

Abstract
There is evidence that vasoactive intestinal polypeptide (VIP) participates in inhibitory neuromuscular transmission (NMT) in the internal anal sphincter (IAS). However, specific details concerning VIP-ergic NMT are limited, largely because of difficulties in selectively blocking other inhibitory neural pathways. The present study used the selective P2Y1 receptor antagonist MRS2500 (1 μm) and the nitric oxide synthase inhibitor NG-nitro-l-arginine (l-NNA; 100 μm) to block purinergic and nitrergic NMT to characterize non-purinergic, non-nitrergic (NNNP) inhibitory NMT and the role of VIP in this response. Nerves were stimulated with electrical field stimulation (0.1–20 Hz, 4–60 s) and the associated changes in contractile and electrical activity measured in non-adrenergic, non-cholinergic conditions in the IAS of wild-type and VIP−/− mice. Electrical field stimulation gave rise to frequency-dependent relaxation and hyperpolarization that was blocked by tetrodotoxin. Responses during brief trains of stimuli (4 s) were mediated by purinergic and nitrergic NMT. During longer stimulus trains, an NNNP relaxation and hyperpolarization developed slowly and persisted for several minutes beyond the end of the stimulus train. The NNNP NMT was abolished by VIP6–28 (30 μm), absent in the VIP−/− mouse and mimicked by exogenous VIP (1–100 nm). Immunoreactivity for VIP was co-localized with neuronal nitric oxide synthase in varicose intramuscular fibres but was not detected in the VIP−/− mouse IAS. In conclusion, this study identified an ultraslow component of inhibitory NMT in the IAS mediated by VIP. In vivo, this pathway may be activated with larger rectal distensions, leading to a more prolonged period of anal relaxation.
Key points
Vasoactive intestinal polypeptide (VIP)-ergic neuromuscular transmission (NMT) was examined in the internal anal sphincter of wild-type mice and compared with that of VIP−/− mice.
Relaxation and hyperpolarization during brief trains of electrical field stimulation (EFS; 4 s) were mediated by purinergic and nitrergic NMT.
During longer stimulus trains, a non-purinergic, non-nitrergic (NNNP) relaxation and hyperpolarization slowly developed and persisted for several minutes beyond the end of the stimulus train.
The NNNP NMT was abolished by VIP receptor antagonists, absent in the VIP−/− mouse internal anal sphincter and mimicked by exogenous VIP.
These data suggest that NNNP NMT gives rise to ultraslow relaxation and hyperpolarization that is mediated by VIP. In vivo, this pathway may be activated with larger rectal distensions, leading to a more prolonged anal relaxation.
Introduction
Vasoactive intestinal polypeptide (VIP) was originally isolated from pig intestinal extracts and found to be a potent vasodilator (Said & Mutt, 1970). It was subsequently identified in nerves of the gastrointestinal (GI) tract (Bryant et al. 1976; Larsson et al. 1976), including some with projections consistent with enteric inhibitory motor neurons (Furness & Costa, 1979). In vivo studies in the 1970s also identified VIP accumulation in the venous drainage of the stomach accompanying activation of vagal afferents either through reflex pathways or via direct electrical stimulation. In both cases, the appearance of VIP correlated with the development of stomach relaxation (Fahrenkrug et al. 1978). In addition, both in vivo and in vitro experiments have reported that a component of nerve-evoked relaxation is blocked by procedures such as immunoneutralization of VIP or breakdown of VIP with peptidases like α-chymotrypsin (Goyal et al. 1980; Grider et al. 1985a,b). Exogenous VIP was also shown to be a potent inhibitor of contraction in various gastrointestinal smooth muscles (e.g. Eklund et al. 1979; Bitar & Makhlouf, 1982). Taken together, these data strongly support a role for VIP in inhibitory neuromuscular transmission (NMT) in the gastrointestinal tract (for review see Fahrenkrug, 1993; Van Geldre & Lefebvre, 2004; Furness, 2006).
Vasoactive intestinal polypeptide has been reported to contribute to inhibitory NMT in the internal anal sphincter (IAS) of various animal species, including humans (Burleigh et al. 1979; Biancani et al. 1985; Nurko & Rattan, 1988; Rattan et al. 2005; Raghavan et al. 2011), along with nitrergic and purineric NMT (Burleigh, 1992; O’Kelly et al. 1993; Rae & Muir, 1996; De Luca et al. 1999; Jones et al. 2003; Opazo et al. 2009, 2011). However, until recently it has been difficult specifically to isolate VIP-ergic NMT, because of the overlapping effects of nitrergic and purinergic NMT and the absence of effective blockers of these pathways. It was not until 1990 and thereafter that nitric oxide was finally recognized as a key component of inhibitory NMT in GI muscles and that blockers became available to eliminate this pathway (see Rand, 1992). Likewise, although ATP was the first of the ‘non-adrenergic, non-cholinergic’ substances proposed as an inhibitory neurotransmitter in the GI tract (for review see Burnstock, 1972), studies of purinergic NMT have historically been complicated by the wide range of purinergic receptor subtypes possibly involved and the absence of selective and efficacious antagonists of these receptors and their pathways. Recently, studies demonstrating the important role of P2Y1 receptors in purinergic NMT in the GI tract have improved the situation, because this receptor can be blocked effectively in some species with the P2Y1 receptor antagonist MRS2179 (Gallego et al. 2006; Wang et al. 2007; Opazo et al. 2009). However, even this antagonist is of limited utility in some animals (e.g. rats and mice). In 2009, the P2Y1 receptor antagonist MRS2500 was identified as a useful alternative to MRS2179 in the rat colon (Grasa et al. 2009) and 2 years later in the rat IAS (Opazo et al. 2011).
In a previous study, we characterized the electrical and contractile events underlying inhibitory NMT in the mouse IAS using the nitric oxide synthase (NOS) inhibitor NG-nitro-l-arginine (l-NNA) and the P2Y1 receptor antagonist MRS2500 (Duffy et al. 2012). A single stimulus was sufficient to activate either nitrergic or purinergic NMT, although maximal responses required additional stimuli. A third non-nitrergic, non-purinergic (NNNP) component was also noted with longer trains of electrical field stimulation (EFS; e.g. 5 Hz for 60 s) but was not examined further in that study. We concluded that inhibitory NMT accompanying brief trains of stimuli (e.g. 5 Hz EFS for 10 s) was mediated by nitrergic and purinergic NMT. This conclusion is in agreement with a previous study of the rat IAS using the same antagonists and similar protocols (Opazo et al. 2011) and is also supported by two earlier studies of the rat and guinea-pig IAS (Rae & Muir, 1996; De Luca et al. 1999). In contrast, a 2005 study (Rattan et al. 2005) concluded that responses elicited with brief trains of stimuli (4 s, 1–20 Hz) were mediated by nitrergic and VIP-ergic NMT in the mouse IAS.
The present study examined the time course and magnitude of nerve-evoked NNNP relaxation and hyperpolarization in the mouse IAS and the stimulus parameters required to elicit this pathway. In addition, some of the experiments described by Duffy et al. (2012) were repeated using the protocol described by Rattan et al. (2005) to determine whether differences between studies could be accounted for by differences in experimental protocol. The role of VIP in NNNP inhibitory NMT was also examined by testing various VIP receptor agonists and antagonists. Stimulus parameters known to elicit NNNP NMT in wild-type (WT) mice were also repeated in the VIP−/− mouse IAS to determine whether this component was modified in the absence of VIP expression. Our results indicate that NNNP inhibitory NMT occurs with longer stimulus trains and higher frequencies of EFS and is likely to be mediated by VIP.
Methods
Tissue preparation
C57BL/6 WT (n = 79) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). JR#9640-B6.129S4-Vip<tm1Clw>/J heterozygote female and homozygote male breeding pairs (Colwell et al. 2003) were also obtained from Jackson Laboratories and were mated to produce homozygote offspring (VIP−/−), confirmed with genotyping (n = 33). All animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, 2011), and all experiments and procedures were approved by the Institutional Animal Use and Care Committee at the University of Nevada. Mice (3–4 months of age) were killed by inhalation of isoflurane (Baxter, Deerfield, IL, USA) followed by cervical dislocation. Overlying tissue was dissected away to permit removal of the rectoanal region. Rectoanal tissue was then pinned in a dissecting dish containing cold Krebs–Ringer buffer solution of the following composition (mm): NaCl, 118.5; KCl, 4.7; CaCl2, 2.5; MgCl2, 1.2; NaHCO3, 23.8; KH2PO4, 1.2; and dextrose, 11.0. The Krebs–Ringer solution was maintained at a pH of 7.4 at 37°C by bubbling to equilibrium with 95%O2–5%CO2. The IAS was identified at the most distal part of the gastrointestinal tract. All adherent skeletal muscle, glands and mucosa were carefully dissected away before carrying out experiments. All experiments were performed in the presence of 1 μm guanethidine and 1 μm atropine (i.e. non-adrenergic, non-cholinergic conditions).
Immunohistochemistry
Cryostat sections
Two-millimetre-wide rectoanal ring preparations were made from C57BL/6 and VIP−/− mice by removing all adherent skeletal muscle and glands whilst keeping the tubular structure of the rectoanal region intact. Preparations were fixed for 15 min with ice-cold paraformaldehyde at 20°C and then washed in 0.1 m phosphate buffer solution (PBS) overnight at 4°C. Rings were dehydrated in graded sucrose solutions (5, 10 and 15% for 15 min each and 20% overnight) and embedded in a 1:1 solution of Tissue Tek OTC compound (Sakura Finetek, Torrance, CA, USA) and 20% sucrose before storage at −80°C. Sections were cut parallel to the circular muscle layer at a thickness of 10–20 μm (thin sections) using a Leica CM 3050 cryostat (Leica Microsystems, Wetzlar, Germany). Tissues were incubated in bovine serum albumin (1% w/v for 1 h at 20°C; Sigma, St Louis, MO, USA) to reduce non-specific antibody binding before incubation with neuronal nitric oxide sythase (nNOS) antibody (1:1200 dilution; gift from Dr Piers Emson, Molecular Science Group, Cambridge, UK) in combination with Triton-X 100 (0.5% solution for 16 h at 4°C). Slides were washed in PBS (0.1 m for 6 h) and then incubated with a donkey anti-sheep secondary antibody (Alexa Fluor 594; Molecular Probes, Eugene, OR, USA) using a working dilution of 1:1000 (1 h at 20°C). Unbound secondary antibody was removed by washing slides in PBS overnight. After a 1 h blocking period (as described above), tissues were incubated with VIP antibody (1:400 dilution; Millipore, Temecula, CA, USA) in combination with Triton-X 100 (0.5% solution for 16 h at 4°C). Slides were then washed in 0.1 m PBS (6 h) before incubation with a donkey anti-rabbit secondary antibody (Alexa Fluor 488; Molecular Probes) using a working dilution of 1:1000 (1 h at 20°C). Tissues were washed in 0.1 m PBS overnight to remove any unbound secondary antibody and subsequently mounted with coverslips using Aquamount mounting medium (Lerner Laboratories, Pittsburgh, PA, USA).
