

Abstract
Gap junctions mediate intercellular signalling in arteries and contribute to endothelium-dependent vasorelaxation, conducted vascular responses and vasomotion. Considering its putative role in vascular dysfunction, mechanistic insights regarding the control of gap junction conductivity are required. Here, we investigated the consequences of endothelial alkalinisation for gap junction communication and endothelium-dependent vasorelaxation in resistance arteries. We studied mesenteric arteries from NMRI mice by myography, confocal fluorescence microscopy and electrophysiological techniques. Removing CO2/HCO3−, reducing extracellular [Cl−] or adding 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid inhibited or reversed Cl−/HCO3− exchange, alkalinised the endothelium by 0.2–0.3 pH units and inhibited acetylcholine-induced vasorelaxation. NO-synthase-dependent vasorelaxation was unaffected by endothelial alkalinisation whereas vasorelaxation dependent on small- and intermediate-conductance Ca2+-activated K+ channels was attenuated by ∼75%. The difference in vasorelaxation between arteries with normal and elevated endothelial intracellular pH (pHi) was abolished by the gap junction inhibitors 18β-glycyrrhetinic acid and carbenoxolone while other putative modulators of endothelium-derived hyperpolarisations – Ba2+, ouabain, iberiotoxin, 8Br-cAMP and polyethylene glycol catalase – had no effect. In the absence of CO2/HCO3−, addition of the Na+/H+-exchange inhibitor cariporide normalised endothelial pHi and restored vasorelaxation to acetylcholine. Endothelial hyperpolarisations and Ca2+ responses to acetylcholine were unaffected by omission of CO2/HCO3−. By contrast, dye transfer between endothelial cells and endothelium-derived hyperpolarisations of vascular smooth muscle cells stimulated by acetylcholine or the proteinase-activated receptor 2 agonist SLIGRL-amide were inhibited in the absence of CO2/HCO3−. We conclude that intracellular alkalinisation of endothelial cells attenuates endothelium-derived hyperpolarisations in resistance arteries due to inhibition of gap junction communication. These findings highlight the role of pHi in modulating vascular function.
Key points
Gap junctions are important for coordination and transfer of signals between cells. Signals initiated in the vascular endothelium can spread through myoendothelial gap junctions and cause relaxation of coupled vascular smooth muscle cells.
The cellular level of acidity is important for control of vascular function. The mechanisms linking disturbed acid–base balance to changes in vascular tone have not been understood in detail.
We show that intracellular alkalinisation of endothelial cells in resistance arteries inhibits myoendothelial coupling and endothelium-dependent vasorelaxation. Although hyperpolarisations are generated in the endothelial cells under alkaline conditions, they are not transferred to the smooth muscle cells. Similarly, dye transfer between endothelial cells is inhibited during intracellular alkalinisation.
These results support the suggestion that intracellular pH is important for control of gap junction conductivity and intercellular communication. We describe a potential new molecular mechanism for development of vascular dysfunction in pathologies (e.g. diabetes, hypertension) involving altered myoendothelial signalling.
Introduction
The vascular endothelium synthesises multiple vasoactive substances and activates vasomodulatory signalling cascades essential for regulation of active tension development. Production of nitric oxide (NO) by the endothelial NO-synthase (eNOS) and vasoactive prostanoids by cyclooxygenase (COX) are well-described catalytic steps involved in endothelium-dependent vasorelaxation. However, even following inhibition of eNOS and COX, endothelium-derived hyperpolarisation (EDH) and relaxation of vascular smooth muscle cells (VSMCs) is readily induced upon activation of the endothelium (Bellien et al. 2008). This NO- and prostanoid-independent vasorelaxant response has been known as the endothelium-derived hyperpolarising factor (EDHF) response and is prominent particularly in resistance arteries (Urakami-Harasawa et al. 1997) where it contributes to control of local tissue perfusion and systemic blood pressure. Several molecular mechanisms have been proposed to mediate EDH, including endothelial production of diffusible factors such as C-type natriuretic peptide (CNP) (Chauhan et al. 2003), 11,12-epoxyeicosatrienoic acid (11,12-EET) (Fisslthaler et al. 1999) and H2O2 (Shimokawa & Matoba, 2004) or generation of a local increase in extracellular [K+] leading to activation of inward rectifier K+ channels (Kir) and the Na+/K+-ATPase (Edwards et al. 1998). Most commonly, however, EDH is ascribed to activation of endothelial K+ channels of small (SK) and intermediate (IK) conductance (Busse et al. 2002; Bellien et al. 2008). This induces endothelial cell (EC) hyperpolarisation, and subsequent electrotonic spread of charge through myoendothelial gap junctions hyperpolarises and relaxes the coupled VSMCs (Edwards et al. 1999; Goto et al. 2002).
Numerous studies have shown that EDH is altered during pathological conditions such as diabetes, hypertension and ischaemia/reperfusion injury (Feletou & Vanhoutte, 2004). Currently, however, not much is known about specific disease mechanisms that modify myoendothelial signalling, and further mechanistic insights are desired to elucidate the link between disease pathogenesis and altered EDH. One potential factor that modifies gap junction communication – and hence possibly EDH – is intracellular pH (pHi). Several studies have shown that gap junction communication is inhibited at low pHi (Turin & Warner, 1977; Spray et al. 1981); and more recently, a bell-shaped relationship between the junctional resistance and pHi has been reported in isolated cardiomyocyte cell pairs with maximal conduction observed around pHi 7.0 (Swietach et al. 2007). As this pH optimum is slightly below resting physiological pHi, gap junction conductivity would be predicted to be particularly sensitive to alkaline shifts in pHi and recently the inhibitory effect of intracellular alkalinisation on gap junction communication was confirmed using cell pairs of the mouse neuroblastoma cell line N2A heterologously expressing connexin 36 (Gonzalez-Nieto et al. 2008). Despite these interesting findings based on isolated cells, the consequences of pHi-mediated changes in gap junction resistance for physiological functions in general, and vascular functions in particular, have not previously been addressed.
Acid–base disturbances are frequently encountered in clinical practice, particularly in patients with renal, pulmonary or metabolic disease. Furthermore, several studies have shown that membrane transport of acid–base equivalents is altered in cardiovascular disease (Boedtkjer & Aalkjaer, 2012). Although it has been realised for more than a century that extracellular acid–base disturbances greatly influence vascular function, the vasomotor effects of intracellular acid–base disturbances have only been addressed more recently (Boedtkjer & Aalkjaer, 2012). We have shown that EC acidification inhibits acetylcholine-induced NO-mediated vasorelaxation (Boedtkjer et al. 2011, 2012) consequent to a strong pH dependency of the eNOS (Fleming et al. 1994; Boedtkjer et al. 2011). In contrast, the vascular consequences of EC alkalinisation have not previously been determined.
Cellular base extrusion in the vascular wall is predominantly mediated by Cl−/HCO3− exchangers (also known as anion exchangers, AEs), which under physiological conditions perform electroneutral exchange of extracellular Cl− for intracellular HCO3− and thus contribute in particular to the recovery of pHi during intracellular alkalinisation. In addition to the dependency on Cl− and HCO3−, the AEs are inhibited by 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS). Both AE2 (slc4a2) and AE3 (slc4a3) have been detected in arteries (Brosius et al. 1997). Under normal circumstances, EC steady-state pHi is established as a balance between the metabolic acid–base production, the net acid-loading activity of the AEs and the net acid-extruding activities of the Na+/H+-exchanger NHE1 and the Na+,HCO3−-cotransporter NBCn1 (Boedtkjer & Aalkjaer, 2012).
In the present study, we used ion substitution and pharmacological inhibition to modify AE activity and investigate functional effects of EC alkalinisation on endothelium-dependent vasorelaxation.
Methods
Male NMRI mice (7–12 weeks old) obtained from Taconic Europe (Ry, Denmark) were killed by cervical dislocation. The whole mesenteric bed was excised and first- or second-order mesenteric arteries were dissected free of surrounding tissue. All animal procedures were approved by the Danish Animal Care and Use Committee.
