

Abstract
Central sleep apnoea is a condition characterized by oscillations between apnoea and hyperpnoea during sleep. Studies in sleeping dogs suggest that withdrawal of peripheral chemoreceptor (carotid body) activation following transient ventilatory overshoots plays an essential role in causing apnoea, raising the possibility that sustaining carotid body activity during ventilatory overshoots may prevent apnoea. To test whether sustained peripheral chemoreceptor activation is sufficient to drive breathing, even in the absence of central chemoreceptor stimulation and vagal feedback, we used a vagotomized, decerebrate dual-perfused in situ rat preparation in which the central and peripheral chemoreceptors are independently and artificially perfused with gas-equilibrated medium. At varying levels of carotid body stimulation (CB PO2/PCO2: 40/60, 100/40, 200/15, 500/15 Torr), we decreased the brainstem perfusate PCO2 in 5 Torr steps while recording phrenic nerve activity to determine the central apnoeic thresholds. The central apnoeic thresholds decreased with increased carotid body stimulation. When the carotid bodies were strongly stimulated (CB 40/60), the apnoeic threshold was 3.6 ± 1.4 Torr PCO2 (mean ± SEM, n = 7). Stimulating carotid body afferent activity with either hypercapnia (60 Torr PCO2) or the neuropeptide pituitary adenylate cyclase-activating peptide restored phrenic activity during central apnoea. We conclude that peripheral stimulation shifts the central apnoeic threshold to very hypocapnic levels that would likely increase the CO2 reserve and have a protective effect on breathing. These data demonstrate that peripheral respiratory chemoreceptors are sufficient to stave off central apnoeas when the brainstem is perfused with low to no CO2.
Key points
Central sleep apnoea is a condition characterized by oscillations between apnoea and hyperpnoea during sleep, which can have many serous health implications.
Each ventilatory overshoot following an apnoea attenuates peripheral chemoreceptor input which, in turn, has the potential to cause a further apnoea.
In a decerebrate, vagotomized, in situ rat preparation, we show that central apnoeas can be overcome both physiologically (with high peripheral CO2) and pharmacologically (with peripheral pituitary adenylate cyclase-activating peptide).
We also show that the central apnoeic threshold, i.e. the CO2 level at which the animal stops breathing, can be lowered by increasing peripheral chemoreceptor stimulation.
These data suggest that stimulation of peripheral chemoreceptors may prevent central apnoeas, re-affirming the peripheral chemoreceptors as possible therapeutic targets for some sleep apnea phenotypes.
Introduction
Inputs from central (brainstem) and peripheral (carotid bodies) respiratory chemoreceptors are integrated within the respiratory control system to protect blood gases against potentially deleterious fluctuations (O’Regan & Majcherczyk, 1982). Respiratory chemoreceptors are important during wakefulness and essential during sleep (Spengler et al. 2001; Dempsey et al. 2004), determining the efficacy of phasic vagal feedback from stretch receptors in the lung, regulating upper and lower airway calibre, and modulating the respiratory rhythm generator and/or respiratory motor neurons to sustain breathing (Kinkead et al. 1994; Spyer & Gourine, 2009; Nattie, 2011). Consequently, abnormalities in brainstem and/or carotid body chemoreflex function might be a primary cause of breathing instability during sleep and a possible target for pharmacotherapeutics for some sleep apnoea phenotypes (Kiwull-Schöne et al. 2008). Here, we use a rodent model to test whether tonic carotid body stimulation is capable of sustaining breathing when the brainstem CO2 chemoreflex is silenced.
Many brainstem areas appear to be endowed with endogenous respiratory CO2 chemosensitivity (ability to detect local changes in PCO2 and initiate ventilatory responses), including the nucleus tractus solitarius, raphe, retrotrapezoid nucleus (RTN), the locus coeruleus and the cerebellar fastigial nucleus (Martino et al. 2007; Guyenet, 2008; Corcoran et al. 2009; Gargaglioni et al. 2010; Nattie, 2011). However, as the central chemoreflex responds slowly to changes in PaCO2 relative to the fluctuations that occur in sleep apnoea, the role of central chemoreceptors in periodic breathing has been questioned (Smith et al. 2003). In contrast to the central chemoreflex, the response of the carotid body-mediated chemoreflex to changes in blood gases occurs within 3–5 s, an order of magnitude faster than that of the brainstem chemoreflex (Carroll et al. 1991).
Since their discovery, the carotid bodies have been considered as the primary oxygen sensors, responding vigorously to precipitous falls in PaO2 that normally accompany asphyxia. In sleep-disordered breathing, such a response may be critical after an apnoea or hypopnea to trigger arousal, open the airways and/or kick-start breathing motor patterns. However, carotid bodies also play an important role in sustaining eupnoeic ventilation under normoxic conditions (Forster et al. 2008; Mouradian et al. 2012) and are reported to respond vigorously to changes in PCO2 (Cunningham, 1987). Thus, elevated CO2 causes increased [Ca2+]i in isolated glomus cells and carotid sinus nerve (CSN) activity in a range of preparations, from the isolated perfused carotid body preparations to in vivo preparations (Iturriaga, 1993; Linton et al. 1995; Cummings & Wilson, 2005). But perhaps most telling, with few exceptions (Lugliani et al. 1971; da Silva et al. 2011), carotid body denervation in awake humans, lambs, goats, ponies, piglets and rats causes a pronounced hypoventilation, with an accompanying increase in PaCO2 of 5–15 Torr (Wade et al. 1970; Bisgard et al. 1976; Olson et al. 1988; Praud et al. 1992; Côtéet al. 1996; Pan et al. 1998; Lowry et al. 1999; Timmers et al. 2003; Mouradian et al. 2012). Despite the increase in PaCO2 being of sufficient magnitude to cause cerebrospinal fluid acidosis, the central chemoreceptors are unable to compensate.