Imaging
Tissues labelled with immunohistochemical techniques were examined with a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss, Thornwood, NY, USA). Images generated using the confocal imaging system are digital composites of Z-series of scans of 0.25–1 μm optical sections through a depth of 0.5–20 μm. Zeiss LSM 5 Image Examiner Software, Adobe Photoshop CS5 Software and CorelDRAW X4 Software were used in the construction of final images.
Contraction experiments
Strips of muscle from the IAS were attached with sutures to a Gould strain gauge and a stable mount and immersed in tissue baths containing 3 ml of oxygenated Krebs-Ringer solution maintained at 37°C. Muscles were initially stretched, followed by a 60 min equilibration period. Nerves were stimulated for 0.5–60 s with EFS using a Grass S48 stimulator (0.1–20 Hz at 15 V and 0.2 ms pulse duration). Platinum inoculation loops (3 mm diameter, separated from one another by 1 cm) were used as stimulating electrodes. Muscle strips were threaded through both loops. Electrical field stimulation gave rise to neural responses that were sensitive to TTX (1 μm). Data were collected, stored and analysed by computer using a data acquisition program (AcqKnowledge 3.9.1; Biopac systems, Inc., Goleta, CA, USA).
Two different protocols were used for preparation and treatment of tissues. The first, based upon Rattan et al. (2005), included creating muscle strips from the final 0.5 mm of the GI tract. Muscles were initially stretched by 0.1 g. Nerves were stimulated for 4 s over a frequency range of 0.5–20 Hz, and NOS was blocked with 300 μm l-NNA. The second protocol (used for most experiments) was based upon our previous studies (Duffy et al. 2012). This protocol included wider muscle strips (i.e. 2 mm) and a broader range of stimulus train lengths (0.5–60 s) at a frequency of either 5 or 20 Hz. The initial stretch of 0.3 g was applied, and a lower concentration of l-NNA (i.e. 100 μm) was used.
Analysis of contraction
Contractile area during EFS was evaluated with AcqKnowledge software by determining the integral of the contractile trace in 10 s increments during long trains of EFS (i.e. 30–60 s) or for 4 s during a brief train of EFS (i.e. 4 s). The area during EFS was then normalized to the prestimulus contractile area and expressed as the percentage change from control activity. Post-stimulus relaxation was determined in 10 s increments following termination of EFS and again normalized to the prestimulus contractile area. Given that post-stimulus relaxation was usually preceded by post-stimulus contraction, relaxation was quantified beginning 10 s after termination of EFS in WT mice and 20 s after termination of EFS in VIP−/− mice. This time difference was necessary because post-stimulus contraction persisted for longer in VIP−/− mice. Baseline (i.e. zero active tension) was determined by the addition of 10 μm sodium nitroprusside (SNP) and 1 μm nifedipine at the end of the experiment. Statistical analysis was performed using one-way ANOVA. Individual data points are expressed as means ± SEM, while data sets (e.g. control vs. MRS2500) were evaluated with Tukey's post hoc multiple comparison Test and considered significantly different when P < 0.05.
Intracellular recordings
Microelectrode experiments
Strips of muscle (2 mm wide) from the IAS were pinned submucosal side up to the base of an electrophysiological chamber. Smooth muscle cell impalements were made with glass microelectrodes filled with 3 m KCl (tip resistances 60–150 MΩ). Nerves were stimulated for 0.5–60 s (0.05–20 Hz; 0.2 ms duration) via platinum electrodes (1 cm length) placed 1 mm away from the muscle on both sides along its entire length. Neural responses elicited in these conditions were TTX sensitive. To maintain impalements, experiments were carried out in the presence of L-type calcium channel inhibitors (3 μm nifedipine, 1 μm nicardipine). In order to examine inhibitory junction potentials (IJPs) elicited with brief (i.e. 4 s) stimulus trains, 300 μm l-NNA was used as required, and impalements were made within the most distal 0.5 mm section of the muscle strip. For all other measurements, 100 μm l-NNA was used as required, and impalements were made within the most distal 1 mm section of the muscle strip. There was no apparent difference in IJPs recorded in these two conditions.
Analysis of intracellular measurements
Inhibitory junction potentials were evaluated in two ways. For brief trains of stimuli (i.e. 4 s; 0.5–20 Hz), IJP area was quantified by determining the integral of electrical activity (mV − s) during 4 s of EFS using AcqKnowledge software. Area values at the lowest frequencies are very small using this approach since hyperpolarization (i.e., the IJP) is balanced by depolarization (i.e., rebound). For the remainder of the experiments, IJP amplitude was measured as the most negative potential occurring at various times either during or after EFS.
Drugs
Atropine sulphate, l-NNA, guanethidine, SNP, TTX, nicardipine and nifedipine were all purchased from Sigma-Aldrich (St Louis, MO, USA). MRS2500, MEN10,627 and L-733-060 were purchased from Tocris Bioscience (Ellisville, MO, USA). Vasoactive intestinal polypeptide was purchased from Bachem (Torrance, CA, USA), and VIP6–28 and VIP10–28 were purchased from Mimotopes (Clayton, Victoria, Australia) and American Peptide Company (Sunnyvale, CA, USA), respectively. Atropine, l-NNA, guanethidine, TTX, SNP, MRS2500, L-733-060, VIP, VIP10–28 and VIP6–28 were dissolved in de-ionized water. Nifedipine and nicardipine were dissolved in ethanol. MEN10,627 was dissolved in DMSO.
Results
Immunohistochemical localization of VIP and nNOS in enteric neurons of the mouse IAS
Expression patterns of VIP and nNOS were examined in the WT and VIP−/− mouse IAS using immunohistochemical labelling methods. In the WT mouse, VIP (Fig. 1Ab) and nNOS (Fig. 1Aa) labelling was present in intramuscular nerve fibres running parallel to smooth muscle cells. Vasoactive intestinal polypeptide was often expressed in a punctuate-like manner compatible with its presence within nerve varicosities (Fig. 1Ab). Neuronal NOS and VIP were co-localized within the same neurons (Fig. 1Ac). In the VIP−/− mouse, labelling of nNOS was present (Fig. 1Ba) but VIP labelling was absent (Fig. 1Bb and Bc), confirming the knock-out status of VIP in this animal.
Figure 1. VIP is present in nNOS+ nerves of WT mice but not VIP−/− mice.
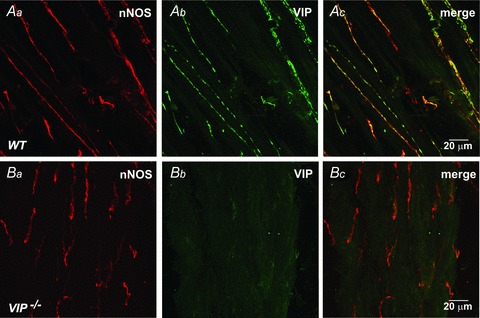
A, Dual labeling of nerves with anti-nNOS (red, Aa) and anti-VIP (green, Ab) antibodies reveals co-localization of VIP and nNOS in WT mice (Ac). Note more punctate nature of VIP labeling. B, nNOS labeling is present in the VIP−/− mouse (Ba,Bc) while VIP labeling is absent (Bb,Bc).
Role of VIP in neural regulation of contractile activity in the mouse IAS
Effect of various blockers on relaxations elicited with brief trains of EFS
Initial studies examined nerve-evoked relaxations elicited with brief trains of EFS (4 s, 0.5–20 Hz) because previous studies suggest that responses to these stimulus parameters are due to nitrergic and VIP-ergic NMT (Rattan et al. 2005). Experiments were carried out using the protocols described by Rattan et al (see Methods). Muscle strips (0.5 mm wide) developed tone and phasic contractile activity over a 10–18 min period following immersion in warm, oxygenated Krebs-Ringer solution. Nerve stimulation (0.5–20 Hz, 4 s) gave rise to frequency-dependent relaxations during EFS (Fig. 2Aa), and in 8 and 75% of muscles a post-stimulus relaxation followed the post-stimulus contraction with 10 and 20 Hz EFS, respectively. Relaxations during EFS were not reduced by the VIP receptor antagonists VIP10–28 or VIP6–28 (30 μm; Fig. 2C), whereas post-stimulus relaxations were abolished (Fig. 2D). Relaxations during EFS were also not reduced by l-NNA alone (300 μm; Fig. 2Ab and C) or MRS2500 alone (1 μm; Fig. 2B and C) but were abolished by combined l-NNA and MRS2500 (0.5–10 Hz, 4 s; Fig. 2Ac and C). In 40% of muscles bathed in MRS2500 and l-NNA, a small relaxation persisted during 20 Hz EFS (see Fig. 2Ac), whereas a small contraction occurred in the rest. Post-stimulus relaxations were not reduced by combined l-NNA and MRS2500 (Fig. 2Ac). These data are in agreement with our previous study and that of Opazo et al. 2011 and suggest that responses elicited with brief trains of EFS are due to nitrergic and purinergic NMT. A third NNNP component was apparent as a post-stimulus relaxation that sometimes followed 10 or 20 Hz EFS.
Figure 2. Brief trains of electrical field stimulation (EFS; i.e. 4 s, 0.5–20 Hz) give rise to nitrergic and purinergic neuromuscular transmission (NMT) in WT mice.