Artery tone
Arteries were mounted in wire myographs (DMT, Aarhus, Denmark) and normalised to 90% of the internal diameter corresponding to a transmural pressure of 100 mmHg as previously described (Mulvany & Halpern, 1977). Following precontraction with noradrenaline to 60–80% of maximal active tension, the response to acetylcholine was investigated. When drugs with reversible actions were used, or consequences of ion substitution were investigated, effects of time were eliminated by alternating the order of the protocol applied to two or four arteries from the same mouse. The average response observed in these arteries was used to represent that animal. When drugs with irreversible actions were employed, arteries were run in parallel as time and vehicle controls.
Endothelial pHi and [Ca2+]i responses
Endothelial pHi and [Ca2+]i responses were evaluated by fluorescence confocal microscopy of mouse mesenteric arteries. Endothelial pHi was measured in 2′,7′-bis-(carboxypropyl)-5(6)-carboxyfluorescein (BCPCF)-loaded inverted arteries as previously described (Boedtkjer & Aalkjaer, 2009). Intracellular [Ca2+] measurements from ECs were performed similar to previous reports (Boedtkjer et al. 2011), using a combination of Calcium Green-1 and Fura Red loaded preferentially into ECs by perfusion of the fluorophores through the lumen of the arteries. Resting endothelial [Ca2+]i levels in the presence and absence of CO2/HCO3− were investigated in paired experiments. Inhibition of the endoplasmic reticulum Ca2+-ATPase by cyclopiazonic acid (CPA) induces a uniform [Ca2+]i rise in the ECs (data not shown), and we analysed the proportion of ECs responding to acetylcholine with an increase in the [Ca2+]-dependent fluorescence ratio of more than 20% of the response to 10 μmol l−1 CPA. We also investigated the effect of CO2/HCO3− on the magnitude of the [Ca2+]i response in ECs that responded to acetylcholine application. For these paired analyses, the order of the protocol (i.e. whether the response to acetylcholine was investigated first under CO2/HCO3−-free or CO2/HCO3−-containing conditions) was alternated between preparations to avoid potential effects of time. The change in [Ca2+]-dependent fluorescence ratio was calculated from the average ratio level measured during the 20 s preceding the application of acetylcholine and during the last 45 s of the 2 min acetylcholine exposure.
Membrane potentials and intercellular dye transfer
Arteries were mounted in a wire myograph (DMT) and normalised as described above. Membrane potential (Vm) measurements were performed using aluminium silicate microelectrodes (WPI, Hitchin, UK) with a resistance of 40–120 MΩ when backfilled with 3 mol l−1 KCl and recorded with an Intra-767 amplifier (WPI), visualised on an oscilloscope (Gould-Nicolet Technologies, Loughton, UK) and continuously stored with a PowerLab system (ADInstruments, Dunedin, New Zealand). Electrode entry into cells resulted in an abrupt drop in voltage followed by a sharp return to baseline upon retraction. To measure membrane potentials from VSMCs, arteries were mounted in normal configuration and the electrode was advanced into the vessel wall from the adventitial side. Membrane potentials from ECs were measured using inverted arteries allowing the electrode to be inserted into ECs from the luminal surface of the arteries. To confirm that these respective methods resulted in measurements from the expected cell types, 5% propidium iodide was included in the electrode solution and the impaled cell visualised at the end of the experiment by confocal microscopy (excitation at 514 nm, emission recorded at wavelengths >560 nm), a principle previously employed by others (Siegl et al. 2005; Haddock et al. 2006; Behringer et al. 2012). If the ECs are dye coupled, the propidium iodide spreads from the impaled EC into neighbouring ECs, which also become fluorescently stained although at a somewhat reduced intensity. Disregarding fluorescence signals more than 50 μm from the nearest stained nucleus, we estimated the degree of cellular coupling by analysing the total number of stained nuclei.
Solutions
The composition of the CO2/HCO3−-containing physiological salt solution (PSS) used in this study was as follows (in mmol l−1): NaCl 114, Hepes 10, NaHCO3 25, MgSO4 1.20, KCl 4.70, glucose 5.50, EDTA 0.026, KH2PO4 1.18, CaCl2 1.60. In CO2/HCO3−-free solutions, NaHCO3 was substituted with an equimolar amount of NaCl. We used two different low [Cl−] solutions. In the first, NaCl was substituted with an equimolar amount of sodium gluconate while CaCl2 was substituted with 8.5 mmol l−1 calcium gluconate to compensate for the Ca2+-chelating action of gluconate, as suggested by others (Swietach et al. 2007). In the other solution, NaCl and KCl were substituted with equimolar amounts of non-chelating sodium methanesulphonate and potassium methanesulphonate, respectively. In all solutions, pH was adjusted to 7.40 at 37°C after bubbling with 5% CO2 balance air (CO2/HCO3−-containing solutions) or nominally CO2-free atmospheric air (CO2/HCO3−-free solutions).
Statistics
Data are expressed as mean ± SEM. Paired two-tailed Student's t test was used when two interventions on the same arteries were compared. When more than two interventions performed on the same arteries were compared, one-way ANOVA for repeated measures was employed followed by Bonferroni post-hoc tests. When successive interventions were compared between groups, two-way ANOVA for repeated measures was used followed by a Bonferroni post-hoc test. When measurements from arteries isolated from different mice were compared, unpaired statistical tests were employed. The number of stained nuclei in dye coupling experiments under different conditions was compared by a non-parametric Mann–Whitney test. Concentration–response relationships were analysed by sigmoidal curve fits and the derived parameters (logEC50 and maximum values) were compared by extra sum-of-squares F-tests. P < 0.05 was considered statistically significant; n equals number of mice (one to four arteries per mouse). Statistical analyses were performed using GraphPad Prism 5.03 software.
Results
Steady-state pHi in mesenteric artery ECs was 7.24 ± 0.09 (n= 19) in the presence of CO2/HCO3−. To study the effects of EC alkalinisation on vasomotor responses, we investigated changes in pHi caused by manoeuvres that inhibit or reverse Cl−/HCO3− exchange. Removing CO2/HCO3−, reducing extracellular [Cl−] to 4.7 mmol l−1 (by substitution with gluconate) or 3.2 mmol l−1 (by substitution with methanesulphonate) or adding 200 μmol l−1 DIDS alkalinised the ECs around 0.2–0.3 pH units (Fig. 1A). Prior application of 200 μmol l−1 DIDS completely abolished the alkalinisation observed when bath [Cl−] was reduced to 4.7 mmol l−1 by substitution with gluconate (ΔpHi=−0.05 ± 0.02, n= 4, P < 0.01 vs. gluconate substitution without DIDS; Supplementary Fig. 1). This is consistent with low bath [Cl−] reversing the DIDS-sensitive AE activity.
Figure 1. Manoeuvres that inhibit or reverse Cl−/HCO3− exchange cause intracellular alkalinisation of ECs. The alkalinisation induced by omission of CO2/HCO3− can be reversed by simultaneous inhibition of Na+/H+-exchange.
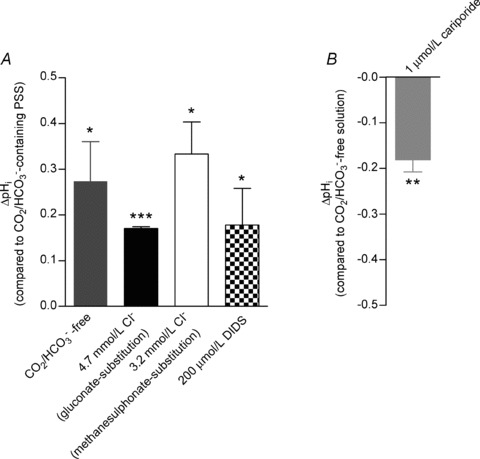
A, mean steady-state pHi changes (obtained 15–30 min after the change of bath solution or addition of drug; n= 4–12) induced by omission of CO2/HCO3−, or by – in the presence of CO2/HCO3−– reducing extracellular [Cl−] or adding 200 μmol l−1 DIDS. Extracellular Cl− was substituted with either methanesulphonate or gluconate. The Ca2+-chelating action of gluconate was compensated for by increasing total [Ca2+] to 8.5 mmol l−1. B, mean steady-state pHi change (n= 4) induced by addition of 1 μmol l−1 of the NHE1-specific inhibitor cariporide in the absence of CO2/HCO3−. Statistical significance was determined by one-sample t tests. *P < 0.05, **P < 0.01, ***P < 0.001 vs. 0.