These observations raise the possibility that, in addition to the carotid bodies’ classic role in terminating apnoeas by triggering vigorous sympathetic and respiratory responses, output from the carotid bodies may also be necessary for preventing apnoeas during eupnoea. Daristotle et al. (1990) addressed this issue directly in goats by selectively making the carotid body hypocapnic using an extracorporeal circuit (carotid bodies isolated from systemic circulation). In awake animals, reducing the carotid body PCO2 by 10.9 Torr decreased ventilation by 24% and increased PaCO2 by 5.6 Torr. In 4 of 14 animals the hypoventilation was accompanied by ventilatory instability, including apnoea (Daristotle et al. 1990). Smith and Dempsey used a similar approach and carotid body denervation to investigate the importance of the carotid body in sustaining breathing during non-rapid eye movement (REM) sleep in conscious dogs. In line with Daristotle's observations, they found that arterial hypocapnia caused by a transient ventilatory overshoot was much more likely to cause apnoea if the hypocapnia was sensed by the carotid body (Nakayama et al. 2003; Smith et al. 2003).
If exposure to hypocapnia causes breathing instability and apnoea by reducing the activity of the carotid body, then sustaining activity of the carotid body should be a rational strategy for preventing apnoeas. This argument is supported by remarkable data obtained by Berkenbosch et al. using an anaesthetized cat preparation that separates perfusion of the pons and brainstem from systemic circulation, thereby allowing blood gases at the carotid bodies and brainstem to be controlled independently (Berkenbosch et al. 1984). Providing the carotid bodies were stimulated, the breathing pattern remained stable even when the PCO2 of the blood supplied to the brainstem was reduced to 3.8 Torr. However, the authors considered it likely that their result was in part caused by the vasoconstriction that accompanies extreme hypocapnia, changing cerebral blood flow and allowing systemic circulation into the medulla. A further caveat was revealed recently with discovery of a population of hypothalamic orexin-containing neurons with exquisite sensitivity to CO2: it is possible that not all central respiratory chemoreceptors are located in the brainstem (Williams et al. 2007).
As pharmacological stimulation of the carotid body might yet provide a therapeutic avenue to treat sleep apnoea in carefully phenotyped patients, the aim of our study was to test whether peripheral respiratory chemoreceptor activation was sufficient to sustain respiratory rhythm generation in the absence of functional central chemoreceptors, vagal feedback and descending projections from higher centres, in a non-anaesthetized preparation. For these experiments, we used a decerebrate, vagotomized dual-perfused rat preparation (illustrated in Fig. 1A) based on the in situ working heart brainstem preparation in which carotid bodies and brainstem are independently perfused with artificial saline (Paton, 1996; Day & Wilson, 2005, 2007, 2009). In this preparation, the circle of Willis is cut preventing the possibility of cross-contamination between the perfusate to the brainstem and carotid bodies. We found that with asphyxia-like (hypoxic-hypercapnia) stimulation of the carotid bodies, breathing motor output persisted even when brainstem perfusion PCO2 approached 0 Torr, demonstrating that peripheral respiratory chemoreceptors are sufficient to stave off central apnoeas caused by severe brainstem hypocapnia. We also show that hypercapnic or pharmacological stimulation of the carotid bodies can overcome central apnoea.
Figure 1. Schematic of the DPP and Protocol 1.
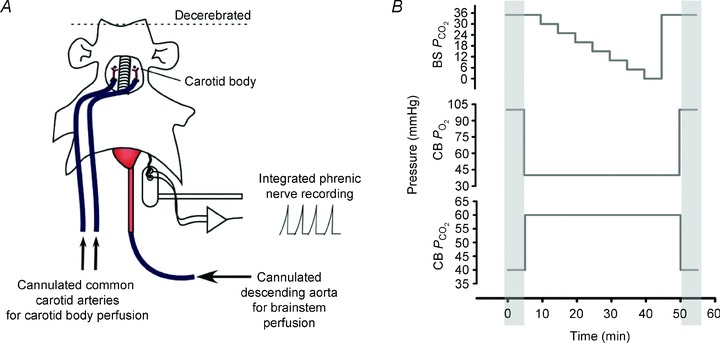
A, the key feature of the DPP used for this study is independent artificial perfusion of the carotid bodies (via ligation, cannulation and perfusion of the common carotid arteries) and brainstem (via cannulation and retrograde perfusion of the descending aorta). The DPP is vagotomized and decerebrated (non-anaesthetized). We use phrenic activity to measure neuronal ventilation. B, illustration of protocol. PCO2 and/or PO2 in the central (brainstem; BS) and peripheral (carotid body; CB) perfusate for the CB 40/60 group. The PCO2 in the brainstem perfusate was balanced with O2. The PCO2 and PO2 in carotid body perfusate were balanced with N2.
Methods
Experimental procedures were approved by the University of Calgary Animal Care Committee, and were in accordance with Canadian law. Experiments were conducted with juvenile male Sprague–Dawley albino rats (weight 100–150 g; ≍ 4–6 weeks old; Charles River, Quebec, Canada).