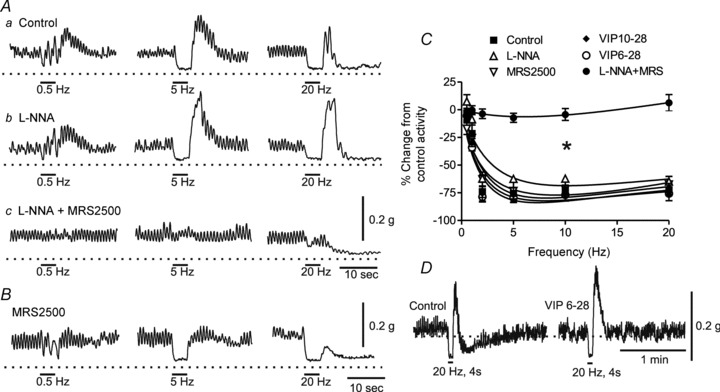
A, sample traces showing responses to EFS in control conditions (Aa), following addition of NG-nitro-l-arginine (l-NNA; 300 μm; Ab), MRS2500 (1 μm; B) or combined addition of l-NNA plus MRS2500 (Ac). C, summary graph of the effect of various drugs on responses to EFS. The EFS gave rise to frequency-dependent contractile inhibition (control, n = 7) that was not reduced by l-NNA (n = 8), MRS2500 (n = 5), VIP6–28 (n = 4) or VIP10–28 (n = 6) but was abolished (*p<0.05) by combined addition of l-NNA and MRS2500 (n = 10). Shown are mean values ± SEM. One-way ANOVA with Tukey's post hoc test. D, sample traces showing the selective effect of VIP6–28 on post-stimulus relaxation but not the relaxation occurring during EFS.
The remaining contractile experiments used our previously described protocol (Duffy et al. 2012), including wider muscle strips (i.e. 2 mm) and a lower concentration of l-NNA (100 μm).
Relationship between stimulus train length and neurally evoked relaxation
In order to examine the stimulus conditions necessary to activate inhibitory NMT further, we compared responses elicited with either 5 or 20 Hz EFS while varying stimulus train lengths between 0.5 and 60 s. In control conditions, 5 Hz EFS reduced spontaneous contractile activity by ∼80%. Peak relaxation developed in <20 s and persisted for the duration of the stimulus train (i.e. 60 s; Fig. 3Aa and Ba). The control relaxation elicited with 20 Hz EFS was not different from that elicited with 5 Hz EFS (Fig. 3Ab and Bb). Relaxations during 5 or 20 Hz EFS were not reduced by MRS2500 (1 μm; Fig. 3Ba and Bb), while post-stimulus contraction was greatly diminished (see Fig. 3Cb) as previously described (Duffy et al. 2012). The combination of MRS2500 (1 μm) and l-NNA (100 μm) abolished relaxation for the first 20 s of EFS at 5 Hz; thereafter, relaxation began to develop and reached 65% of maximum by the end of the stimulus train (i.e. 5 Hz, 60 s; Fig. 3Aa and Ba). With 20 Hz EFS, the NNNP response began with contraction followed by relaxation that reached ∼49% of maximum by the end of the stimulus train (i.e. 30 s; Fig. 3Ab and Bb).
Figure 3. Non-nitrergic, non-purinergic (NNNP) relaxation develops slowly during longer stimulus trains in WT mice and persists beyond the end of the stimulus train.
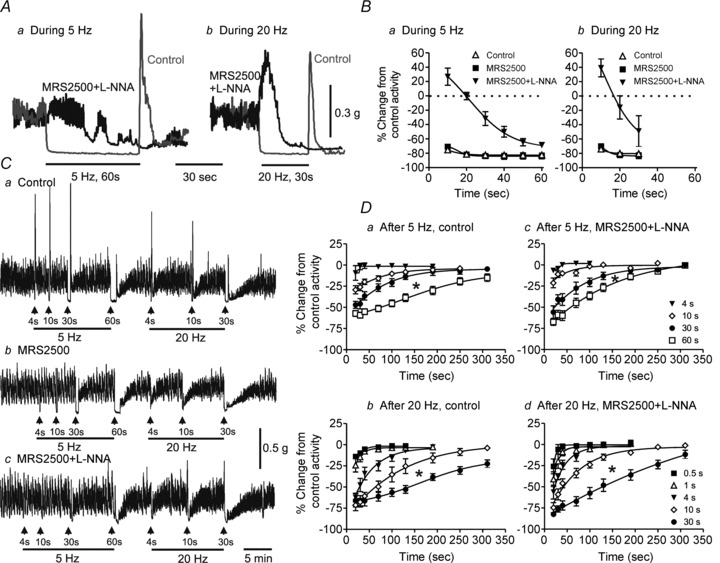
A, sample traces showing inhibition of contraction with 5 Hz EFS for 60 s (Aa) or 20 Hz EFS for 30 s (Ab) in control conditions (grey traces) and following addition of l-NNA (100 μm) plus MRS2500 (1 μm; black traces). B, summary graphs of the effects during 5 Hz EFS for 60 s (Ba) or during 20 Hz EFS for 30 s (Bb) on contractile activity in control conditions (5 Hz, n = 9; 20 Hz, n = 8), with MRS2500 (5 Hz, n = 5; 20 Hz, n = 5) or with combined addition of l-NNA and MRS2500 (5 Hz, n = 9; 20 Hz, n = 9). In the presence of l-NNA and MRS2500, relaxation during EFS begins after approximately 20 s of EFS at 5 Hz and 15 s of EFS at 20 Hz. C, sample traces of post-stimulus relaxations following 5 Hz (left) or 20 Hz (right) EFS with increasing stimulus train length. Neither the control relaxation during EFS nor the post-stimulus relaxation following EFS (Ca) is reduced by MRS2500 (1 μm) alone (Cb), while l-NNA (100 μm) plus MRS2500 greatly attenuates relaxation during EFS but not post-stimulus relaxation (Cc). D, summary graphs of post-stimulus relaxations following various stimulus train lengths in control conditions at 5 Hz (Da, n = 5–11) or 20 Hz (Db, n = 7–9) or in the presence of l-NNA plus MRS2500 (Dc, 5 Hz, n = 11–13; Dd, 20 Hz, n = 6–11). Post-stimulus relaxation significantly increased (*p<0.05) with increasing stimulus train length (one-way ANOVA with Tukey's post hoc test). Shown are mean values ± SEM.
The relationship between stimulus train length and post-stimulus relaxation was also examined. In control conditions, post-stimulus relaxation began to appear with 5 Hz EFS for 10 s (Fig. 3Da) and with 20 Hz EFS for 0.5 s (Fig. 3Db). With increasing stimulus train length, the amplitude and duration of post-stimulus relaxation increased. Post-stimulus relaxation was not reduced by combined l-NNA and MRS2500 (Fig. 3Cc, Dc and Dd). The NNNP relaxation during and after EFS was abolished by the neurotoxin TTX (1 μm, n = 3), confirming its neural origin.
Changes in nerve-evoked relaxation in the VIP−/− mouse and with VIP6–28 in WT mice
To address the role of VIP in NNNP inhibitory NMT, contractile responses to EFS were examined in the VIP−/− mouse. In control conditions, relaxations during 5 and 20 Hz EFS (Fig. 4A and D) were not different from those in WT mice (see Fig. 3), while post-stimulus relaxation was absent (Fig. 4A and E). As in WT mice, MRS2500 did not reduce relaxations during EFS (Fig. 4B and D). However, following combined addition of MRS2500 and l-NNA the relaxation during EFS was absent in VIP−/− mice, and a non-cholinergic contraction was observed (Fig. 4C and D). The contraction with 20 Hz EFS was significantly reduced by either the NK1 receptor antagonist L-733,060 (79.6 ± 4% of control, n = 4) or the NK2 receptor antagonist MEN10,627 (78.0 ± 4.2% of control, n = 3) and further reduced with combined drugs (67.2 ± 2.4% of control, n = 7), suggesting a role for tachykinins in this response. The incomplete block of contraction may be due either to limited efficacy of tachykinin receptor antagonists or, alternatively, to participation of some additional contractile substance and/or receptor (e.g. a P2Y receptor other than P2Y1; Zizzo et al. 2007).
Figure 4. Non-nitrergic, non-purinergic (NNNP) relaxation is absent from the VIP−/− mouse IAS.
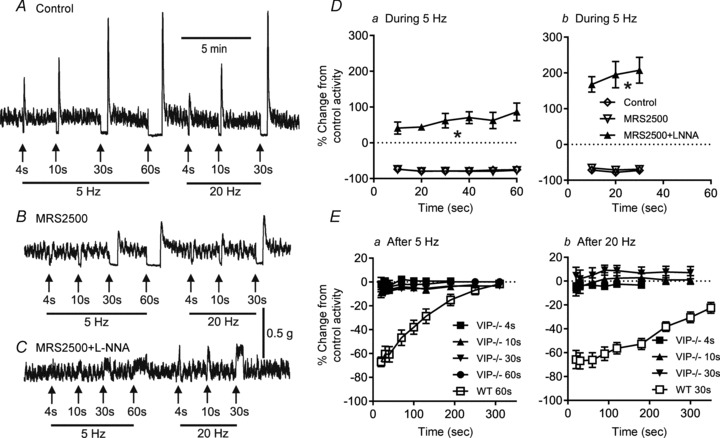
A–C, sample traces showing responses to 5 Hz (left) or 20 Hz EFS (right) with increasing stimulus train length in control conditions (A), following the addition of MRS2500 (1 μm; B) and with l-NNA (100 μm) plus MRS2500 (C) in VIP−/− mice. The NNNP relaxation observed following EFS in WT mice is absent from the VIP−/− mouse IAS. A non-cholinergic contraction occurs during EFS in the presence of MRS2500 and l-NNA that is ∼27% of a contraction in response to 60 mm KCl (C). D, summary graphs of responses during 5 Hz (Da, n = 8–9) or 20 Hz EFS (Db, n = 8) in VIP−/− mice. An ∼80% maximal relaxation occurred in both VIP−/− (D; open diamonds and inverted open triangles) and WT mice (Fig. 3B, open triangles and filled squares), whereas addition of MRS2500 + l-NNA significantly changed the response (*p<0.05) to contraction during EFS (filled triangles) in VIP−/− mice (Da and Db). E, summary graphs of activity following various stimulus train lengths at 5 Hz (Ea, n = 8) or 20 Hz (Eb, n = 6) in control conditions in VIP−/− mice (filled symbols) compared with that of WT mice with 20 Hz EFS for 30 s (open squares). The post-stimulus relaxation that is present in WT mice is absent in VIP−/− mice. One-way ANOVA with Tukey's post hoc multiple comparison. Shown are mean values ± SEM.