In the absence of CO2/HCO3−, EC steady-state pHi was 7.53 ± 0.05 (n= 39). The pHi change caused by omission of CO2/HCO3− was reversible upon restoration of CO2/HCO3− (Supplementary Fig. 2A and B). Both omission and restoration of CO2/HCO3− elicited a fast transient change in pHi of large magnitude which within 5–10 min resulted in a stable steady-state change in pHi (Supplementary Fig. 2A and B) of the magnitude reported in Fig. 1A. Upon addition of 1 μmol l−1 of the NHE1-specific inhibitor cariporide in the absence of CO2/HCO3−, EC pHi declined and within approximately 5 min reached a new steady-state level close to that seen in the presence of CO2/HCO3− (Fig. 1B and Supplementary Fig. 3).
Vasomotor responses
To investigate the consequences of EC alkalinisation for endothelium-dependent regulation of artery tone, we studied the response of noradrenaline-precontracted mouse mesenteric arteries to acetylcholine. The effects of the interventions were investigated when pHi after an intervention (e.g. solution change or addition of drug) had reached a new steady-state level (see, for example, Supplementary Fig. 2). Under control conditions, arteries investigated in the presence of CO2/HCO3− relaxed around 70–80% when exposed to 0.5 or 3 μmol l−1 acetylcholine (Fig. 2A). This acetylcholine-induced vasorelaxation was reduced to around 50% in the absence of CO2/HCO3− (Fig. 2A). The difference between arteries investigated in the presence and absence of CO2/HCO3− was maintained after eNOS inhibition with 100 μmol l−1 N-nitro-l-arginine methyl ester (l-NAME; Fig. 2A). In contrast, the l-NAME-sensitive component of the vasorelaxant response to acetylcholine was unaffected by omission of CO2/HCO3− (Fig. 2A). After combined inhibition of COX (with 3 μmol l−1 indomethacin) and eNOS, the difference in vasorelaxation to acetylcholine was still observed between arteries investigated in the presence and absence of CO2/HCO3− (Fig. 2B and C). Representative traces of the l-NAME- and indomethacin-insensitive vasorelaxation to acetylcholine are shown in Fig. 2B and its sensitivity to IK and SK inhibitors (50 nmol l−1 apamin plus 50 nmol l−1 charybdotoxin or 1 μmol l−1 TRAM-34) is shown in Fig. 2D and E. Together, these results demonstrate that the EDH-type vasorelaxation in mouse mesenteric arteries is inhibited in the absence of CO2/HCO3−. The effect of removing CO2/HCO3− on endothelium-dependent vasorelaxation was reversible upon restoration of CO2/HCO3− and the order of the protocol was alternated between preparations to eliminate potential effects of time (Supplementary Fig. 2C).
Figure 2. EDH-type vasorelaxation is reduced by omission of CO2/HCO3− in mesenteric arteries.
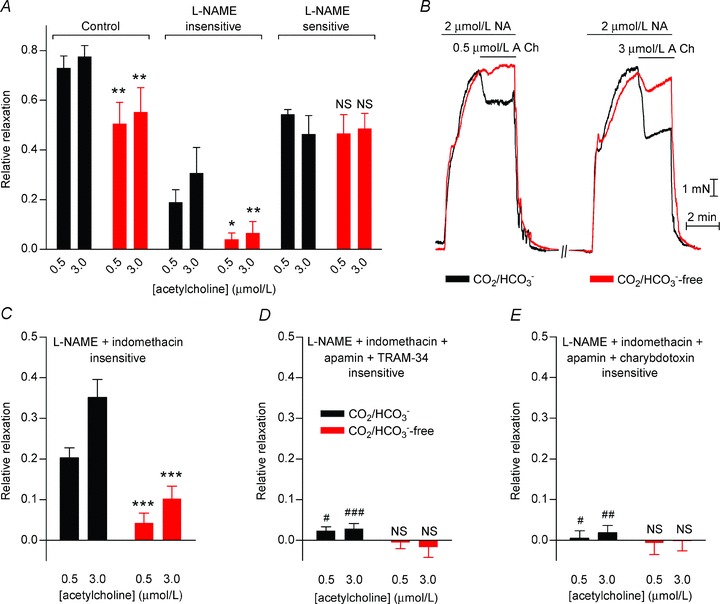
A, vasorelaxation to acetylcholine was attenuated in the absence of CO2/HCO3− both before and after treatment with 100 μmol l−1 l-NAME (n= 5). The effect of omitting or restoring CO2/HCO3− was investigated 30 min after the buffer change to allow a new steady-state pHi to be reached (Supplementary Fig. 2A and B). In this and the following panels, the black bars and traces represent experiments performed in the presence of CO2/HCO3−, while the red bars and traces represent experiments performed under CO2/HCO3−-free conditions. B, representative force recordings showing the relaxation of noradrenaline (NA)-precontracted arteries to acetylcholine (ACh) in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin with or without CO2/HCO3−. C, vasorelaxation to acetylcholine after treatment with 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin was reduced in the absence of CO2/HCO3− (n= 12). The effect of omitting or restoring CO2/HCO3− was investigated 30 min after the buffer change to allow a new steady-state pHi to be reached. D, vasorelaxation to acetylcholine in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin was abolished by 50 nmol l−1 apamin and 1 μmol l−1 TRAM-34 (n= 5). E, vasorelaxation to acetylcholine in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin was abolished by 50 nmol l−1 apamin and 50 nmol l−1 charybdotoxin (n= 5). Comparisons were made with two-way ANOVA followed by Bonferroni post-hoc tests. *P < 0.05, **P < 0.01, ***P < 0.001, NS: not significantly different vs. CO2/HCO3−. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. CO2/HCO3− in C.
We hypothesised that EC alkalinisation was the cause of the attenuated EDH-type vasorelaxation and investigated whether other manoeuvres that alkalinise the endothelium (Fig. 1A) could replicate the effect of removing CO2/HCO3−. Indeed, the response to acetylcholine was greatly attenuated when bath [Cl−] was reduced to 4.7 mmol l−1 by substitution with gluconate (Fig. 3A) or 3.2 mmol l−1 by substitution with methanesulphonate (Fig. 3B). Also, 200 μmol l−1 DIDS, a non-specific AE inhibitor, reduced EDH-type vasorelaxation (Fig. 3C). To further support that pHi is the mechanistic link between omission of CO2/HCO3− and reduced EDH-type vasorelaxation, we investigated whether restoring EC pHi in the absence of CO2/HCO3− would restore the EDH. Indeed, addition of 1 μmol l−1 cariporide not only returned EC pHi to near-normal levels (Fig. 1 and Supplementary Fig. 3), it also rescued the EDH-type vasorelaxation (Fig. 3D).
Figure 3. DIDS and low extracellular [Cl−] inhibit EDH-type vasorelaxation, while the Na+/H+ exchange inhibitor cariporide, which restores EC pHi, also rescues the EDH-type vasorelaxation in the absence of CO2/HCO3−.