Dual-perfused preparation (DPP)
Rats were weighed, lightly anaesthetized in a bell jar with halothane till recumbent and then prepared for experimentation. In the initial experiments, animals were given an intra-peritoneal injection of sodium pentobarbital (30–50 mg kg−1) or intubated and ventilated with 2% halothane. The efficacy of anaesthesia was assessed by testing for absence of response to noxious tail pinch and corneal reflex. In rapid succession, rats were cut in half just below the diaphragm and placed in ice-cold perfusate containing (in mm): NaCl, 115; NaHCO3, 24; KCl, 4; CaCl2, 2; MgSO4, 1; NaH2PO4, 1.25; glucose, 10; sucrose, 12; equilibrated with 95% O2–5% CO2. Next, the skin from the top of the head was removed. Animals were then placed in the experimental chamber in a supine position and fixed with ear bars. The descending aorta was cannulated with a double-lumen catheter. One lumen of the catheter was connected to a peristaltic pump (Gilson Minipuls 3) and used to perfuse the descending aorta retrogradely with warm (33°C) perfusate, equilibrated with 40 Torr PCO2 in O2 (central perfusion). The other lumen was attached to a pressure transducer to monitor perfusion pressure. Once cannulated, the speed of the peristaltic pump was increased so as to ramp perfusion pressure to 60 mmHg over the first few minutes using a custom-built computer-controlled feedback system. Next, the preparation was decerebrated at the mid-collicular level (approximate level of lambda). All tissue rostral to the decerebration was removed, as was the remaining cortex dorsal to the colliculi rendering the preparation insentient. The face and any remaining skin were also removed. The common carotid arteries were exposed, the jugular veins and vagus nerves sectioned, and the arteries tied off above the clavicles and cannulated. More recent experiments used a slightly different approach to preparing the DPP because we found it significantly increased the yield of viable preparations. In this revised approach, anaesthesia was maintained via a facemask supplied with 2–4% halothane. The efficacy of anaesthesia was assessed by testing for the absence of response to noxious tail pinch and corneal reflex. The animal was then placed in ice-cold perfusate and the skin removed rostrally from the abdomen to the cranium. While still breathing, the preparation was decerebrated, as above, at the mid-collicular level. All tissue rostral to the decerebration was removed, as was the remaining cortex dorsal to the colliculi. The animal was then rapidly sectioned just below the diaphragm, placed in the experimental chamber and fixed in a supine position with ear bars. The descending aorta was cannulated, as above, and the perfusion pressure gradually increased to 60 mmHg. Both vagi were sectioned at the cervical level, followed by cannulation of the carotid arteries.
A separate peristaltic pump (Gilson Minipuls 3) was used to perfuse both common carotid arteries (peripheral perfusion). Following cannulation, the flow through each common carotid artery was ramped up to 15 ml min−1.
Up to this stage in the dissection, central and peripheral perfusions were from the same tonometer (300 ml). Next, carotid and brainstem compartments were supplied with perfusate from separate tonometers with different gas compositions (carotid body: 100 Torr PO2, 35 Torr PCO2 in N2; brainstem: 35 Torr PCO2 in O2). Perfusate exiting the preparation was collected in the base of the recording chamber and returned to the tonometers for re-equilibration and recirculation. The central perfusion pressure was ramped to 90 mmHg.
Electrophysiology
The left phrenic nerve was located and dissected free of surroundings tissues, leaving a small portion of diaphragm attached at the distal end. The nerve was placed in a custom holder that prevented the nerve from drying out and facilitated long-term extracellular recordings. The phrenic neurogram was amplified (A-M Systems Differential AC Amplifier Model 1700), filtered (low cut-off, 300 Hz; high cut-off, 5 kHz), rectified and integrated (Amplitude Demodulator; Saga Tech), computer archived (Axon Instruments Digidata 1322A and Axoscope 9.0) at a sampling rate of 50 Hz, and analysed off-line.
Experimental protocols
Protocol 1
A graphic description of the protocol can be found in Fig. 1B: 35–40 min following dissection and pressure equilibration, the baseline condition was established: brainstem perfusate 35 Torr PCO2 in O2 and carotid bodies perfusate, 40 Torr PCO2 and 100 Torr PO2 in N2. Then, the carotid bodies were stimulated with 60 Torr PCO2 and 40 Torr PO2 in N2 for 5 min without any change in the perfusate to the brainstem (stimulated conditions). Next, with the carotid bodies stimulated, brainstem perfusate PCO2 was decreased by 5 Torr (balance with O2) every 5 min to find the central apnoeic threshold, defined as the brainstem perfusate PCO2 at or below which phrenic bursts were abolished (Dempsey & Skatrud, 1986; Boden et al. 1998). In some cases, phrenic bursts continued for 5 min even when brainstem perfusate PCO2 was 0 Torr. Once the apnoeic threshold was obtained, or brainstem perfusate PCO2 was 0 Torr but phrenic bursts persisted, the PCO2 of the brainstem perfusate was returned to baseline conditions for 5 min (35 Torr PCO2 in O2), while maintaining stimulation to the carotid bodies. Finally, the baseline conditions used at the start of the protocol were re-imposed: brainstem perfusate 35 Torr PCO2 in O2 and carotid bodies perfusate, 40 Torr PCO2 and 100 Torr PO2 in N2. This protocol was repeated in separate preparations where the carotid body perfusate was maintained at 100 Torr PO2 with 40 Torr PCO2, as well as both 200 and 500 Torr PO2 with 15 Torr PCO2 (n = 6/group; see Figs 3 and 4).
Figure 3. Respiratory rate, nVT and 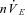 at different levels of brainstem PCO2.
at different levels of brainstem PCO2.