Additional experiments were undertaken with the VIP receptor antagonist VIP6–28 in WT mice. VIP6–28 (30 μm) abolished the NNNP relaxation during (Fig. 5A and Ba) and after EFS (Fig. 5A and Bb). During EFS, relaxation was replaced with a non-cholinergic contraction (Fig. 5Ab and Ba). Thus, the NNNP responses observed following addition of VIP6–28 were very similar to those observed in the VIP−/− mouse IAS (compare Fig. 5B with Fig. 4Db and Eb). Taken together, these data provide strong support for the hypothesis that NNNP inhibitory NMT is mediated by VIP.
Figure 5. The VIP receptor antagonist VIP6–28 abolishes NNNP relaxation in WT mice to reveal non-cholinergic contraction.
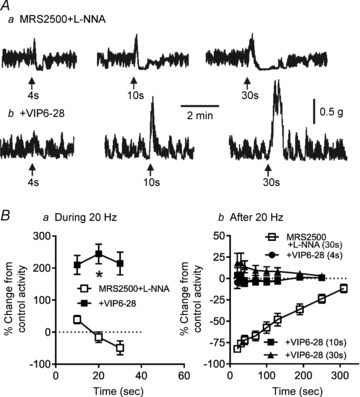
Aa, sample traces showing NNNP relaxations elicited with 20 Hz EFS for 4–30 s in WT mice. Ab, sample traces from the same muscle following addition of 30 μm VIP6–28. The NNNP relaxation is blocked, revealing non-cholinergic contraction. B, summary graph of the contractile activity during (Ba) and after 20 Hz EFS (4–30 s; Bb) in WT muscles. The relaxation occurring in the presence of MRS2500 and l-NNA (n = 9) was blocked by VIP6–28 (n = 5) and replaced with contraction (Ba; *p<0.05). Post-stimulus relaxation (n = 11) was also blocked by VIP6–28 (n = 5; Bb). One-way ANOVA with Tukey's post hoc multiple comparison. Shown are mean values ± SEM. Both 100 μm l-NNA and 1 μm MRS2500 were present throughout.
Effect of exogenous VIP on contractile activity in WT and VIP−/− mice
Exogenous VIP was applied to muscles to determine whether this peptide mimics the contractile effects of NNNP inhibitory NMT. Cumulative addition of VIP (0.1–30 nm) gave rise to concentration-dependent relaxations in WT and VIP−/− mice, with the VIP−/− mouse being slightly more sensitive to VIP than the WT mouse (Fig. 6A, C and E). These relationships were shifted to the right by the VIP receptor antagonist VIP6–28 (30 μm; Fig. 6B, D and E).
Figure 6. Vasoactive intestinal polypeptide relaxes the WT and VIP−/− mouse IAS in a concentration-dependent manner and this is reduced by VIP6–28.
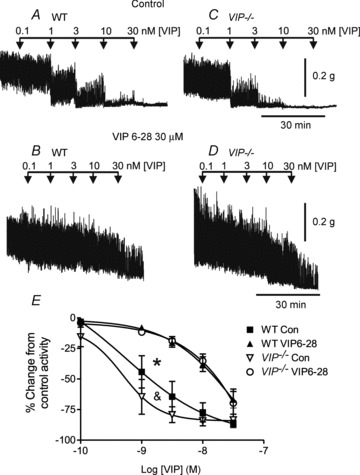
A–D, sample traces showing the response to VIP in the absence (A and C) and presence (B and D) of VIP6–28. E, summary graph of the concentration–response relationship for VIP in the absence and presence of VIP6–28 (30 μm). There is a small but significant (& p<0.05) difference in the VIP–relaxation relationship in the VIP−/− (n = 4) vs. the WT mouse (n = 5) in control conditions, whereas both relationships shift significantly (*p<0.05) to the right in the presence of VIP6–28. One-way ANOVA with Tukey's post hoc test. Shown are mean values ± SEM. Both MRS2500 (1 μm) and l-NNA (100 μm) were present throughout.
Role of VIP in neural regulation of electrical activity in the mouse IAS
Effect of various blockers on the IJPs elicited with brief trains of EFS
Membrane potential (Em) was measured to identify the electrical events underlying inhibitory NMT. Initial experiments examined responses to brief trains of EFS (i.e. 4 s, 0.5–20 Hz) as described for contractile experiments with brief trains of EFS (see Fig. 2). A single stimulus gave rise to a brief duration (i.e. <1.5 s), maximal amplitude IJP (i.e. 26.2 ± 0.9 mV, n = 8), while trains of stimuli at ≥5 Hz produced continuous hyperpolarization (Fig. 7Aa). NG-Nitro-l-arginine (l-NNA; 300 μm) did not reduce IJPs (Fig. 7Ab and C), whereas both the single IJP (i.e. 5.1 ± 0.6 mV) and composite IJPs were substantially reduced by MRS2500 (1 μm; see Fig. 7B and C). The combination of l-NNA and MRS2500 abolished IJPs during 0.5–10 Hz EFS (4 s), while a small transient hyperpolarization sometimes occurred during 20 Hz EFS (Fig. 7Ac and C). The initial transient hyperpolarization at 20 Hz was blocked with further increases of MRS2500 (i.e. 3 μm), indicating that it was due to incomplete block of the purinergic NMT. These data agree with our contractile measurements (Fig. 2), suggesting that responses to brief trains of EFS (4 s) are due to the combined effects of nitrergic and purinergic NMT.
Figure 7. Brief trains of EFS (i.e. 4 s, 0.5–20 Hz) give rise to inhibitory junction potentials (IJPs) mediated by nitrergic and purinergic NMT in WT mice.
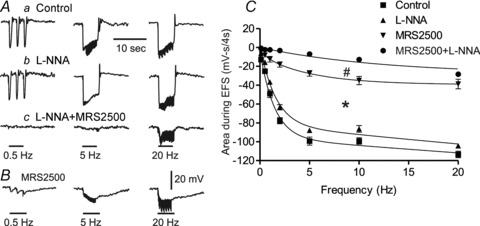
A, sample traces showing IJPs at 0.5, 5 and 20 Hz in control conditions (Aa), following addition of l-NNA (300 μm; Ab) and with MRS2500 (1 μm) plus l-NNA (Ac). B, sample trace from a different muscle showing the IJPs generated in the presence of MRS2500 alone. C, summary graph of area during EFS (mV-s(4 s)-1) in control conditions (filled squares), following addition of l-NNA (filled triangles) or MRS2500 (inverted filled triangles) or with combined MRS2500 and l-NNA (filled circles). Inhibitory junction potentials were significantly reduced in the presence of MRS2500 alone (*p<0.05) and further reduced with combined MRS2500 and l-NNA (#p<0.05). Shown are mean values ± SEM; n = 5–8. One-way ANOVA with Tukey's post hoc test.
Relationship between stimulus train length and neurally evoked hyperpolarization in WT and VIP−/− mice
Contractile measurements revealed that NNNP relaxations required higher frequencies and/or longer stimulus trains. Additional experiments were undertaken to determine whether NNNP IJPs require similar stimulus conditions. Resting Em in WT muscles was −45.8 ± 0.3 mV (n = 12). In control conditions, during 5 Hz EFS for 60 s there was an initial fast hyperpolarization followed by a period of steep repolarization and then a slower period of repolarization (Fig. 8Aa, left trace). After 30 s at 5 Hz, the IJP had declined in amplitude by ∼29%. Similar composite IJPs were observed with 20 Hz EFS (Fig. 8Aa, right trace, and Cb). MRS2500 (1 μm) abolished the initial fast IJP and reduced the remaining IJP elicited with either 5 or 20 Hz EFS to ∼10 mV (i.e. ∼62% smaller than the control peak hyperpolarization). This smaller IJP persisted for the duration of the stimulus train (Fig. 8Ab and C). The combined l-NNA (100 μm) and MRS2500 abolished all hyperpolarization during the initial phase of stimulation (Fig. 8Ac and C). After ∼30 s at 5 Hz and ∼10 s at 20 Hz, hyperpolarization began to develop (Fig. 8Ac and C). Greater NNNP hyperpolarization occurred with 20 Hz EFS (i.e. 6.0 ± 1.4 mV at 30 s, n = 4) than with 5 Hz EFS (i.e. 1.2 ± 0.5 mV at 30 s and 3.1 ± 0.5 mV at 60 s, n = 4).
Figure 8. An NNNP hyperpolarization develops during EFS with longer stimulus trains in WT but not VIP−/− mice.
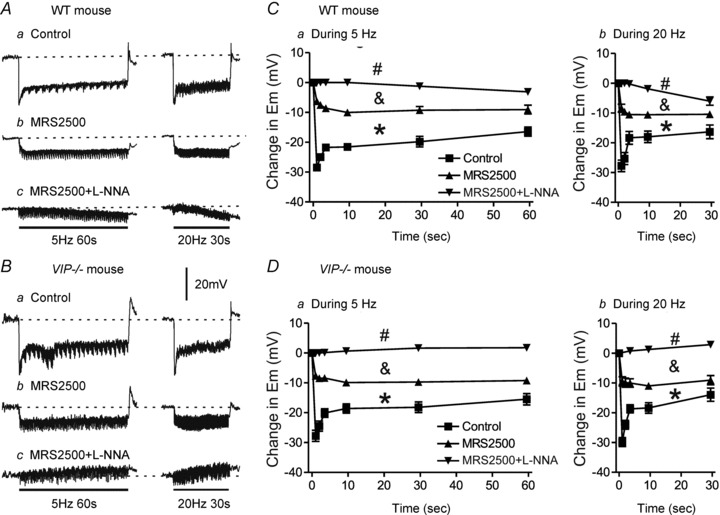
A and B, sample traces showing IJPs elicited with 5 Hz EFS for 60 s (left traces) or 20 Hz EFS for 30 s (right traces) in WT (A) and VIP−/− mice (B). Inhibitory junction potentials were reduced by MRS2500 (1 μm) in WT (Ab) and VIP−/− mice (Bb), while a small hyperpolarization developed in WT mice (Ac) and a small depolarization developed in VIP−/− mice (Bc) following combined addition of MRS2500 and l-NNA (100 μm). C and D, summary graphs of IJP amplitude with time during 5 Hz (n = 4–6) or 20 Hz EFS (n = 4–5) in WT mice (C) and 5 Hz (n = 7–10) and 20 Hz (n = 7–10) EFS in VIP−/− mice (D). Control IJPs in WT and VIP−/− mice were not significantly different. Inhibitory junction potentials were significantly reduced (*p<0.05) by MRS2500 alone in both WT and VIP−/− mice, but remaining IJPs were still not different from one another. Combined MRS2500 plus l-NNA further reduced (& p<0.05) IJPs in both WT and VIP−/− mice, but in this case the remaining electrical events in WT mice were significantly different from those in VIP−/− mice (#p<0.05). One-way ANOVA with Tukey's post hoc test. Shown are mean values ± SEM.