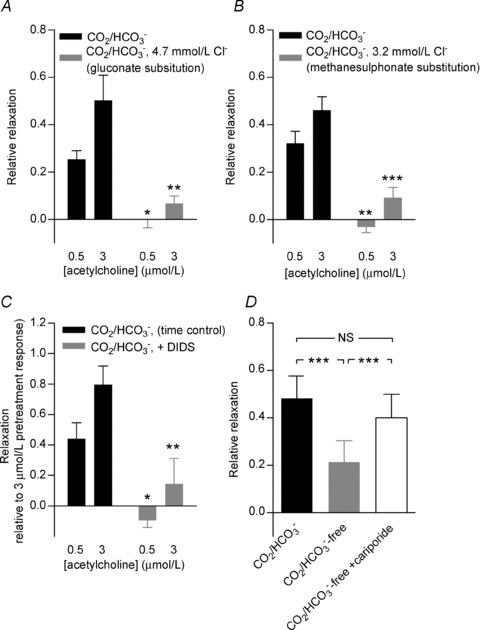
A, reduction of extracellular [Cl−] to 4.7 mmol l−1 by substitution with gluconate inhibits EDH-type vasorelaxation (n= 4). The effect of reducing or restoring extracellular [Cl−] was investigated 15 min after the solution change to allow pHi to reach a new steady-state level (Supplementary Fig. 1). B, reduction of extracellular [Cl−] to 3.2 mmol l−1 by substitution with methanesulphonate inhibits EDH-type vasorelaxation (n= 5). The effect of reducing or restoring extracellular [Cl−] was investigated 15 min after the solution change to allow pHi to reach a new steady-state level (Supplementary Fig. 1). C, addition of 200 μmol l−1 DIDS inhibits EDH-type vasorelaxation. The relaxation relative to a pre-treatment control relaxation is shown together with a time control run in parallel (n= 5). The effect of DIDS was investigated 20 min after addition of the drug to allow time for pHi to reach a new steady-state level. D, EDH-type vasorelaxation evoked by 3 μmol l−1 acetylcholine was restored by addition of 1 μmol l−1 cariporide in the absence of CO2/HCO3− (n= 8). The effect of cariporide was investigated 15 min after addition of the drug, when a new steady-state pHi level had been reached (Supplementary Fig. 3). Comparisons were made with a two-way ANOVA followed by a Bonferroni post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. CO2/HCO3− or as specified. NS, not significantly different.
To confirm that the attenuated EDH-type vasorelaxation was not due to a general inhibition of VSMC relaxation, we finally investigated the response to the endothelium-independent vasodilator S-nitroso-N-acetylpenicillamine (SNAP). We found no significant difference in the sensitivity (logEC50=−5.78 ± 0.06 with CO2/HCO3− and logEC50=−5.80 ± 0.10 without CO2/HCO3−; n= 6; P= 0.81, extra sum-of-squares F-test) or maximum response (maximum relative relaxation was 102 ± 4% with CO2/HCO3− and 94 ± 6% without CO2/HCO3−; n= 6; P= 0.30, extra sum-of-squares F-test) to the NO donor SNAP between arteries investigated in the presence and nominal absence of CO2/HCO3− (Supplementary Fig. 4).
VSMC membrane potential responses
To investigate the mechanistic background for the altered vascular responsiveness to acetylcholine during endothelial alkalinisation, we investigated whether it was associated with changes in VSMC membrane potential. The resting VSMC membrane potential was similar in the presence (Vm=−47.1 ± 1.5 mV; n= 11) and absence (Vm=−46.3 ± 1.1 mV; n= 9) of CO2/HCO3−. We showed also in paired experiments that omission of CO2/HCO3− did not change the VSMC membrane potential (ΔVm=−0.2 ± 2.5 mV; n= 5; P= 0.93, one-sample two-tailed t test).
We next measured VSMC membrane potential responses to acetylcholine in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin. In the presence of CO2/HCO3−, application of 3 μmol l−1 acetylcholine hyperpolarised VSMCs by 10.8 ± 2.0 mV (Fig. 4A and B). In contrast, when arteries were investigated in the absence of CO2/HCO3−, the VSMC membrane potential was unaffected by application of acetylcholine (ΔVm=−1.0 ± 1.0 mV; Fig. 4A and B).
Figure 4. EDH-type VSMC hyperpolarisations to acetylcholine and SLIGRL-amide are attenuated by omission of CO2/HCO3− while the response to pinacidil is unaffected.
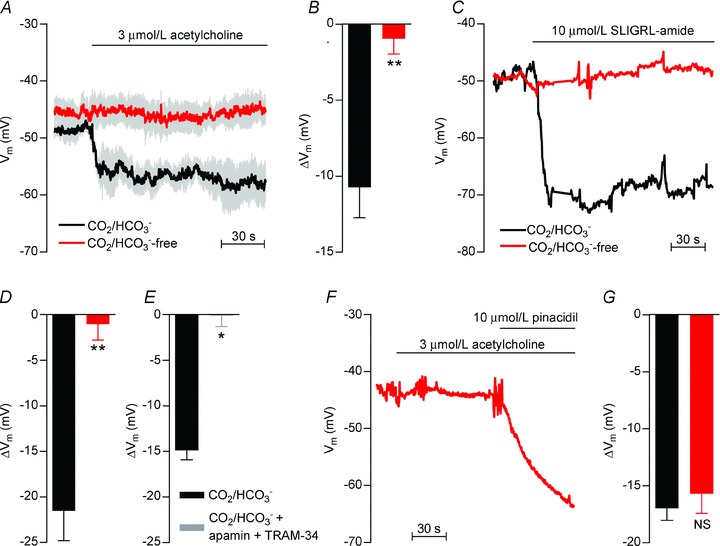
A, average traces (n= 4–5) of the VSMC membrane potential response to acetylcholine in the presence and absence of CO2/HCO3−. All experiments were performed in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin. Vertical grey lines represent SEM. Membrane potentials were measured at least 30 min after the change of bath solution to allow pHi to reach a new steady-state level. B, average effect (n= 4–5) of 3 μmol l−1 acetylcholine on VSMC membrane potential in the presence and absence of CO2/HCO3−. All experiments were performed in the presence of 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin. C, original traces showing the VSMC membrane potential response to 10 μmol l−1 SLIGRL-amide in the presence and absence of CO2/HCO3−. The experiments were performed in the presence of 100 nmol l−1 iberiotoxin, 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin. Membrane potentials were measured at least 30 min after the change of bath solution to allow pHi to reach a new steady-state level. D, average effect (n= 4) of 10 μmol l−1 SLIGRL-amide on VSMC membrane potential in the presence and absence of CO2/HCO3−. All experiments were performed in the presence of 100 nmol l−1 iberiotoxin, 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin. E, average effect (n= 3) of 10 μmol l−1 SLIGRL-amide on VSMC membrane potential in the presence and absence of 50 nmol l−1 apamin and 1 μmol l−1 TRAM-34. All experiments were performed in the presence of CO2/HCO3−, 100 μmol l−1 l-NAME, 3 μmol l−1 indomethacin and 100 nmol l−1 iberiotoxin. F, original trace of the VSMC membrane potential response to 3 μmol l−1 acetylcholine and 10 μmol l−1 pinacidil in the absence of CO2/HCO3−. G, average VSMC membrane potential responses (n= 2–3) to 10 μmol l−1 pinacidil applied in the presence or absence of CO2/HCO3−. Membrane potentials were measured at least 30 min after the change of bath solution to allow pHi to reach a new steady-state level. The comparisons were performed by paired or unpaired Student's t tests. *P < 0.05, **P < 0.01, NS: not significantly different vs. CO2/HCO3−.
To investigate whether another agonist which activates EDH shows a similar dependency on CO2/HCO3−, we measured the VSMC membrane potential response to 10 μmol l−1 of the proteinase-activated receptor-2 (PAR2) agonist SLIGRL-amide (McGuire et al. 2002). Initial experiments indicated that SLIGRL-amide in addition to endothelium-dependent effects acted directly on the VSMCs through activation of large conductance (BK) Ca2+-activated K+ channels; and hence, all experiments in addition to 100 μmol l−1 l-NAME and 3 μmol l−1 indomethacin were performed in the presence of 100 nmol l−1 of the BK-selective antagonist iberiotoxin. As shown in Fig. 4C and D, SLIGRL-amide induced a strong VSMC hyperpolarisation in the presence of CO2/HCO3− (ΔVm=−21.5 ± 3.3 mV). This response was completely abolished in the nominal absence of CO2/HCO3− (ΔVm=−1.0 ± 1.8 mV; Fig. 4C and D). Supporting the EDH-type nature of the response to SLIGRL-amide under the given conditions, we showed that the hyperpolarisation to 10 μmol l−1 SLIGRL-amide in the presence of CO2/HCO3− was completely inhibited after application of 50 nmol l−1 apamin and 1 μmol l−1 TRAM-34 (Fig. 4E).