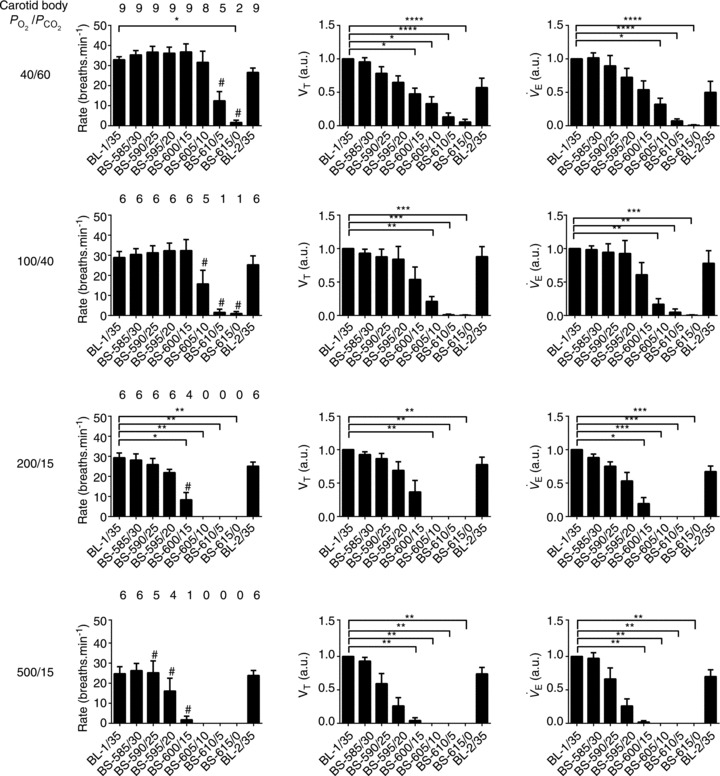
Bins represent average values of respiratory variables over 60 s (between minutes 4 and 5, i.e. the last 60 s bin) of each experimental condition. The numbers above rate data represent the number of animals in which phrenic bursts persisted at each condition. nVT: amplitude of phrenic in arbitrary units (a.u.); 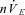 : nVT multiplied by breaths per minute in arbitrary units (a.u.). Error bars represent SEM. Friedman test with Dunn's multiple comparisons test, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Wilcoxon signed rank test (hypothetical mean of 0), #P > 0.05, thus not significantly different to 0 (apnoea). BL, baseline; BS-585/30, brainstem perfusate gas pressure of 585 Torr PO2 and 30 Torr PCO2.
: nVT multiplied by breaths per minute in arbitrary units (a.u.). Error bars represent SEM. Friedman test with Dunn's multiple comparisons test, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Wilcoxon signed rank test (hypothetical mean of 0), #P > 0.05, thus not significantly different to 0 (apnoea). BL, baseline; BS-585/30, brainstem perfusate gas pressure of 585 Torr PO2 and 30 Torr PCO2.
Figure 4. TI, TE and EI at different levels of brainstem PCO2.
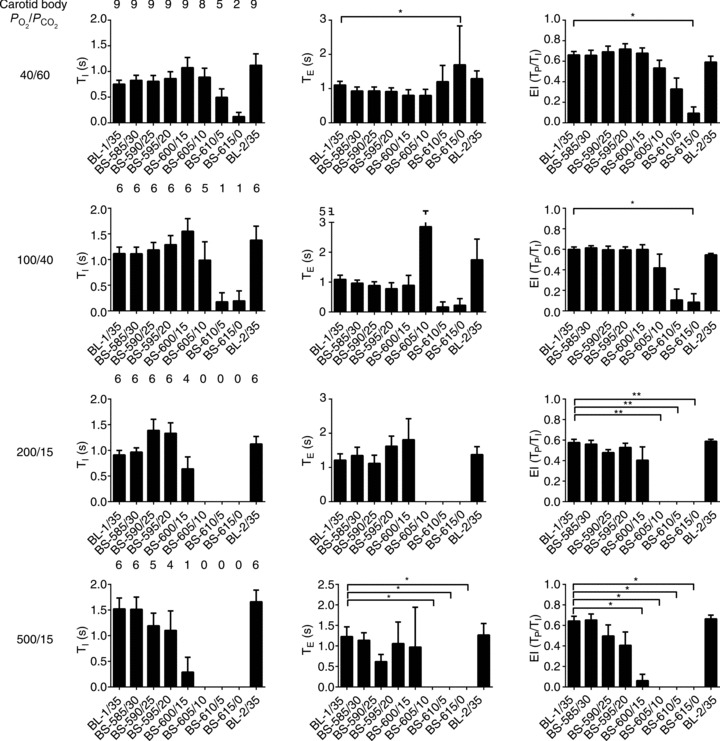
Bins represent average values of respiratory variables over 60 s (between minutes 4 and 5, i.e. the last 60 s bin) of each experimental condition. TI: duration of phrenic burst; TE: interburst interval; Tp: time from onset of phrenic burst to burst peak; EI: eupnoeic index (Tp/TI) where, at the extremes, a value of 0 represents a rapid-onset decrementing burst and a value of 1 is an augmenting burst with rapid offset. Error bars represent SEM. Friedman test with Dunn's multiple comparisons test, *P ≤ 0.05; **P ≤ 0.01.
Only preparations in which phrenic discharges returned when brainstem perfusate PCO2 was increased (following determination of the apnoeic threshold) were included in the analysis. Every phrenic burst over the protocol period was analysed.
Protocol 2
Following dissection, baseline conditions were established as in Protocol 1. Next, the gas equilibrating the tonometer supplying the brainstem was made severely hypocapnic (10 Torr PCO2, in O2) without changing the gas equilibrating the carotid bodies (100 Torr PO2, 40 Torr PCO2 in N2). After apnoea had developed, a 1 min bolus of pituitary adenylate cyclase-activating peptide (PACAP)-38 (final concentration: 100 nm) was added to the peripheral perfusate before it reached the preparation. Six preparations were used for this protocol, three with CSN intact and the remainder with CSN denervated.
Protocol 3
Baseline conditions were established as in Protocol 1, and the gas equilibrating the tonometer supplying the brainstem was made severely hypocapnic (10 Torr PCO2, in O2) without changing the gas equilibrating the carotid bodies (100 Torr PO2, 40 Torr PCO2 in N2). After apnoea had developed, the gas equilibrating the carotid bodies was made hypercapnic (60 Torr PCO2). Both brainstem and carotid body perfusate were returned to baseline gas values 5 min after breathing had resumed.
Phrenic neurogram analysis
The following respiratory variables were quantified from the integrated phrenic neurogram using custom-written software (RJAW): period (TTOT), respiratory rate (fR, 60 times the inverse of the period), time to peak (Tp), inspiratory duration (TI), expiratory duration (TE), neural tidal volume (nVT, the peak phrenic amplitude), neural minute ventilation (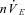 , the product of fR and nVT) and eupnoeic index (EI, Tp divided by TI). A schematic of this quantification has been previously published (Day & Wilson, 2005). Respiratory variables were averaged in 60 s bins. nVT and
, the product of fR and nVT) and eupnoeic index (EI, Tp divided by TI). A schematic of this quantification has been previously published (Day & Wilson, 2005). Respiratory variables were averaged in 60 s bins. nVT and 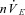 were normalized to the last minute of the initial baseline section (baseline 1: minutes 4–5) for graphical representation.
were normalized to the last minute of the initial baseline section (baseline 1: minutes 4–5) for graphical representation.