In order to examine the role of VIP in NNNP inhibitory NMT further, we compared the IJPs elicited during EFS in WT mice with those occurring in VIP−/− mice. The membrane potential in VIP−/− mice was slightly less negative than in WT mice (i.e. −45.8 ± 0.3 mV in WT vs.−43.5 ± 0.4 mV VIP−/− mice, n = 12, P < 0.05), while the IJP elicited with a single stimulus was not different (i.e. −27.3 ± 0.5 in WT vs.−28.0 ± 0.8 in VIP−/− mice, n = 8). In addition, IJPs elicited during repetitive EFS at 5 and 20 Hz were not significantly different in control conditions or in the presence of MRS2500 (Fig. 8Ba, Bb and D). However, following combined addition of l-NNA and MRS2500 an NNNP hyperpolarization was not observed during EFS in the VIP−/− mouse IAS (Fig. 8Bc and D). Instead, a small depolarization developed slowly during EFS (Fig. 8Bc and D).
The post-stimulus hyperpolarization following 5 or 20 Hz EFS applied for various lengths of time was also evaluated in WT and VIP−/− mice (with 100 μm l-NNA and 1 μm MRS2500 present throughout). A small post-stimulus hyperpolarization followed 5 Hz EFS for 60 s or 20 Hz EFS for 4 s, and this persisted for ∼2 min (Fig. 9A, C and E). Interestingly, the post-stimulus hyperpolarization sometimes continued to develop for up to 30 s after the end of the stimulus train. As the stimulus train length was increased, the amplitude and duration of post-stimulus hyperpolarization also increased. Thus, 20 Hz EFS for 30 s gave rise to a post-stimulus hyperpolarization that persisted for ∼4 min, reaching a maximal amplitude of ∼8 mV (see Fig. 9A and E). In contrast, post-stimulus hyperpolarization was absent in the VIP−/− mouse IAS (Fig. 9B, D and F). The post-stimulus hyperpolarization accompanying 20 Hz EFS for 30 s in control conditions in WT mice was not different from that observed in the presence of MRS2500 plus l-NNA (control, 7.2 ± 1.2, 6.6 ± 1.4 and 5.2 ± 1.1 mV, n = 4 vs. MRS2500+l-NNA, 7.9 ± 1.9, 7.1 ± 2.4 and 6.9 ± 24 mV, n = 5 for 30, 60 and 90 s post-stimulus hyperpolarization, respectively; Fig. 10). These data indicate that the time- and stimulus-dependent features of NNNP hyperpolarization are similar to those of NNNP relaxation. The neural origin of NNNP hyperpolarization was confirmed with TTX (n = 3).
Figure 9. Non-nitrergic, non-purinergic (NNNP) relaxation post-stimulus hyperpolarization develops following longer stimulus trains in WT but not VIP−/− mice.
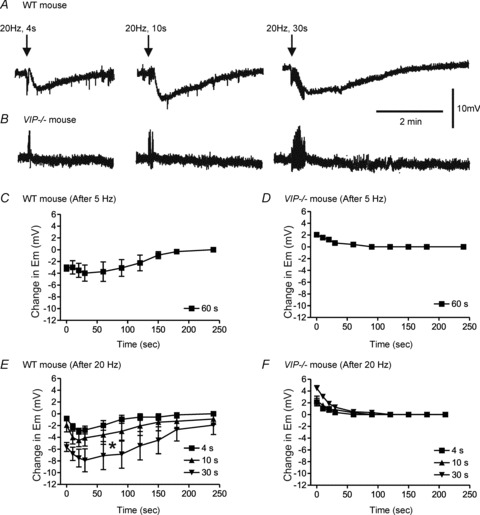
A and B, sample traces showing an NNNP post-stimulus hyperpolarization following 20 Hz EFS in the WT (A) but not VIP−/− mice (B). C–F, summary graphs of the changes in membrane potential (Em) following stimulus trains of various lengths at 5 Hz (C, n = 4; D, n = 5) or 20 Hz (E, n = 5; F, n = 6) in WT (C and E) and VIP−/− mice (D and F). Post-stimulus hyperpolarization developed following EFS in WT mice. At 20 Hz EFS, the amount of hyperpolarization significantly (*p<0.05) increased with longer stimulus trains (E). Post-stimulus hyperpolarization was absent in VIP−/− mice. Rather, Em repolarized towards baseline for 10–30 s following EFS (D and F). One-way ANOVA with Tukey's post hoc test. Shown are mean values ± SEM.
Figure 10. Non-nitrergic, non-purinergic (NNNP) hyperpolarization is independent of nitrergic and purinergic NMT.
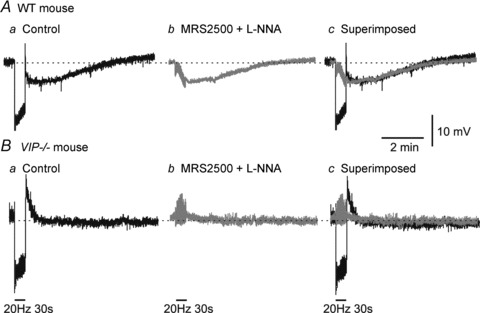
A, sample traces showing the various junction potentials associated with 20 Hz EFS for 30 s in a WT mouse IAS. In control conditions, a faster purinergic/nitrergic IJP is followed by a slower post-stimulus hyperpolarization (Aa; black trace). Following addition of MRS2500 (1 μm) and l-NNA (100 μm) the purinergic/nitrergic IJP is blocked, while the slower component remains (Ab; grey trace). Superimposing these traces shows that the NNNP hyperpolarization remains unchanged in the absence of the purinergic/nitrergic IJP (Ac). B, in the VIP−/− mouse IAS, the purinergic/nitrergic IJP is present, but post-stimulus hyperpolarization is not (Ba). Following addition of MRS2500 and l-NNA, no further hyperpolarization occurs (Bb and Bc).
Effects of VIP6–28 and VIP on electrical activity
Additional experiments were undertaken in WT mice to examine the effects of the VIP receptor antagonist VIP6–28 on NNNP IJPs elicited with 20 Hz EFS for 10 s. The VIP6–28 (30 μm, n = 3) abolished the NNNP hyperpolarization elicited with EFS in control muscles (Fig. 11Ab). In order to determine whether exogenous VIP mimics the electrical events underlying NNNP inhibitory NMT, we also measured the effects of VIP on Em in WT mice. Vasoactive intestinal polypeptide (1–100 nm) gave rise to a concentration-dependent hyperpolarization averaging ∼13 mV with 30 nm VIP (Fig. 11B and C).
Figure 11. Non-nitrergic, non-purinergic (NNNP) hyperpolarization is abolished by VIP6–28 and mimicked by exogenous VIP.
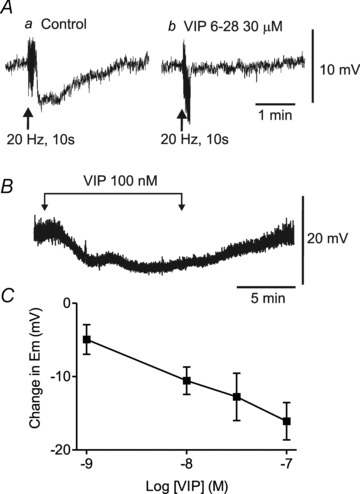
A, sample traces showing the post-stimulus hyperpolarization accompanying 20 Hz EFS for 10 s (Aa). This hyperpolarization is blocked following 17 min superfusion with VIP6–28 (30 μm; Ab). B, sample trace showing the hyperpolarization elicited with superfusion of 100 nm VIP. C, summary graph of the concentration-dependent effects of VIP (1–100 nm, n = 3–7) on membrane potential (Em). Shown are mean values ± SEM. Atropine, guanethidine, 1 μm MRS2500 and 100 μm l-NNA were present throughout.
Discussion
The present study investigated NNNP inhibitory NMT in the mouse IAS. This pathway was activated with repetitive stimulation of nerves and gave rise to a slowly developing relaxation and hyperpolarization. It was TTX sensitive, blocked by VIP receptor antagonists, absent in the VIP−/− mouse and mimicked by exogenous VIP. Taken together, these data strongly suggest that the slow component of inhibitory NMT in the mouse IAS is mediated by VIP.
Vasoactive intestinal polypeptide and a second peptide designated peptide histidine isoleucine or PHI are derived from a 170-amino-acid precursor molecule (i.e. prepro-VIP). These peptides are packaged together in large dense-core vesicles in the GI tract (Agoston et al. 1985). While VIP and PHI each have biological activity, VIP is significantly more active than PHI (for review see Fahrenkrug, 2010). Neuropeptides are also located in large dense-core vesicles in the central nervous system, and here vesicle fusion and release can occur outside of the synaptic cleft, resulting in more slowly developing, diffuse, protracted effects (Agnati et al. 1995; Fahrenkrug, 2010). In addition, fusion and release of neurotransmitter from large dense-core vesicles often requires multiple stimuli, probably because of a dependence upon larger and more sustained increases in intracellular calcium in the presynaptic terminal (Sudhof & Malenka, 2008). In peripherial organs, neuropeptides are also commonly associated with ‘slow neurotransmission’, requiring multiple stimuli for release (Burnstock, 2004). The NNNP NMT in the mouse IAS also required multiple stimuli, developing slowly during EFS and persisting well beyond the period of nerve stimulation, i.e. features consistent with peptidergic neurotransmission. Studies of other GI muscles (e.g. stomach and colon) have also reported a requirement for multiple stimuli to elicit VIP-ergic NMT (Boeckxstaens et al. 1992; Takahashi & Owyang, 1995; Tonini et al. 2000; Mule & Serio, 2003; El-Mahmoudy et al. 2006). This conclusion differs somewhat from several earlier studies reporting that responses elicited in vitro with the fewest number of stimuli (i.e. one or two) were most sensitive to blockade with VIP antiserum (e.g. Grider et al. 1985a,b), as were reflex responses elicited with the smallest amount of distension (Grider & Makhlouf, 1986; Grider, 1989).