Importantly, although no hyperpolarisation was seen to the endothelium-dependent agonists acetylcholine and SLIGRL-amide in the absence of CO2/HCO3−, a normal hyperpolarisation was evoked when pinacidil (10 μmol l−1) was applied under the same conditions to open ATP-sensitive K+ channels (KATP channels) in the VSMCs and thus alter VSMC membrane potential independently of the endothelium (Fig. 4F and G).
The mechanism of EDH
As previously described, the mechanism responsible for EDH remains controversial, and probably varies between vascular beds. Thus, we investigated which mechanism(s) was responsible for the acetylcholine-induced EDH-type vasorelaxation in the investigated mouse mesenteric arteries (Fig. 5). No effect of the Kir channel inhibitor Ba2+ (30 μmol l−1; Fig. 5A), the BK channel-specific inhibitor iberiotoxin (100 nmol l−1; Fig. 5B) or the H2O2 scavenging enzyme polyethylene glycol (PEG)-catalase (250 U ml−1; Fig. 5C) was seen. The EDH-type vasorelaxation, however, was inhibited by application of the gap junction inhibitors carbenoxolone (100 μmol l−1; Fig. 5D) or 18β-glycyrrhetinic acid (30 μmol l−1; Fig. 5E). Although these widely used gap junction inhibitors have additional unspecific effects (Matchkov et al. 2004; Behringer et al. 2012), our findings are consistent with the EDH-type vasorelaxation in mouse mesenteric small arteries being mediated by electrotonic spread of current through myoendothelial gap junctions.
Figure 5. EDH-type vasorelaxation is inhibited by gap junction inhibitors but unaffected by other putative modulators of the EDH.
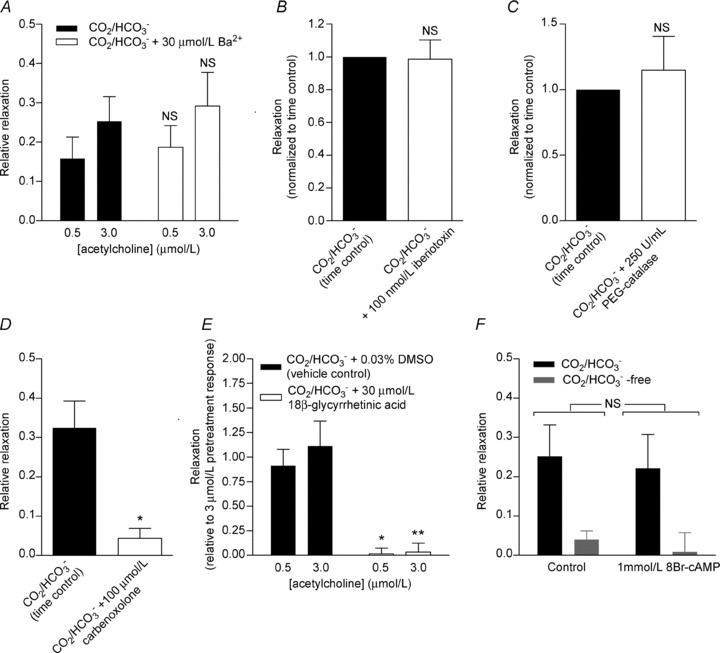
A, mean acetylcholine-induced relaxation of noradrenaline-precontracted arteries (n= 4) in CO2/HCO3−-containing PSS under control conditions or treated with 30 μmol l−1 Ba2+ to inhibit Kir channels. B, average relaxation to 3 μmol l−1 acetylcholine of noradrenaline-precontracted arteries (n= 4) in CO2/HCO3−-containing PSS under control conditions or treated with 100 nmol l−1 iberiotoxin to inhibit BK channels. C, mean relaxation to 3 μmol l−1 acetylcholine of noradrenaline-precontracted arteries (n= 4) in CO2/HCO3−-containing PSS under control conditions or treated with 250 U ml−1 of the H2O2 scavenger PEG-catalase. D, mean relaxation to 3 μmol l−1 acetylcholine of noradrenaline-precontracted arteries (n= 4) in CO2/HCO3−-containing PSS under control conditions or treated with 100 μmol l−1 of the gap junction inhibitor carbenoxolone. E, mean relaxation of noradrenaline-precontracted arteries to acetylcholine (n= 4) in CO2/HCO3−-containing PSS under control conditions or treated with 30 μmol l−1 of the gap junction inhibitor 18β-glycyrrhetinic acid. F, mean acetylcholine-induced relaxation of noradrenaline-precontracted arteries (n= 4) in the presence and absence of CO2/HCO3− under control conditions or treated with 1 mmol l−1 8Br-cAMP. The effect of omitting or restoring CO2/HCO3− was investigated 30 min after the buffer change to allow a new steady-state pHi to be reached. Comparisons in A, E and F were performed by two-way ANOVA followed by Bonferroni post-hoc tests. Comparisons in B, C and D were performed by paired Student's t test. *P < 0.05, **P < 0.01 vs. CO2/HCO3−. NS, not significantly different from CO2/HCO3− or as indicated.
It has previously been suggested that cAMP can facilitate EDH-type vasorelaxation by enhancing conduction through gap junctions (Griffith et al. 2002). We tested whether reduced cAMP levels could be responsible for the inhibitory action of high EC pHi on the EDH-type response. The cell-permeable cAMP analogue 8Br-cAMP (1 mmol l−1), however, had no effect on the vasorelaxation to acetylcholine in the presence or in the absence of CO2/HCO3− (Fig. 5F).
Endothelial [Ca2+]i responses
To further investigate the mechanistic explanation for the inhibition of the EDH during EC alkalinisation, we examined the EC [Ca2+]i response to acetylcholine (Fig. 6). The resting baseline level of the [Ca2+]-sensitive fluorescence ratio in the absence of CO2/HCO3− was 95 ± 4% of that seen in the presence of CO2/HCO3− (n= 5; P= 0.26; paired two-tailed Student's t test) suggesting that the resting level of [Ca2+]i in the ECs does not differ between arteries in the presence and absence of CO2/HCO3−. Upon application of 3 μmol l−1 acetylcholine, around 50% of the ECs responded with an increase in the [Ca2+]-dependent fluorescence ratio greater than 20% of the response to 10 μmol l−1 of the endoplasmic reticulum Ca2+-ATPase inhibitor CPA (Fig. 6A). The distribution of ECs based on the magnitude of their Ca2+ response to acetylcholine was not significantly different in the presence and absence of CO2/HCO3− (Fig. 6A). Furthermore, no difference in the [Ca2+]-dependent fluorescence ratio was observed when responses to acetylcholine were elicited in the same ECs in the presence and absence of CO2/HCO3− (Fig. 6B). The order of the experimental protocol was alternated between preparations to avoid possible effects of time. Taken together, these findings suggest that activation of ECs by acetylcholine and the associated [Ca2+]i response are not affected by an alkaline shift in EC pHi.
Figure 6. Endothelial [Ca2+]i responses to acetylcholine stimulation are unaffected by intracellular alkalinisation.
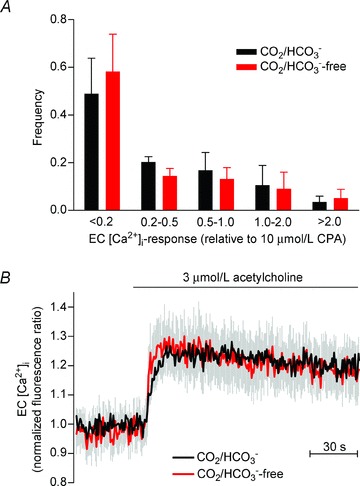
A, bar chart showing the fraction of ECs responding with a given magnitude of Ca2+i response relative to the average CPA response. All ECs loaded with fluorescent dye within the field of view were analysed. No significant effect of CO2/HCO3− on the frequency distribution was observed (n= 6; P= 0.90; two-way ANOVA). B, mean traces (n= 6) of the changes in EC [Ca2+]i-dependent fluorescence during acetylcholine stimulation. The traces represent paired analyses of the same ECs investigated both in the presence and in the absence of CO2/HCO3−. Cells that responded during the first stimulation to acetylcholine were analysed. The Ca2+i response in the absence of CO2/HCO3− was not significantly different from the Ca2+i response in the presence of CO2/HCO3− (P= 0.70; paired two-tailed Student's t test). The effect of omitting or restoring CO2/HCO3− was investigated 30 min after the buffer change to allow a new steady-state pHi to be reached (Supplementary Fig. 2A and B).