Statistical analysis
For statistical tests of each respiratory variable, the last time bin (between minutes 4 and 5) of each experimental condition was analysed. The data were not normally distributed owing to the occurrence of apnoea, so we used non-parametric statistics. We used the Friedman's two-way ANOVA by ranks test to determine the merit of the null hypothesis that respiratory variables were the same between conditions. To determine at what condition apnoea was statistically present, we used a series of one-sample Wilcoxon signed rank tests to test the null hypothesis that respiratory rate during each condition had a value of zero. Grouped data are given as mean ± SEM.
Results
Central apnoeic threshold at varying levels of carotid body activity
Examples showing the effects of lowering brainstem perfusate PCO2 on phrenic nerve activity when the carotid bodies are stimulated with perfusate containing PO2/PCO2 of 40/60, 100/40 and 500/15 Torr are illustrated in Fig. 2. As can be seen, rate and neural tidal volume decrease with brainstem perfusate PCO2 but, in the case of the hypoxic-hypercapnic carotid body, apnoea is not apparent until the brainstem perfusate is severely hypocapnic (nominally 0 Torr PCO2; see Wilson et al. 2001). A similar degree of resistance to central apnoea was apparent across all preparations in which the carotid bodies were stimulated with hypoxic-hypercapnia (Fig. 3). Thus, in two of nine preparations, rhythmic phrenic bursts continued even when brainstem perfusate PCO2 was zero. In the remaining seven preparations, progressive reduction in brainstem perfusate PCO2 eventually caused cessation of phrenic activity, but the PCO2 reached when cessation occurred (approximating the central apnoeic threshold) was 3.57 ± 1.43 Torr. In three of these seven preparations, phrenic activity only stopped when brainstem perfusate PCO2 was nominally 0 Torr. The highest central apnoeic threshold in the seven preparations was between 10 and 15 Torr PCO2. To statistically evaluate the level of brainstem perfusate PCO2 required to obtain apnoea across all nine preparations, we used a series of one-sample Wilcoxon signed rank tests (null hypothesis: breaths per minute equals zero). Accordingly, from a statistical perspective, when the carotid body was stimulated by 40 Torr PO2 and 60 Torr PCO2, a brainstem perfusate PCO2 of 10 Torr or more was sufficient to prevent apnoea. When output from the carotid body was attenuated by increasing carotid body PO2 and decreasing PCO2, the mean central apnoeic thresholds increased. Thus, with CB PO2/PCO2 of 100/40, 200/15 and 500/15, apnoeic thresholds were 7.0 ± 0.5, 11.7 ± 0.4 and 16.7 ± 0.9 Torr PCO2, respectively. From a statistical perspective, the brainstem perfusate PO2 required to prevent apnoea for each of these levels of carotid body stimulation was 15, 20 and 30 Torr, respectively (n = 6/group).
Figure 2. Stimulating the carotid body staves off apnoeas.
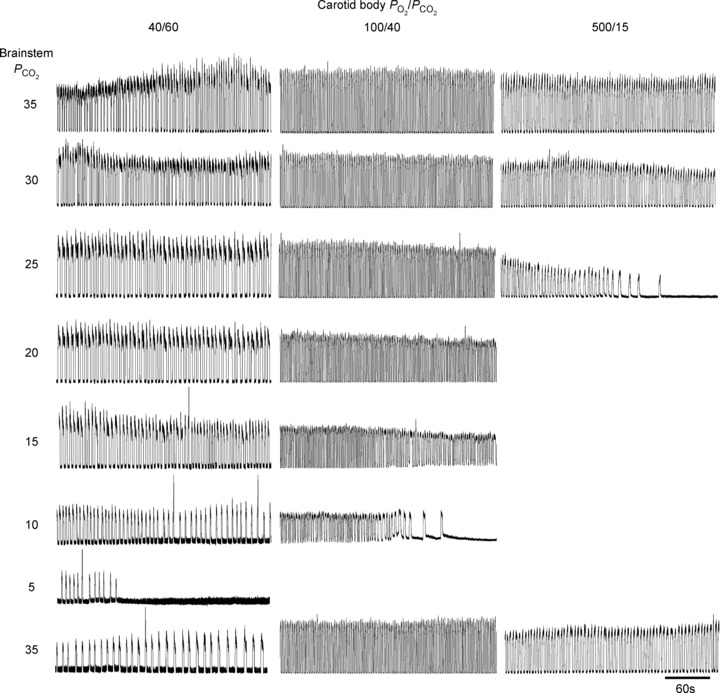
Rectified integrated phrenic neurograms of representative preparations subjected to Protocol 1. With removal of carotid body stimulation, the apnoeic threshold is raised in a linear fashion.