The expression pattern of nNOS and VIP in myenteric neurons has been examined in a number of studies. Percentages reported for nNOS expression in intestinal myenteric neurons vary between 25 and 48%, while 27–93% of these have also been reported to coexpress VIP. These cell populations represent more than only inhibitory motor neurons; for instance, VIP is present in some neurons that innervate the mucosa as well as some interneurons (see Van Geldre & Lefebvre, 2004; Furness, 2006; Qu et al. 2008). Myenteric neurons expressing VIP and nNOS have also been identified in the mouse IAS, although percentages were not included (Rattan et al. 2005). A few studies have also examined co-localization of nNOS and VIP within the muscularis of the guinea-pig distal colon (Lomax & Furness, 2000) and mouse small intestine (Qu et al. 2008). There is general agreement between those two studies and the present study that nearly all nNOS nerve fibres also express VIP. Thus, VIP is present at the site of inhibitory NMT and is likely to be released along with nitric oxide when nerves are activated.
Previous studies of the mouse IAS (Rattan et al. 2005) proposed that inhibitory NMT elicited with brief trains of EFS (i.e. 4 s, 0.5–20 Hz) was mediated by nitrergic and VIP-ergic NMT, while purinergic NMT was not considered. In contrast, in agreement with studies of the rat IAS (Opazo et al. 2011), we previously reported that inhibitory NMT in the mouse IAS elicited with 5 Hz EFS for 10 s was mediated by nitrergic and purinergic NMT (Duffy et al. 2012). To reconcile these differences, we used the experimental conditions described by Rattan et al. (2005), including the following factors: (i) a wider range of stimulus parameters (0.5–20 vs. 5 Hz); (ii) a shorter train length (4 vs. 10 s); (iii) thinner muscle strips (0.5 vs. 2 mm); and (iv) a higher concentration of l-NNA (300 vs. 100 μm). Using these conditions, we found, as previously reported (Duffy et al. 2012), that relaxations during EFS at ≤10 Hz were abolished by combined l-NNA and MRS2500, making it unlikely that VIP-ergic NMT contributes to these responses. We also found, in contrast to Rattan et al. (2005), that responses during EFS were not reduced by either VIP10–28 or VIP6–28, further discounting a role for VIP during brief trains of EFS (i.e. 4 s). In contrast, the post-stimulus relaxation that sometimes followed 10 or 20 Hz EFS was selectively blocked by VIP receptor antagonists, indicating that these blockers were capable of blocking VIP-ergic NMT if present and when not accompanied by other inhibitory neural pathways (see Fig. 2D). Given that the amplitude of post-stimulus relaxation was usually smaller than the relaxation that preceded it during EFS, this inhibition would not be accounted for by measuring ‘peak’ relaxation, as described by Rattan et al. (2005). In summary, these data support our previous study and that of Opazo et al. (2011) and suggest that responses during brief trains of EFS (e.g. 4 s, 0.5–10 Hz) are due to the combined effects of nitrergic and purinergic NMT without an apparent role for VIP-ergic NMT. The discrepancies between mouse IAS studies cannot be accounted for by differences in stimulus parameters, muscle width or the concentration of l-NNA applied. The remainder of our study therefore focused upon characterizing the third component of inhibitory NMT.
In order to determine the optimal conditions for activation of NNNP inhibitory NMT, we examined responses elicited with either 5 or 20 Hz EFS with various stimulus train lengths between 0.5 and 60 s. The NNNP relaxation occurred with fewer stimuli at 20 Hz than at 5 Hz, i.e. a small relaxation was detected following 10 stimuli at 20 Hz, whereas ∼50 stimuli were required to elicit a similar response with 5 Hz EFS. As discussed above, this frequency dependence may be related to the optimal conditions necessary for activation and release of peptides from large dense-core vesicles.
Interestingly, while purinergic and nitrergic NMT predominate during EFS, post-stimulus relaxation was largely dependent upon NNNP inhibitory NMT. Thus, post-stimulus relaxation was absent in the VIP−/− mouse IAS and blocked with VIP6–28 in WT mice. A noteworthy characteristic of post-stimulus relaxation was the extent to which it continued beyond the period of EFS (i.e. 2.5 min following 4 s EFS at 20 Hz) and the extent to which it could be further prolonged with relatively small increments in stimulus train length (i.e. 6 min following 30 s of EFS at 20 Hz). This persistence of neurally evoked inhibition is also seen in studies of VIP-ergic NMT in other GI regions (Fahrenkrug et al. 1978; Boeckxstaens et al. 1992; Takahashi & Owyang, 1995; Tonini et al. 2000; Mule & Serio, 2003). In vivo, it is possible that VIP participates in the neural control of the IAS by prolonging relaxation in response to larger rectal distending pressures. In keeping with this proposal, studies of the rectoanal inhibitory reflex in both the mouse and the human reveal more prolonged anal relaxations accompanying larger rectal distensions (de Lorijn et al. 2005; Cheeney et al. 2012). Studies of the nNOS−/− mouse also showed that the rectoanal inhibitory reflex was reduced but not abolished in this animal, suggesting a role for other neurotransmitters besides NO in this response (de Lorijn et al. 2005). Further studies are required to address the possible role of VIP in the regulation of anal pressure in vivo.
An additional non-cholinergic excitatory neural pathway was apparent in VIP−/− mice in the presence of atropine, guanethidine, l-NNA and MRS2500 and in WT mice when VIP6–28 was added to this list of drugs. In contrast to NNNP inhibitory NMT, non-cholinergic excitatory NMT was largely confined to the period of EFS. Our results suggest that tachykinins participate in this response, because contraction was reduced by either NK1 or NK2 antagonists and further reduced by combined addition of drugs. These data are in keeping with previous studies suggesting the involvement of both NK1 and NK2 receptors in tachykinergic NMT (for review see Shimizu et al. 2008). Excitatory motor innervation to the mouse IAS is predominantly cholinergic (Duffy et al. 2012), and tachykinins are usually co-localized with acetylcholine in cholinergic motor neurons (Qu et al. 2008; Shimizu et al. 2008). Thus, the identification of a tachykinergic component of excitatory NMT in the mouse IAS is not particularly surprising. It should be noted however that excitatory motor innervation in the mouse IAS differs significantly from that of the monkey and dog IAS, in which extrinsic sympathetic neural pathways predominate (Tichenor et al. 2002; Cobine et al. 2007).
In order to evaluate the neural pathways underlying brief trains of EFS further, we measured the electrical events associated with 4 s EFS at 0.5–20 Hz. In keeping with our contractile experiments, we found that the IJPs elicited during EFS at ≤10 Hz were entirely abolished by combined addition of MRS2500 and l-NNA, again suggesting that they are due to nitrergic and purinergic NMT. Results of experiments with longer trains of EFS were also in agreement with contractile measurement, in that high-frequency EFS (i.e. 20 Hz) was more effective than low-frequency EFS (i.e. 5 Hz) in eliciting NNNP hyperpolarization. We also observed that post-stimulus hyperpolarization (like post-stimulus relaxation) continued for several minutes after the end of the stimulus train. A number of possible mechanisms could account for the extended nature of VIP-ergic NMT, including the following: (i) continued release of VIP; (ii) limited degradation and/or reuptake of VIP; and (iii) prolonged effector pathways initiated by VIP. Additional studies are required to address these possibilities.
The above results reveal that the electrical and contractile events accompanying NNNP inhibitory NMT have a similar dependence upon stimulus train length and frequency as well as a similar time course. However, these events were not correlated in a strict 1:1 manner, i.e. NNNP relaxation developed with fewer stimuli and persisted for longer than NNNP hyperpolarization. Thus, in addition to hyperpolarization-induced closing of L-type voltage-dependent calcium channels (VDCCs), other pathways are likely to contribute to VIP-induced relaxation, such as the following pathways: (i) direct inhibition of VDCCs, e.g. the predicted downstream mediator of VIP, i.e. cAMP-dependent protein kinase (PKA; e.g. Hagen et al. 2006) can directly inhibit VDCCs in some smooth muscles (Zhu et al. 2005); (ii) decreased sensitivity of the myofilaments to Ca2+ via PKA (Somlyo & Somlyo, 2003); and (iii) increased Ca2+ uptake and efflux from smooth muscle cells, again via PKA (Morgado et al. 2012).
A simple 1:1 correlation between electrical and contractile events was also not apparent in experiments with the P2Y1 receptor antagonist MRS2500, i.e. this antagonist reduced IJPs to a much greater extent than relaxation (compare Fig. 2C with Fig. 7C). This disparity has been addressed in our previous study of the mouse IAS (Duffy et al. 2012) and is briefly summarized here. Spontaneous contractile activity in the mouse IAS is highly dependent upon VDCCs. These channels have a discrete threshold potential for activation. Nitrergic NMT by itself is capable of producing sufficient hyperpolarization to close VDCCs and thus block electromechanical coupling. Although a larger hyperpolarization is produced with purinergic NMT, this ‘additional’ hyperpolarization does not produce further inhibition of VDCCs. From this standpoint, the two pathways are not strictly additive but rather redundant. With regard to the present study (as well as similar studies by others), it is likely that both additive as well as redundant pathways are involved when multiple neurotransmitters are released. As a result of this, interpreting the possible role of a neurotransmitter based solely upon what happens when only one neurotransmitter pathway is blocked is not very informative.