Endothelial membrane potential responses
Next, we aimed to investigate whether the inhibited EDH-type vasorelaxation seen during EC alkalinisation was consequent to a blunted endothelial hyperpolarisation to acetylcholine or due to reduced transfer of the response to the VSMCs. We measured EC membrane potentials in the presence and absence of CO2/HCO3− and investigated the membrane potential response to acetylcholine (Fig. 7). The ability to impale ECs and VSMCs selectively was confirmed by including propidium iodide in the electrode solution and evaluating the shape and orientation of the stained cell nuclei (Fig. 8A and Supplementary Fig. 5).
Figure 7. The hyperpolarisation of ECs upon acetylcholine stimulation is unaffected by intracellular alkalinisation.
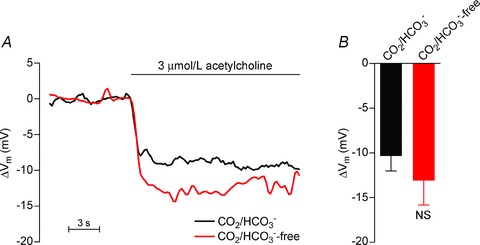
A, original traces of the EC membrane potential response to acetylcholine stimulation in the presence and absence of CO2/HCO3−. Membrane potentials were measured at least 30 min after the change of bath solution to allow pHi to reach a new steady-state level. B, mean peak membrane potential changes in ECs (n= 4–5) following stimulation with 3 μmol l−1 acetylcholine in the presence and absence of CO2/HCO3−. The comparison was performed using unpaired, two-tailed Student's t test. NS, not significantly different vs. CO2/HCO3−.
Figure 8. Intercellular dye coupling (propidium iodide) is attenuated at high EC pHi.
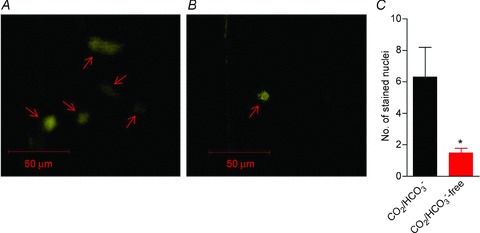
A, representative example of the staining pattern seen after impalement of an EC in the presence of CO2/HCO3−. As exemplified, the dye has spread from the impaled cell to at least four additional ECs (arrows) with a somewhat reduced staining intensity in the coupled cells compared with the impaled cell. B, in the absence of CO2/HCO3−, intercellular dye coupling was greatly attenuated. In the example illustrated, only the impaled EC (arrow) was stained. C, mean number of stained cells after EC impalement was significantly lower in the absence than in the presence of CO2/HCO3−, indicating reduced intercellular dye coupling (n= 3–4). The comparison was performed by a non-parametric Mann–Whitney test. *P < 0.05 vs. CO2/HCO3−.
The resting EC membrane potential was −42.9 ± 2.5 mV (n= 5) in the absence of CO2/HCO3−, which was not significantly different from that recorded in the presence of CO2/HCO3− (−42.7 ± 4.8 mV; n= 4; P= 0.97, unpaired two-tailed Student's t test). Furthermore, application of 3 μmol l−1 acetylcholine hyperpolarised ECs to the same extent in the presence (ΔVm=−10.4 ± 1.7 mV) and absence (ΔVm=−13.1 ± 2.7 mV) of CO2/HCO3− (Fig. 7A and B). These findings suggest that reduced current transfer across the myoendothelial gap junctions is responsible for the attenuation of EDH-type vasorelaxation during EC alkalinisation.
Intercellular dye coupling
To further examine the intercellular communication at normal and high endothelial pHi, we investigated the level of dye coupling in the presence and absence of CO2/HCO3− (Fig. 8). In the presence of CO2/HCO3−– consistent with reports from others (Behringer et al. 2012) – we observed dye coupling between ECs with a mean of 6.3 ± 1.9 stained nuclei (Fig. 8A and C). This intercellular dye coupling was greatly reduced in the absence of CO2/HCO3− when only 1.5 ± 0.3 stained nuclei were seen (Fig. 8B and C), signifying that the dye was almost completely restricted to the impaled EC.
Discussion
In the current study, we investigated the effects of disturbed intracellular acid–base balance on integrated vascular function and show for the first time that EC alkalinisation inhibits gap junction communication and myoendothelial signalling. Based on ion imaging in combination with pharmacological, electrophysiological and dye coupling experiments, we propose that a pHi-induced inhibition of myoendothelial gap junction communication is responsible for the attenuation of the EDH observed at high EC pHi. Reduced gap junction conductivity at high pHi has previously been reported for isolated ventricular cardiomyocyte cell pairs (Swietach et al. 2007) and mouse neuroblastoma N2A cell pairs heterologously expressing connexin 36 (Gonzalez-Nieto et al. 2008) but the applicability in the vascular wall and its implications for vasomotor function have not previously been determined.
We investigated effects of manoeuvres that inhibit Cl−/HCO3− exchange on EC pHi and [Ca2+]i by fluorescence confocal microscopy of isolated mesenteric arteries (Boedtkjer & Aalkjaer, 2009; Boedtkjer et al. 2011). The steady-state resting pHi recorded with this technique in the presence of CO2/HCO3− varies between 7.05 and 7.35 (Boedtkjer & Aalkjaer, 2009; Boedtkjer et al. 2011, 2012) in arteries from mice on different genetic backgrounds (NMRI, FVB, C57BL/6J), which is in good agreement with measurements from other EC preparations (Fleming et al. 1994; Sun et al. 1999; Taylor et al. 2006).
We provide three independent pieces of evidence for inhibition of gap junctions in mesenteric arteries during endothelial alkalinisation. (1) The EDH-type vasorelaxation – which in this preparation is sensitive to gap junction inhibition – and the associated VSMC hyperpolarisation are attenuated at high EC pHi despite a normal hyperpolarisation of ECs. (2) Intercellular dye coupling is reduced at high pHi. (3) In the presence of CO2/HCO3− when the EDH is intact, ECs (7.24 ± 0.09) and VSMCs (7.13 ± 0.03; Boedtkjer et al. 2006) have fairly similar pHi levels. In the absence of CO2/HCO3− when EDH is inhibited, a pH difference of approximately 0.8 pH units exists between the ECs (pHi= 7.53 ± 0.05) and the VSMCs (pHi= 6.73 ± 0.03; Boedtkjer et al. 2006). The opposite pHi changes observed in ECs and VSMCs upon omission of CO2/HCO3− probably reflect their different rates of Cl−/HCO3− exchange relative to Na+,HCO3− cotransport and hence the net direction of HCO3− transport. Regardless, it is difficult to envisage that a pH difference of 0.8 units can be maintained between cell types connected by low resistance couplings as gap junctions have previously been shown to be permeable to protons carried on diffusible buffers (Zaniboni et al. 2003). Together, these findings strongly imply that gap junctions in the artery wall are inhibited at high EC pHi and that EDH-type vasorelaxation is consequently attenuated. That pHi is the link between altered AE activity and reduced EDH-type vasorelaxation is confirmed by the finding that the NHE1 inhibitor cariporide restored not only EC pHi but also the vasorelaxant response to acetylcholine when applied in the absence of CO2/HCO3−.
We induced intracellular alkalinisation of ECs by inhibiting or reversing Cl−/HCO3− exchange activity through several different means, including omission of CO2/HCO3−. The crucial role of the CO2/HCO3− buffer system for myoendothelial signalling is consistent with a previous report that substitution of CO2/HCO3− with artificial buffers (Tris or Hepes) attenuates endothelium-dependent relaxation of the rabbit aorta (Hayashi & Hester, 1987) although this previous study did not investigate the mechanistic background (or the role of pHi) for the change in vasorelaxation.