Effect on rate versus neuronal tidal volume
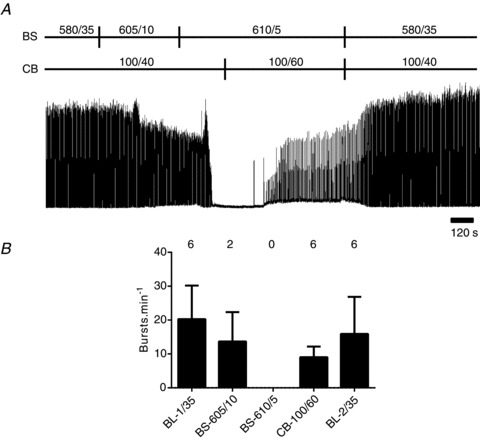
Although changing brainstem perfusate PCO2 affected all respiratory variables (Friedman's two-way ANOVA by rank, P < 0.001 for all variables), respiratory variables were not equally affected. With the carotid bodies stimulated with hypoxic-hypercapnia, both nVT and 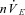 were approximately proportional to brainstem perfusate PCO2, with both timing and volume variables affected (Fig. 3). Whereas the latter was influenced progressively by a decrease in brainstem perfusate PCO2, timing variables were relatively intransient compared with nVT until severe levels of hypocapnia were reached. At these severe levels, both TI and TE were affected. Specifically, as brainstem perfusate PCO2 decreased, TI first increased above baseline levels (from 0.75 ± 0.03 s to 1.07 ± 0.07 s at 15 Torr brainstem perfusate PCO2) before falling to zero (Fig. 4). This trend was apparent in seven of the nine preparations with the carotid bodies stimulated with hypoxic-hypercapnia, as well as in all preparations with carotid body PO2/PCO2 levels of 100/40 and 200/15 Torr, but was absent in the 500/15 Torr group.
were approximately proportional to brainstem perfusate PCO2, with both timing and volume variables affected (Fig. 3). Whereas the latter was influenced progressively by a decrease in brainstem perfusate PCO2, timing variables were relatively intransient compared with nVT until severe levels of hypocapnia were reached. At these severe levels, both TI and TE were affected. Specifically, as brainstem perfusate PCO2 decreased, TI first increased above baseline levels (from 0.75 ± 0.03 s to 1.07 ± 0.07 s at 15 Torr brainstem perfusate PCO2) before falling to zero (Fig. 4). This trend was apparent in seven of the nine preparations with the carotid bodies stimulated with hypoxic-hypercapnia, as well as in all preparations with carotid body PO2/PCO2 levels of 100/40 and 200/15 Torr, but was absent in the 500/15 Torr group.
To determine the effects of brainstem hypocapnia on burst shape, which in turn may indicate oxygenation status of brainstem tissue, we quantified a eupnoeic index (EI; see Materials and methods for derivation). An EI of 0 indicates a rapid-onset decrementing burst, whereas an EI of 1 indicates a ramping burst with a sudden offset. We consider bursts with EI below 0.5 to be gasp-like, and those above 0.5 to be eupnoeic. Changing brainstem perfusate PCO2 had a significant effect on the EI (Friedman's two-way ANOVA by rank: P < 0.01; Fig. 4). On average, with the carotid bodies stimulated, bursts remained eupnoeic even with brainstem perfusate PCO2 as low as 5 Torr (EI was 0.59 ± 0.06; with only one preparation having an EI below 0.5). Nonetheless, between 15 and 0 Torr, EI diminished such that at 0 Torr the EI of the two preparations that continued to generate bursts was 0.42 ± 0.05. All other groups followed a broadly similar pattern, with EI diminishing at higher brainstem perfusate PCO2, in line with the relative apnoeic threshold/decreasing carotid body stimulation, i.e. the lower the level of carotid body activity, the higher the level of central CO2 at which EI diminished, whilst exhibiting a similar pattern.
Pharmacological stimulation of the carotid body during central apnoea
To determine whether pharmacological stimulation of the carotid body is sufficient to overcome central apnoea, we perfused the common carotid artery with the stress peptide PACAP (Fig. 5). We used six preparations for these experiments, three with the CSN intact and three with it denervated. In all preparations, with the carotid body perfusate having a PO2 and PCO2 of 100 and 40 Torr, lowering the brainstem perfusate PCO2 to 10 Torr caused a long-duration apnoea. In CSN intact preparations, adding a 1 min bolus of PACAP to the peripheral perfusate, giving a final concentration of 100 nm, promptly terminated the apnoea by causing a pronounced and long-lived increase in phrenic nerve activity. In preparations with the CSN denervated prior to PACAP application, application of PACAP had no immediate effect: apnoea persisted during the 3rd min of PACAP exposure. Thus, the stimulatory effects of PACAP over the first 3 min were largely dependent on an intact CSN (+CSN/–CSN: P < 0.001). These data suggest that pharmacological stimulation of the carotid body is a viable method of overcoming central apnoeas.
Figure 5. Excitation of carotid body with stress peptide pituitary adenylate cyclase-activating peptide (PACAP) overcomes central apnoeas.
In all preparations, central apnoea was produced by reducing brainstem PCO2 from 35 to 10 Torr. A, rectified integrated phrenic neurogram of a representative preparation exposed to PACAP during apnoea with carotid sinus nerve (CSN) intact. Note the rapid, sustained reversal of central apnoea on application of PACAP. B, rectified integrated phrenic neurogram of a representative preparation exposed to PACAP during apnoea with CSN denervated. Note the lack of immediate response to PACAP; the response to PACAP only occurs after several minutes and is not sustained. C, summary data. I: brainstem normocapnic; II: brainstem hypocapnic; III: third minute after perfusing the carotid bodies with 100 nm PACAP; IV: period of washout used for quantification. The carotid body was maintained normocapnic and normoxic throughout the protocol. Error bars represent SEM. **Indicates significant difference (P < 0.001) between carotid body intact versus carotid body denervated preparations. BS, brainstem; CB, carotid body; DPP, dual-perfused preparation.
Interestingly, while PACAP failed to restore phrenic activity within the first 3 min in all denervated preparations tested, a transient recovery from apnoea (lasting 3–4 min) occurred in all denervated preparations sometime thereafter (4–6 min; Fig. 5B). This suggests that a second component of the respiratory controller is also stimulated by PACAP. The considerable delay in response to PACAP with CSN denervated suggests this second component may be located within central respiratory or autonomic nuclei (Farnham et al. 2008; Peña, 2010; Inglott et al. 2011).
Hypercapnic stimulation of the carotid body during central apnoea
To determine whether hypercapnic stimulation of the carotid body is sufficient to overcome central apnoea, we perfused the common carotid artery with normoxic-hypercapnic solution. In all preparations, with the carotid body perfusate having a PO2 and PCO2 of 100 and 40 Torr, lowering the brainstem perfusate PCO2 to between 5 and 10 Torr caused a long-duration apnoea. Upon stimulation of the carotid body with perfusate containing PO2 and PCO2 of 100 and 60 Torr, phrenic activity promptly resumed in all preparations (Fig. 6).