An important aim of the present study was to determine whether NNNP inhibitory NMT was mediated by VIP. In order to assign a neurotransmitter role to a substance confidently, a variety of criteria must be satisfied (see Van Geldre & Lefebvre, 2004). The present study fulfils several of these criteria. Our immunohistochemical studies revealed that VIP was co-localized with nNOS in varicose nerve fibres within the musculature, indicating that VIP is physically located near the site of NMT. Our functional studies demonstrated that nerve-evoked relaxation and hyperpolarization were mimicked by exogenous VIP, fulfilling the criteria of mimicry. The NNNP inhibitory NMT and the response to exogenous VIP were also blocked by the VIP receptor antagonist VIP6–28, fulfilling the criteria of ‘parallel antagonism’.
Particularly compelling evidence for VIP as a neurotransmitter was obtained by measuring responses to nerve stimulation in the IAS of the VIP−/− mouse (Colwell et al. 2003). The intestine of these mice is heavier and shorter than that of littermate controls, owing to a thickening of the muscularis propria (including the circular muscle layer) as well as increased length of the villi and goblet cell density. These differences are more pronounced in the small intestine than in the large intestine, although the IAS was not studied directly. Of particular interest, these mice have impaired intestinal transit and in some cases die during the first year of life due to stenosis of the gut (Lelievre et al. 2007). The present study is the first to use this animal to examine the role of VIP in inhibitory NMT in the GI tract. Vasoactive intestinal polypeptide-immunopositive fibres were not detected in the muscularis of the VIP−/− mouse IAS, providing anatomical evidence that VIP expression is absent. When the stimulus protocols used in WT mice were repeated in the VIP−/− mouse, NNNP inhibitory NMT was absent, while purinergic and nitrergic NMT were intact. Essentially, responses to EFS in the VIP−/− mouse IAS were the same as those observed in WT mice following exposure to the VIP receptor antagonist VIP6–28. These data all support the hypothesis that NNNP inhibitory NMT is mediated by VIP. Given that VIP and PHI are derived from the same gene, we cannot exclude a possible additional role for PHI, but the results do suggest that pituitary adenylate cyclase- activating peptide (PACAP) is unlikely to be involved, because PACAP is derived from a different gene (Fahrenkrug, 2010).
The VIP−/− mouse also provided us with the opportunity to re-examine the serial cascade model for VIP and NO (e.g. Jin et al. 1996). Our results revealed that nitrergic NMT was not reduced in the VIP−/− mouse IAS, indicating that VIP was not required for this pathway. The VIP-ergic NMT was also unchanged in WT mice following NOS blockade (e.g. Fig. 3D and 10A), suggesting that NO was not required. Taken together, these data provide additional support for the hypothesis that NO and VIP act as neurotransmitters in parallel (Keef et al. 1994; Bayguinov et al. 1999) rather than in series (for review see Van Geldre & Lefebvre, 2004).
In summary, a third component of inhibitory NMT was isolated and characterized in the mouse IAS using selective blockers of nitrergic and purinergic NMT. This component of NMT developed slowly with repetitive EFS and persisted well beyond the end of nerve stimulation. It was blocked by VIP receptor antagonists, mimicked by exogenous VIP and absent from the VIP−/− mouse IAS. These data all support the hypothesis that NNNP inhibitory NMT is VIP-ergic and represent the first characterization of this pathway in the mouse IAS. While the role of VIP in vivo is still unclear, it is possible that VIP-ergic NMT is activated with larger rectal distensions, leading to a more prolonged period of anal relaxation.
Acknowledgments
This study was supported by NIH grant DK078736 to K.D.K. and an imaging core sponsored by NIH Program Project Grant DK41315. We would also like to extend our appreciation to Dr Kenton Sanders for critical evaluation of this manuscript. The project described was supported by grants from the National Center for Research Resources (5P20RR016464) and the National Institute of General Medical Sciences (8 P20 GM103440)
Glossary
- EFS
electrical field stimulation
- Em
membrane potential
- GI
gastrointestinal
- IAS
internal anal sphincter
- IJP
inhibitory junction potential
- l-NNA
NG-nitro-l-arginine
- NK
neurokinin
- NMT
neuromuscular transmission
- nNOS
neuronal nitric oxide synthase
- NNNP
non-nitrergic, non-purinergic
- NO
nitric oxide
- NOS
nitric oxide synthase
- PACAP
pituitary adenylate cyclase-activating peptide
- PBS
phosphate buffer solution
- PHI
peptide histidine isoleucine
- PKA
cAMP-dependent protein kinase
- SNP
sodium nitroprusside
- VDCC
voltage-dependent calcium channel
- VIP
vasoactive intestinal polypeptide
- WT
wild-type
Author contributions
These experiments were performed in the laboratory of K.D.K. Both K.D.K. and C.A.C. were involved in all aspects of the study, including design, collection, analysis, interpretation of data and writing the manuscript. S.N.S., R.A.M., R.E.K. and A.M.D. all participated in data collection and analysis. All authors have approved this manuscript.
References
- Agnati LF, Zoli M, Strömberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience. 1995;69:711–726. doi: 10.1016/0306-4522(95)00308-6. [DOI] [PubMed] [Google Scholar]
- Agoston DV, Ballmann M, Conlon JM, Dowe GH, Whittaker VP. Isolation of neuropeptide-containing vesicles from the guinea pig ileum. J Neurochem. 1985;45:398–406. doi: 10.1111/j.1471-4159.1985.tb04001.x. [DOI] [PubMed] [Google Scholar]
- Bayguinov O, Keef KD, Hagen B, Sanders KM. Parallel pathways mediate inhibitory effects of vasoactive intestinal polypeptide and nitric oxide in canine fundus. Br J Pharmacol. 1999;126:1543–1552. doi: 10.1038/sj.bjp.0702450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancani P, Walsh J, Behar J. Vasoactive intestinal peptide: a neurotransmitter for relaxation of the rabbit internal anal sphincter. Gastroenterology. 1985;89:867–874. doi: 10.1016/0016-5085(85)90585-2. [DOI] [PubMed] [Google Scholar]
- Bitar KN, Makhlouf GM. Relaxation of isolated gastric smooth muscle cells by vasoactive intestinal peptide. Science. 1982;216:531–533. doi: 10.1126/science.6176025. [DOI] [PubMed] [Google Scholar]
- Boeckxstaens GE, Pelckmans PA, De Man JG, Bult H, Herman AG, Van Maercke YM. Evidence for a differential release of nitric oxide and vasoactive intestinal polypeptide by nonadrenergic noncholinergic nerves in the rat gastric fundus. Arch Int Pharmacodyn Ther. 1992;318:107–115. [PubMed] [Google Scholar]
- Bryant MG, Polak MM, Modlin I, Bloom SR, Albuquerque RH, Pearse AG. Possible dual role for vasoactive intestinal peptide as gastrointestinal hormone and neurotransmitter substance. Lancet. 1976;1:991–993. doi: 10.1016/s0140-6736(76)91863-8. [DOI] [PubMed] [Google Scholar]
- Burleigh DE. NG-Nitro-L-arginine reduces nonadrenergic, noncholinergic relaxations of human gut. Gastroenterology. 1992;102:679–683. doi: 10.1016/0016-5085(92)90120-n. [DOI] [PubMed] [Google Scholar]
- Burleigh DE, D’Mello A, Parks AG. Responses of isolated human internal anal sphincter to drugs and electrical field stimulation. Gastroenterology. 1979;77:484–490. [PubMed] [Google Scholar]
- Burnstock G. Purinergic nerves. Pharmacol Rev. 1972;24:509–581. [PubMed] [Google Scholar]
- Burnstock G. Cotransmission. Curr Opin Pharmacol. 2004;4:47–52. doi: 10.1016/j.coph.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Cheeney G, Nguyen M, Valestin J, Rao SS. Topographic and manometric characterization of the recto-anal inhibitory reflex. Neurogastroenterol Motil. 2012;24:e147–e154. doi: 10.1111/j.1365-2982.2011.01857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobine CA, Fong M, Hamilton R, Keef KD. Species dependent differences in the actions of sympathetic nerves and noradrenaline in the internal anal sphincter. Neurogastroenterol Motil. 2007;19:937–945. doi: 10.1111/j.1365-2982.2007.00982.x. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R939–R949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- de Lorijn F, de Jonge WJ, Wedel T, Vanderwinden JM, Benninga MA, Boeckxstaens GE. Interstitial cells of Cajal are involved in the afferent limb of the rectoanal inhibitory reflex. Gut. 2005;54:1107–1113. doi: 10.1136/gut.2004.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Li CG, Rand MJ. Nitrergic and purinergic mechanisms and their interactions for relaxation of the rat internal anal sphincter. J Auton Pharmacol. 1999;19:29–37. doi: 10.1046/j.1365-2680.1999.00114.x. [DOI] [PubMed] [Google Scholar]
- Duffy AM, Cobine CA, Keef KD. Changes in neuromuscular transmission in the W/Wv mouse internal anal sphincter. Neurogastroenterol Motil. 2012;24:e41–e55. doi: 10.1111/j.1365-2982.2011.01806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund S, Jodal M, Lundgren O, Sjöqvist A. Effects of vasoactive intestinal polypeptide on blood flow, motility and fluid transport in the gastrointestinal tract of the cat. Acta Physiol Scand. 1979;105:461–468. doi: 10.1111/j.1748-1716.1979.tb00111.x. [DOI] [PubMed] [Google Scholar]
- El-Mahmoudy A, Khalifa M, Draid M, Shiina T, Shimizu Y, El-Sayed M, Takewaki T. NANC inhibitory neuromuscular transmission in the hamster distal colon. Pharmacol Res. 2006;54:452–460. doi: 10.1016/j.phrs.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Fahrenkrug J. Transmitter role of vasoactive intestinal peptide. Pharmacol Toxicol. 1993;72:354–363. doi: 10.1111/j.1600-0773.1993.tb01344.x. [DOI] [PubMed] [Google Scholar]
