Google Scholar]
- 15.Matrisciano F, et al. Defective group-II metaboropic glutamate receptors in the hippocampus of spontaneously depressed rats. Neuropharmacology. 2008;55:525–531. doi: 10.1016/j.neuropharm.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Neugebauer V, Galhardo V, Maione S, Mackey SC. Forebrain pain mechanisms. Brain Res Brain Res Rev. 2009;60:226–242. doi: 10.1016/j.brainresrev.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson MJ, et al. Depression and pain. Front Biosci. 2009;14:5031–5051. doi: 10.2741/3585. [DOI] [PubMed] [Google Scholar]
- 18.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan V, Nestler EJ. Linking molecules to mood: New insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 21.Covington HE, 3rd, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morishima Y, et al. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc Natl Acad Sci USA. 2005;102:4170–4175. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Bodinat C, et al. Agomelatine, the first melatonergic antidepressant: Discovery, characterization and development. Nat Rev Drug Discov. 2010;9:628–642. doi: 10.1038/nrd3140. [DOI] [PubMed] [Google Scholar]
- 24.McEwen BS, et al. The neurobiological properties of tianeptine (Stablon): From monoamine hypothesis to glutamatergic modulation. Mol Psychiatry. 2010;15:237–249. doi: 10.1038/mp.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiménez-Vasquez PA, et al. Electroconvulsive stimuli selectively affect behavior and neuropeptide Y (NPY) and NPY Y(1) receptor gene expressions in hippocampus and hypothalamus of Flinders Sensitive Line rat model of depression. Eur Neuropsychopharmacol. 2007;17(4):298–308. doi: 10.1016/j.euroneuro.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Pucilowski O, Overstreet DH, Rezvani AH, Janowsky DS. Chronic mild stress-induced anhedonia: Greater effect in a genetic rat model of depression. Physiol Behav. 1993;54:1215–1220. doi: 10.1016/0031-9384(93)90351-f. [DOI] [PubMed] [Google Scholar]
- 27.Fritz EB, McEwen BS. Effect of carnitine on fatty-acid oxidation by muscle. Science. 1959;128:334. doi: 10.1126/science.129.3345.334. [DOI] [PubMed] [Google Scholar]
- 28.Costell M, Miguez MP, O’Connor JE, Grisolin SC. Effect of hyperammonemia on the levels of carnitine in mice. Neurology. 1987;37:804–808. doi: 10.1212/wnl.37.5.804. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu E, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- 30.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications. Biol Psychiatry. 2008;64:527–532. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjørnebekk A, Mathé AA, Gruber SH, Brené S. Housing conditions modulate escitalopram effects on antidepressive-like behavior and brain neurochemistry. Int J Neuropsychopharmacol. 2008;11:1135–1147. doi: 10.1017/S1461145708008912. [DOI] [PubMed] [Google Scholar]
- 32.Elfving B, et al. Inverse correlation of brain and blood BDNF levels in a genetic rat model of depression. Int J Neuropsychopharmacol. 2010;13:563–572. doi: 10.1017/S1461145709990721. [DOI] [PubMed] [Google Scholar]
- 33.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowy MT, Wittenberg L, Yamamoto BK. Effect of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats. J Neurochem. 1995;65:268–274. doi: 10.1046/j.1471-4159.1995.65010268.x. [DOI] [PubMed] [Google Scholar]
- 35.Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: Implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51:775–787. doi: 10.1016/s0006-3223(01)01362-2. [DOI] [PubMed] [Google Scholar]
- 36.Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: Effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- 37.Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of superfusion. Neurochem Res. 2000;25:1265–1274. doi: 10.1023/a:1007648229795. [DOI] [PubMed] [Google Scholar]
- 38.Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 39.Kurita M, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaki S, et al. mGlu2/3 and mGlu5 receptors: Potential targets for novel antidepressants. Neuropharmacology. 2013;66:40–52. doi: 10.1016/j.neuropharm.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 41.Kovačević T, et al. Reduced metabotropic glutamate receptor 5 in the Flinders Sensitive Line of rats, an animal model of depression: an autoradiographic study. Brain Res Bull. 2012;87:406–412. doi: 10.1016/j.brainresbull.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 43.Yasuhara A, et al. Prodrugs of 3-(3,4-dichlorobenzyloxy)-2-amino-6-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (MGS0039): a potent and orally active group II mGluR antagonist with antidepressant-like potential. Bioorg Med Chem. 2006;14:4193–4207. doi: 10.1016/j.bmc.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186:587–593. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- 45.Fell MJ, et al. N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl) phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/antidepressant properties: In vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J Pharmacol Exp Ther. 2011;336:165–177. doi: 10.1124/jpet.110.172957. [DOI] [PubMed] [Google Scholar]
- 46.Tsunoka T, et al. Association analysis of group II metabotropic glutamate receptor genes (GRM2 and GRM3) with mood disorders and fluvoxamine response in a Japanese population. Prog Neuropsychopharmacol Biol. 2009;33:875–879. doi: 10.1016/j.pnpbp.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Zanardi R, Smeraldi E. A double-blind, randomised, controlled clinical trial of acetyl-L-carnitine vs. amisulpride in the treatment of dysthymia. Eur Neuropsychopharmacol. 2006;16:281–287. doi: 10.1016/j.euroneuro.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Garzya G, et al. Evaluation of the effects of L-acetylcarnitine on senile patients suffering from depression. Drugs Exp Clin Res. 1990;16:101–106. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.