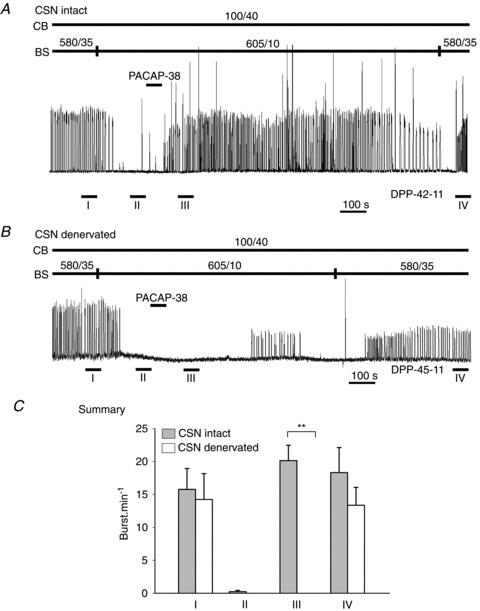
Figure 6. Excitation of carotid body with hypercapnia overcomes central apnoeas.
In all preparations, central apnoea was produced by reducing brainstem PCO2 from 35 to between 5 and 10 Torr. A, rectified integrated phrenic neurogram of a representative preparation exposed to 60 Torr PCO2 during apnoea. B, summary data. Note that breathing resumed in all cases with hypercapnic stimulation of the carotid body. Error bars represent SEM. BL, baseline; BS, brainstem; CB, carotid body.
Discussion
Our data demonstrate that providing the peripheral chemoreceptors are stimulated, respiratory rhythm generation is highly resilient to brainstem perfusion with low PCO2 even in the absence of descending input from the cerebrum, vagal feedback and anaesthetics. When the carotid bodies are stimulated, the main effect of brainstem PCO2 on phrenic activity is a reduction in burst amplitude. Only at extreme levels of brainstem perfusate hypocapnia (less than 10 Torr PCO2) is frequency and shape affected: as PCO2 approaches zero, frequency slows and bursts transition from incrementing to gasp-like. These data suggest that: (a) the peripheral chemoreceptors are sufficient to drive breathing through an excitatory pathway onto the respiratory rhythm generator that does not require central chemosensitivity; and (b) stimulating the carotid body may provide a therapeutic avenue to stave off apnoea caused by central hypocapnia. In addition, we demonstrate that specific carotid body hypercapnia is sufficient to overcome central apnoea, complicating interpretation of several recent studies suggesting that rodent carotid bodies do not contribute to CO2 chemosensitivity (Mouradian et al. 2012).
Our data are corroborated by data from an anaesthetized cat model in which the brainstem was perfused independently of systemic circulation, albeit with intact vagal input from the lungs and the cerebrum present (Berkenbosch et al. 1984). Those data also suggest the resilience of the respiratory rhythm generator to severe central hypocapnia when peripheral chemoreceptors are stimulated. In the cat preparation, extreme hypocapnia may have caused changes in cerebral circulation resulting in compromised independence of brainstem and systemic perfusate; the authors considered it likely that brainstem chemoreceptors had been exposed to the hypercapnic and hypoxic systemic perfusate used to stimulate the peripheral chemoreceptors. In addition, artificial perfusion in the cat model was limited to the brainstem and did not include recently discovered hypothalamic neurons that exhibit an exquisite CO2 sensitivity on par with that of RTN neurons (Williams et al. 2007). The in situ DPP escapes both these confounders as the circle of Willis is sectioned, preventing cross-contamination between peripheral and central perfusate. Given the gross differences between the cat preparation and the DPP (cat versus rat, normothermia versus mild hypothermia, baroreflex and heart intact versus artificial pump and pressure feedback system, blood versus artificial perfusate, vagus intact versus vagotomized, and anaesthetized versus decerebrate), the similar observations give credence to the notion that central chemosensitivity is not absolutely necessary for breathing per se, while peripheral chemoreceptors alone are sufficient to maintain activity of the respiratory rhythm generator, at least under normoxic conditions. This suggestion is further endorsed by the recent findings of Ramanantsoa et al. (2011). They showed that normal, rhythmic breathing can persist in transgenic mice deficient in RTN neurons and effectively lacking respiratory CO2 sensitivity. This rhythm is, however, severely disrupted by ‘physiological denervation’ of the carotid body, whereby hyperoxia essentially silences the peripheral chemoreceptors, resulting in periodic breathing (deep breathing interrupted by prolonged apnoeas). This suggests that although the peripheral chemoreceptors are capable of supporting a normal, rhythmic breathing pattern in the absence of central chemoreceptors, central chemoreceptors are required to support breathing under certain conditions.
Caveats and comparison of results to conscious animals
In situ perfused brainstem preparations are growing in popularity as models for investigating respiratory control because they produce a eupnoeic-like motor pattern similar to that seen in vivo, have open autonomic and respiratory feedback loops, lack confounding influences from higher brain centres and anaesthetics, and allow precise experimental control of arterial gases and pressure (Dutschmann et al. 2000; Wilson et al. 2001). As with any reduced preparation, these advantages have to be carefully weighed against a number of caveats (for an extensive discussion of the merits of the DPP see Day & Wilson (2005, 2007)). In the context of the current study, two key issues are: one, whether brainstem tissue is adequately oxygenated when severely hypocapnic; and two, whether a decerebrate preparation is more or less relevant than data from other preparations.