Changes in pHi have multiple cellular targets and modulate enzymatic activities, intracellular Ca2+ handling and ion channel function, among others (Boedtkjer & Aalkjaer, 2012).
In the current study, we show that EC Ca2+ handling is unaffected by the pHi disturbance induced by omission of CO2/HCO3−. This is probably due to the prolonged nature of the pHi disturbance: in VSMCs, it has been shown that while acute changes in pHi greatly alter Ca2+ signalling, sustained changes in pHi have no effect on intracellular Ca2+ handling (Boedtkjer & Aalkjaer, 2012). Acute changes in pHi have been suggested to interfere with cellular Ca2+ handling by modifying the membrane potential, inhibiting Ca2+ transport across the plasmalemma or the endoplasmic reticulum membrane or by competition between H+ and cytosolic Ca2+ for buffer binding (Boedtkjer & Aalkjaer, 2012). The apparent Ca2+-independent nature of the inhibition of intercellular coupling by high EC pHi is consistent with previous findings from isolated cardiomyocyte cell pairs demonstrating that alkaline shifts in pHi reduce gap junction conductivity even in the presence of high intracellular concentrations of the Ca2+ buffer BAPTA (Swietach et al. 2007).
Effects of pHi on ion channel function have been suggested to play a role in vascular cells (Boedtkjer & Aalkjaer, 2012). In the context of the present study, it is relevant to note that most K+ channels are inhibited at low rather than high pHi (Xu et al. 1996; Schubert et al. 2001; Mao et al. 2003; Van Slyke et al. 2012) and hence inhibition of IK and SK channels is an unlikely explanation for the reduced EDH. This conclusion is further supported by our observation that the EC hyperpolarisation upon acetylcholine stimulation was not significantly affected by CO2/HCO3−-free conditions.
The inhibition of EDH described in the present study could in principle be caused by a change in either ECs or VSMCs. Lowering the extracellular concentration of Cl− alkalinises both ECs and VSMCs (Aalkjaer & Hughes, 1991), but the other interventions differ in their effect on pHi between ECs and VSMCs. As such, even though both omission of CO2/HCO3− and addition of DIDS alkalinise ECs, removal of CO2/HCO3− causes an approximately 0.4 pH unit intracellular acidification of mesenteric artery VSMCs from NMRI mice (Boedtkjer et al. 2006) while addition of 200 μmol l−1 DIDS has no significant effect on VSMC pHi (ΔpHi=−0.07 ± 0.06; n= 5; P= 0.37; our unpublished observation). As noted above, the contrasting effects of CO2/HCO3−-free conditions and addition of DIDS on EC and VSMC pHi probably reflect that in VSMCs Na+,HCO3− cotransport activity is larger than Cl−/HCO3− exchange activity (Boedtkjer et al. 2006), while in the ECs the relationship appears to be the opposite. Supporting a primary role for changes in the intracellular environment of ECs rather than VSMCs, we furthermore demonstrated that dye coupling between ECs was reduced in the absence of CO2/HCO3−.
As mentioned above, previous studies have described changes in cellular coupling induced by an alkaline shift in pHi based on studies with isolated cardiomyocyte cell pairs which primarily express connexin 43 (Swietach et al. 2007) and N2A cell pairs heterologously expressing connexin 36 (Gonzalez-Nieto et al. 2008). In the vasculature, the myoendothelial gap junctions have been shown to consist of connexin 37 and connexin 40 (Haddock et al. 2006) while connexin 43 expression has also been reported in ECs (Hill et al. 2002). The inhibition of gap junction communication by high pHi in diverse experimental preparations with different expression patterns of connexin proteins may suggest that regulation of intercellular coupling by pHi in the alkaline range is a general phenomenon although additional studies are required to investigate the effects of pHi on other connexin isoforms and cellular coupling in other cell and tissue types.
The molecular mechanism responsible for inhibition of gap junction-dependent cellular coupling by high pHi has not been conclusively determined. In previous studies using isolated cell pairs, it has been argued that a chemical gating mechanism is more likely than a change in membrane expression of connexin proteins based on the fast and reversible inhibition of cellular coupling following an increase in pHi (Swietach et al. 2007). Due to the more complex nature of our preparation and the difficulty of maintaining cell impalements for electrical recordings during solution changes, we did not investigate the detailed time course of the change in cellular coupling. Instead we studied the changes in mechanical and electrical properties when pHi after a change of bath solution or addition of drug had reached a new steady-state level after 15–30 min. Consistent with previous findings from ventricular cardiomyocyte cell pairs (Swietach et al. 2007), we showed that the inhibition of myoendothelial coupling was reversible. Intracellular trafficking of connexin 43 proteins (labelled with green fluorescent protein) heterologously expressed in A7r5 rat VSMCs following treatment with ouabain has previously been shown to be a slow process beginning within 1–2 h and completed after approximately 4 h (Martin et al. 2003). The relatively fast inhibition of cellular coupling observed in the present study and its reversible nature are consistent with the previous proposition that a high pHi inhibits cellular coupling via a chemical gating mechanism although at this point we cannot exclude that a change in membrane expression of gap junction proteins may also play a role.
Interestingly, although we have previously shown that EC acidification inhibits NO synthesis (Boedtkjer et al. 2011, 2012), we show in the current study that NO dependent vasorelaxation is unaffected by a 0.2–0.3 pH unit alkalinisation. This finding is in agreement with previous in vitro kinetic analyses showing that the isolated eNOS has a bell-shaped pH dependency with maximal enzymatic activity around pH 7.5 (Fleming et al. 1994). As resting EC pHi is slightly below this pH optimum, one would indeed predict that the changes in enzymatic activity caused by a moderate acidification would be far greater than the changes in enzymatic activity observed during an alkalinisation of similar magnitude.
In conclusion, we propose that disturbed pHi regulation is an important parameter for modulating integrated vascular responses. In particular, myoendothelial signalling is inhibited by moderate EC alkalinisation and this may be a mechanism for altered EDH during pathological conditions.
Acknowledgments
The Water and Salt Research Center at Aarhus University was established and supported by the Danish National Research Foundation. This work was supported by the Danish Council for Independent Research (grant nos. 10-094816 to E.B. and 271-06-0472 to C.A.), the Danish Heart Foundation (grant no. 08-10-R68-A2179-B719-22494 to E.B.) and the Lundbeck Foundation (grant no. R93-A8859 to E.B.).
Glossary
- 11,12-EET
11,12-epoxyeicosatrienoic acid
- AE
anion exchanger
- BCPCF
2′,7′-bis-(carboxypropyl)-5(6)-carboxyfluorescein
- BK channels
large-conductance Ca2+-activated K+ channels
- CNP
C-type natriuretic peptide
- COX
cyclooxygenase
- CPA
cyclopiazonic acid
- DIDS
4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid
- EC
endothelial cell
- eNOS
endothelial NO-synthase
- EDH
endothelium-derived hyperpolarisation
- EDHF
endothelium-derived hyperpolarising factor
- IK channels
intermediate-conductance Ca2+-activated K+ channels
- KATP channels
ATP-sensitive K+ channels
- Kir channels
inward rectifier K+ channels
- l-NAME
N-nitro-l-arginine methyl ester
- PAR2
proteinase-activated receptor 2
- PEG
polyethylene glycol
- pHi
intracellular pH
- PSS
physiological salt solution
- SK channels
small-conductance Ca2+-activated K+ channels
- SNAP
S-nitroso-N-acetylpenicillamine
- Vm
membrane potential
- VSMC
vascular smooth muscle cell
Author contributions
Experiments were performed at the Department of Biomedicine, Aarhus University, Denmark. E.B. and C.A. conceived the project, designed the experiments and interpreted the data. E.B. and S.K. collected the data. E.B. analysed the data and wrote the manuscript. All authors revised the manuscript for important intellectual content and approved the final version.