- Fahrenkrug J. VIP and PACAP. Results Probl Cell Differ. 2010;50:221–234. doi: 10.1007/400_2009_24. [DOI] [PubMed] [Google Scholar]
- Fahrenkrug J, Haglund U, Jodal M, Lundgren O, Olbe L, de Muckadell OB. Nervous release of vasoactive intestinal polypeptide in the gastrointestinal tract of cats: possible physiological implications. J Physiol. 1978;284:291–305. doi: 10.1113/jphysiol.1978.sp012541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness JB. The Enteric Nervous System. Oxford: Blackwell Publishing; 2006. [Google Scholar]
- Furness JB, Costa M. Projections of intestinal neurons showing immunoreactivity for vasoactive intestinal polypeptide are consistent with these neurons being the enteric inhibitory neurons. Neurosci Lett. 1979;15:199–204. doi: 10.1016/0304-3940(79)96113-5. [DOI] [PubMed] [Google Scholar]
- Gallego D, Hernández P, Clavé P, Jiménez M. P2Y1 receptors mediate inhibitory purinergic neuromuscular transmission in the human colon. Am J Physiol Gastrointest Liver Physiol. 2006;291:G584–G594. doi: 10.1152/ajpgi.00474.2005. [DOI] [PubMed] [Google Scholar]
- Goyal RK, Rattan S, Said SI. VIP as a possible neurotransmitter of non-cholinergic non-adrenergic inhibitory neurones. Nature. 1980;288:378–380. doi: 10.1038/288378a0. [DOI] [PubMed] [Google Scholar]
- Grasa L, Gil V, Gallego D, Martín MT, Jiménez M. P2Y1 receptors mediate inhibitory neuromuscular transmission in the rat colon. Br J Pharmacol. 2009;158:1641–1652. doi: 10.1111/j.1476-5381.2009.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grider JR. Identification of neurotransmitters regulating intestinal peristaltic reflex in humans. Gastroenterology. 1989;97:1414–1419. doi: 10.1016/0016-5085(89)90384-3. [DOI] [PubMed] [Google Scholar]
- Grider JR, Cable MB, Bitar KN, Said SI, Makhlouf GM. Vasoactive intestinal peptide: relaxant neurotransmitter in tenia coli of the guinea pig. Gastroenterology. 1985a;89:36–42. [PubMed] [Google Scholar]
- Grider JR, Cable MB, Said SI, Makhlouf GM. Vasoactive intestinal peptide as a neural mediator of gastric relaxation. Am J Physiol Gastrointest Liver Physiol. 1985b;248:G73–G78. doi: 10.1152/ajpgi.1985.248.1.G73. [DOI] [PubMed] [Google Scholar]
- Grider JR, Makhlouf GM. Colonic peristaltic reflex: identification of vasoactive intestinal peptide as mediator of descending relaxation. Am J Physiol Gastrointest Liver Physiol. 1986;251:G40–G45. doi: 10.1152/ajpgi.1986.251.1.G40. [DOI] [PubMed] [Google Scholar]
- Hagen BM, Bayguinov O, Sanders KM. VIP and PACAP regulate localized Ca2+ transients via cAMP-dependent mechanism. Am J Physiol Cell Physiol. 2006;291:C375–C385. doi: 10.1152/ajpcell.00495.2005. [DOI] [PubMed] [Google Scholar]
- Jin JG, Murthy KS, Grider JR, Makhlouf GM. Stoichiometry of neurally induced VIP release, NO formation, and relaxation in rabbit and rat gastric muscle. Am J Physiol Gastrointest Liver Physiol. 1996;271:G357–G369. doi: 10.1152/ajpgi.1996.271.2.G357. [DOI] [PubMed] [Google Scholar]
- Jones OM, Brading AF, Mortensen NJ. Role of nitric oxide in anorectal function of normal and neuronal nitric oxide synthase knockout mice: a novel approach to anorectal disease. Dis Colon Rectum. 2003;46:963–970. doi: 10.1007/s10350-004-6694-y. [DOI] [PubMed] [Google Scholar]
- Keef KD, Shuttleworth CWR, Xue C, Bayguinov O, Publicover NG, Sanders KM. Relationship between nitric oxide and vasoactive intestinal polypeptide in enteric inhibitory neurotransmission. Neuropharmacology. 1994;33:1303–1314. doi: 10.1016/0028-3908(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Larsson LI, Fahrenkrug J, Schaffalitzky De Muckadell O, Sundler F, Håkanson R, Rehfeld JR. Localization of vasoactive intestinal polypeptide (VIP) to central and peripheral neurons. Proc Natl Acad Sci U S A. 1976;73:3197–3200. doi: 10.1073/pnas.73.9.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre V, Favrais G, Abad C, Adle-Biassette H, Lu Y, Germano PM, Cheung-Lau G, Pisegna JR, Gressens P, Lawson G, Waschek JA. Gastrointestinal dysfunction in mice with a targeted mutation in the gene encoding vasoactive intestinal polypeptide: a model for the study of intestinal ileus and Hirschsprung's disease. Peptides. 2007;28:1688–1699. doi: 10.1016/j.peptides.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax AE, Furness JB. Neurochemical classification of enteric neurons in the guinea-pig distal colon. Cell Tissue Res. 2000;302:59–72. doi: 10.1007/s004410000260. [DOI] [PubMed] [Google Scholar]
- Morgado M, Cairrão E, Santos-Silva AJ, Verde I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci. 2012;69:247–266. doi: 10.1007/s00018-011-0815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mule F, Serio R. NANC inhibitory neurotransmission in mouse isolated stomach: involvement of nitric oxide, ATP and vasoactive intestinal polypeptide. Br J Pharmacol. 2003;140:431–437. doi: 10.1038/sj.bjp.0705431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurko S, Rattan S. Role of vasoactive intestinal polypeptide in the internal anal sphincter relaxation of the opossum. J Clin Invest. 1988;81:1146–1153. doi: 10.1172/JCI113429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kelly T, Brading A, Mortensen N. Nerve mediated relaxation of the human internal anal sphincter: the role of nitric oxide. Gut. 1993;34:689–693. doi: 10.1136/gut.34.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo A, Lecea B, Admella C, Fantova MJ, Jiménez M, Martí-Ragué J, Clavé P. A comparative study of structure and function of the longitudinal muscle of the anal canal and the internal anal sphincter in pigs. Dis Colon Rectum. 2009;52:1902–1911. doi: 10.1007/DCR.0b013e3181b160be. [DOI] [PubMed] [Google Scholar]
- Opazo A, Lecea B, Gil V, Jiménez M, Clavé P, Gallego D. Specific and complementary roles for nitric oxide and ATP in the inhibitory motor pathways to rat internal anal sphincter. Neurogastroenterol Motil. 2011;23:e11–e25. doi: 10.1111/j.1365-2982.2010.01602.x. [DOI] [PubMed] [Google Scholar]
- Qu ZD, Thacker M, Castelucci P, Bagyanszki M, Epstein ML, Furness JB. Immunohistochemical analysis of neuron types in the mouse small intestine. Cell Tissue Res. 2008;334:147–161. doi: 10.1007/s00441-008-0684-7. [DOI] [PubMed] [Google Scholar]
- Rae MG, Muir TC. Neuronal mediators of inhibitory junction potentials and relaxation in the guinea-pig internal anal sphincter. J Physiol. 1996;493:517–527. doi: 10.1113/jphysiol.1996.sp021400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S, Gilmont RR, Miyasaka EA, Somara S, Srinivasan S, Teitelbaum DH, Bitar KN. Successful implantation of bioengineered, intrinsically innervated, human internal anal sphincter. Gastroenterology. 2011;141:310–319. doi: 10.1053/j.gastro.2011.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand MJ. Nitrergic transmission: nitric oxide as a mediator of non-adrenergic, non-cholinergic neuro-effector transmission. Clin Exp Pharmacol Physiol. 1992;19:147–169. doi: 10.1111/j.1440-1681.1992.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Rattan S, Regan RF, Patel CA, De Godoy MA. Nitric oxide not carbon monoxide mediates nonadrenergic noncholinergic relaxation in the murine internal anal sphincter. Gastroenterology. 2005;129:1954–1966. doi: 10.1053/j.gastro.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Said SI, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science. 1970;169:1217–1218. doi: 10.1126/science.169.3951.1217. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Matsuyama H, Shiina T, Takewaki T, Furness JB. Tachykinins and their functions in the gastrointestinal tract. Cell Mol Life Sci. 2008;65:295–311. doi: 10.1007/s00018-007-7148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Malenka RC. Understanding synapses: past, present, and future. Neuron. 2008;60:469–476. doi: 10.1016/j.neuron.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Owyang C. Vagal control of nitric oxide and vasoactive intestinal polypeptide release in the regulation of gastric relaxation in rat. J Physiol. 1995;484:481–492. doi: 10.1113/jphysiol.1995.sp020680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichenor SD, Buxton IL, Johnson P, O’Driscoll K, Keef KD. Excitatory motor innervation in the canine rectoanal region: role of changing receptor populations. Br J Pharmacol. 2002;137:1321–1329. doi: 10.1038/sj.bjp.0704987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonini M, De Giorgio R, De Ponti F, Sternini C, Spelta V, Dionigi P, Barbara G, Stanghellini V, Corinaldesi R. Role of nitric oxide- and vasoactive intestinal polypeptide-containing neurones in human gastric fundus strip relaxations. Br J Pharmacol. 2000;129:12–20. doi: 10.1038/sj.bjp.0702977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Geldre LA, Lefebvre RA. Interaction of NO and VIP in gastrointestinal smooth muscle relaxation. Curr Pharm Des. 2004;10:2483–2497. doi: 10.2174/1381612043383890. [DOI] [PubMed] [Google Scholar]
- Wang GD, Wang XY, Hu HZ, Liu S, Gao N, Fang X, Xia Y, Wood JD. Inhibitory neuromuscular transmission mediated by the P2Y1 purinergic receptor in guinea pig small intestine. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1483–G1489. doi: 10.1152/ajpgi.00450.2006. [DOI] [PubMed] [Google Scholar]
- Zhu HL, Hirst GD, Ito Y, Teramoto N. Modulation of voltage-dependent Ba2+ currents in the guinea-pig gastric antrum by cyclic nucleotide-dependent pathways. Br J Pharmacol. 2005;146:129–138. doi: 10.1038/sj.bjp.0706295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzo MG, Mulè F, Serio R. Inhibitory purinergic transmission in mouse caecum: role for P2Y1 receptors as prejunctional modulators of ATP release. Neuroscience. 2007;150:658–664. doi: 10.1016/j.neuroscience.2007.09.055. [DOI] [PubMed] [Google Scholar]