Supplementary material
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3
Supplementary Fig. 4
Supplementary Fig. 5
References
- Aalkjaer C, Hughes A. Chloride and bicarbonate transport in rat resistance arteries. J Physiol. 1991;436:57–73. doi: 10.1113/jphysiol.1991.sp018539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Socha MJ, Polo-Parada L, Segal SS. Electrical conduction along endothelial cell tubes from mouse feed arteries: confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol. 2012;166:774–787. doi: 10.1111/j.1476-5381.2011.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellien J, Thuillez C, Joannides R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam Clin Pharmacol. 2008;22:363–377. doi: 10.1111/j.1472-8206.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Aalkjaer C. Insulin inhibits Na+/H+ exchange in vascular smooth muscle and endothelial cells in situ: involvement of H2O2 and tyrosine phosphatase SHP-2. Am J Physiol Heart Circ Physiol. 2009;296:H247–H255. doi: 10.1152/ajpheart.00725.2008. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Aalkjaer C. Intracelullar pH in the resistance vasculature: regulation and functional implications. J Vasc Res. 2012;49:479–496. doi: 10.1159/000341235. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Damkier HH, Aalkjaer C. NHE1 knockout reduces blood pressure and arterial media/lumen ratio with no effect on resting pHi in the vascular wall. J Physiol. 2012;590:1895–1906. doi: 10.1113/jphysiol.2011.227132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Aalkjaer C. NBCn1 (slc4a7) mediates the Na+-dependent bicarbonate transport important for regulation of intracellular pH in mouse vascular smooth muscle cells. Circ Res. 2006;98:515–523. doi: 10.1161/01.RES.0000204750.04971.76. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Fuchtbauer EM, Aalkjaer C. Disruption of Na+,HCO3− cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca2+-sensitivity and hypertension development in mice. Circulation. 2011;124:1819–1829. doi: 10.1161/CIRCULATIONAHA.110.015974. [DOI] [PubMed] [Google Scholar]
- Brosius FC, III, Pisoni RL, Cao X, Deshmukh G, Yannoukakos D, Stuart-Tilley AK, Haller C, Alper SL. AE anion exchanger mRNA and protein expression in vascular smooth muscle cells, aorta, and renal microvessels. Am J Physiol. 1997;273:F1039–F1047. doi: 10.1152/ajprenal.1997.273.6.F1039. [DOI] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Chauhan SD, Nilsson H, Ahluwalia A, Hobbs AJ. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc Natl Acad Sci U S A. 2003;100:1426–1431. doi: 10.1073/pnas.0336365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. EDHF: new therapeutic targets. Pharmacol Res. 2004;49:565–580. doi: 10.1016/j.phrs.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- Fleming I, Hecker M, Busse R. Intracellular alkalinization induced by bradykinin sustains activation of the constitutive nitric oxide synthase in endothelial cells. Circ Res. 1994;74:1220–1226. doi: 10.1161/01.res.74.6.1220. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Nieto D, Gomez-Hernandez JM, Larrosa B, Gutierrez C, Munoz MD, Fasciani I, O’Brien J, Zappala A, Cicirata F, Barrio LC. Regulation of neuronal connexin-36 channels by pH. Proc Natl Acad Sci U S A. 2008;105:17169–17174. doi: 10.1073/pnas.0804189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Fujii K, Kansui Y, Abe I, Iida M. Critical role of gap junctions in endothelium-dependent hyperpolarization in rat mesenteric arteries. Clin Exp Pharmacol Physiol. 2002;29:595–602. doi: 10.1046/j.1440-1681.2002.03689.x. [DOI] [PubMed] [Google Scholar]
- Griffith TM, Chaytor AT, Taylor HJ, Giddings BD, Edwards DH. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc Natl Acad Sci U S A. 2002;99:6392–6397. doi: 10.1073/pnas.092089799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddock RE, Grayson TH, Brackenbury TD, Meaney KR, Neylon CB, Sandow SL, Hill CE. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am J Physiol Heart Circ Physiol. 2006;291:H2047–H2056. doi: 10.1152/ajpheart.00484.2006. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Hester RK. Endothelium-dependent relaxations in rabbit aorta are depressed in artificial buffered solutions. J Pharmacol Exp Ther. 1987;242:523–530. [PubMed] [Google Scholar]
- Hill CE, Rummery N, Hickey H, Sandow SL. Heterogeneity in the distribution of vascular gap junctions and connexins: implications for function. Clin Exp Pharmacol Physiol. 2002;29:620–625. doi: 10.1046/j.1440-1681.2002.03699.x. [DOI] [PubMed] [Google Scholar]
- Mao J, Wu J, Chen F, Wang X, Jiang C. Inhibition of G-protein-coupled inward rectifying K+ channels by intracellular acidosis. J Biol Chem. 2003;278:7091–7098. doi: 10.1074/jbc.M211461200. [DOI] [PubMed] [Google Scholar]
- Martin PE, Hill NS, Kristensen B, Errington RJ, Griffith TM. Ouabain exerts biphasic effects on connexin functionality and expression in vascular smooth muscle cells. Br J Pharmacol. 2003;140:1261–1271. doi: 10.1038/sj.bjp.0705556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchkov VV, Rahman A, Peng H, Nilsson H, Aalkjaer C. Junctional and nonjunctional effects of heptanol and glycyrrhetinic acid derivates in rat mesenteric small arteries. Br J Pharmacol. 2004;142:961–972. doi: 10.1038/sj.bjp.0705870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JJ, Hollenberg MD, Andrade-Gordon P, Triggle CR. Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase-activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol. 2002;135:155–169. doi: 10.1038/sj.bjp.0704469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Schubert R, Krien U, Gagov H. Protons inhibit the BKCa channel of rat small artery smooth muscle cells. J Vasc Res. 2001;38:30–38. doi: 10.1159/000051027. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Matoba T. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pharmacol Res. 2004;49:543–549. doi: 10.1016/j.phrs.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Siegl D, Koeppen M, Wolfle SE, Pohl U, De Wit C. Myoendothelial coupling is not prominent in arterioles within the mouse cremaster microcirculation in vivo. Circ Res. 2005;97:781–788. doi: 10.1161/01.RES.0000186193.22438.6c. [DOI] [PubMed] [Google Scholar]
- Spray DC, Harris AL, Bennett MV. Gap junctional conductance is a simple and sensitive function of intracellular pH. Science. 1981;211:712–715. doi: 10.1126/science.6779379. [DOI] [PubMed] [Google Scholar]
- Sun B, Vaughan-Jones RD, Kambayashi JI. Two distinct HCO3−-dependent H+ efflux pathways in human vascular endothelial cells. Am J Physiol. 1999;277:H28–H32. doi: 10.1152/ajpheart.1999.277.1.H28. [DOI] [PubMed] [Google Scholar]
- Swietach P, Rossini A, Spitzer KW, Vaughan-Jones RD. H+ ion activation and inactivation of the ventricular gap junction: a basis for spatial regulation of intracellular pH. Circ Res. 2007;100:1045–1054. doi: 10.1161/01.RES.0000264071.11619.47. [DOI] [PubMed] [Google Scholar]
- Taylor CJ, Nicola PA, Wang S, Barrand MA, Hladky SB. Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol. 2006;576:769–785. doi: 10.1113/jphysiol.2006.117374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turin L, Warner A. Carbon dioxide reversibly abolishes ionic communication between cells of early amphibian embryo. Nature. 1977;270:56–57. doi: 10.1038/270056a0. [DOI] [PubMed] [Google Scholar]
- Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest. 1997;100:2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Slyke AC, Cheng YM, Mafi P, Allard CR, Hull CM, Shi YP, Claydon TW. Proton block of the pore underlies the inhibition of hERG cardiac K+ channels during acidosis. Am J Physiol Cell Physiol. 2012;302:C1797–C1806. doi: 10.1152/ajpcell.00324.2011. [DOI] [PubMed] [Google Scholar]
- Xu Z, Patel KP, Rozanski GJ. Intracellular protons inhibit transient outward K+ current in ventricular myocytes from diabetic rats. Am J Physiol. 1996;271:H2154–H2161. doi: 10.1152/ajpheart.1996.271.5.H2154. [DOI] [PubMed] [Google Scholar]
- Zaniboni M, Rossini A, Swietach P, Banger N, Spitzer KW, Vaughan-Jones RD. Proton permeation through the myocardial gap junction. Circ Res. 2003;93:726–735. doi: 10.1161/01.RES.0000093986.47383.CE. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.