Although the artificial perfusate supplied to the brainstem of the in situ preparation has a high PO2 (PO2 > 500 Torr), it has a much lower oxygen-carrying capacity than blood. During normocapnia, tissue PO2 measurements suggest that the brainstem is hyperoxic, but with the cerebral vasoconstriction associated with severe hypocapnia, the possibility exists that the tissue becomes hypoxic (Wilson et al. 2001). However, we observed a stable eupnoeic index until the PCO2 in the brainstem perfusate dropped below 10 Torr (see Fig. 4), suggesting that respiratory centres were oxygenated sufficiently so as not to cause rapid-onset, decrementing (‘gasp-like’) phrenic bursts that usually go hand-in-hand with brainstem hypoxia (Fewell, 2005). Some preparations produced gasp-like bursts as perfusate PCO2 approached zero. Thus, when the carotid bodies are stimulated, the demise of respiratory bursts at low PCO2 may be as much a consequence of indirect effects of low CO2 on cerebrovascular resistance resulting in hypoxia, as directly caused by loss of CO2 stimulation of brainstem respiratory chemoreceptors.
A main advantage of the DPP is that it reduces the respiratory chemoreflex control system to its most fundamental components, chiefly brainstem and carotid body, and therefore one might expect insights very different from data derived solely from conscious animals that are influenced by higher command centres that coordinate diverse behaviours. As we have reviewed previously, CO2 chemosensitivity is shifted in the hypocapnic direction in all decerebrate preparations, including the dog, cat and rat (Day & Wilson, 2005, 2007). Currently, the factor(s) that causes this shift is unknown, but we assume it relates to ‘descending wakefulness drive’ and possibly involves orexin neurons that project to the brainstem from the hypothalamus (Williams et al. 2007; Gestreau et al. 2008; Lazarenko et al. 2011; Nattie, 2011). Loss of such an input with decerebration may make the brainstem circuit abnormally sensitive to peripheral chemoreceptor input, explaining the current results. However, the fact that denervation of the carotid body causes pronounced hypoventilation in conscious animals (see Introduction) suggests that the brainstem respiratory control is always heavily dependent on carotid body input even in conscious animals when the carotid bodies and chemosensitivity in general are considered to serve a lesser role. In this respect, we note that at altitude, where the carotid body is stimulated by hypoxia (and CO2 is ‘blown off’), ventilation continues despite often intense hypocapnia. While other factors may be at play in this example, including hypothalamic, descending and adrenal-dependent influences, the most parsimonious explanation is that, in some cases at least, the peripheral chemoreflex helps sustain ventilation during times of diminished central chemoreflex (Duffin, 2010).
Effect of low brainstem PCO2 on respiratory burst frequency and amplitude
With carotid body stimulation, timing variables reflecting the output of the respiratory rhythm generation (i.e. TI, TE, fR) were constant over a large range of brainstem perfusate PCO2: only when PCO2 approached the apnoeic threshold were such variables affected. In contrast, nVT and 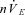 decreased linearly with hypocapnia. Thus, in the vagotomized in situ preparation, when the carotid bodies are stimulated, the carotid bodies are the dominant influence on the respiratory rhythm generator, and the role of brainstem PCO2 is largely restricted to the modulation of downstream mechanisms that determine the amplitude of motor neuron output. If central chemosensitivity is largely mediated by one population of neurons, such as the RTN as some have speculated (Guyenet, 2008; Guyenet et al. 2010), then the central chemoreceptors would not appear to contribute substantially to respiratory rhythm generation. On the other hand, if central chemosensitivity is shared by multiple sites (for example, the RTN, raphe, nucleus tractus solitarius and/or locus coeruleus; Nattie & Li, 2009), then it is possible that endowing the brainstem with excitation from the carotid body saturates the activity of populations that modulate rhythm generation (e.g. RTN), but has only mild effects on the dynamic range of the populations that control burst pattern (amplitude).
decreased linearly with hypocapnia. Thus, in the vagotomized in situ preparation, when the carotid bodies are stimulated, the carotid bodies are the dominant influence on the respiratory rhythm generator, and the role of brainstem PCO2 is largely restricted to the modulation of downstream mechanisms that determine the amplitude of motor neuron output. If central chemosensitivity is largely mediated by one population of neurons, such as the RTN as some have speculated (Guyenet, 2008; Guyenet et al. 2010), then the central chemoreceptors would not appear to contribute substantially to respiratory rhythm generation. On the other hand, if central chemosensitivity is shared by multiple sites (for example, the RTN, raphe, nucleus tractus solitarius and/or locus coeruleus; Nattie & Li, 2009), then it is possible that endowing the brainstem with excitation from the carotid body saturates the activity of populations that modulate rhythm generation (e.g. RTN), but has only mild effects on the dynamic range of the populations that control burst pattern (amplitude).
Stimulating the carotid body as a therapeutic strategy for central sleep apnoea?
During non-REM sleep, animals and humans are critically dependent upon chemoreceptor feedback to protect blood gases (Spengler et al. 2001; Dubreuil et al. 2008). Control theory and experimental data suggest that increases in peripheral and central chemoreflex gain may be primary factors in causing some forms of sleep-disordered breathing, destabilizing breathing by contributing to ventilatory overshoots in response to blood gas perturbations (Longobardo et al. 1982; Cherniack, 1984; Khoo, 2000; Kiwull-Schöne et al. 2008).
Smith and Dempsey have proposed that ventilatory sensitivity to PCO2 below eupnoeic levels may be a key determinant of ventilatory instability during sleep (Dempsey et al. 2004). According to the Smith and Dempsey doctrine, the high gain chemoreflexes associated with sleep apnoea lead to oscillations in PaCO2. PaCO2 perturbations below eupnoeic levels diminish carotid body activity, decreasing 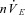 , and if PaCO2 is reduced far enough, apnoea results (i.e. Dempsey et al. 2004; Dempsey, 2005). In support of their doctrine, Smith and Dempsey used an extracorporally perfused carotid body conscious dog model to demonstrate that the withdrawal of capnic stimuli from the carotid body during hyperpnoea likely plays a critical role in causing apnoeas, leading to further respiratory oscillations (Nakayama et al. 2003; Smith et al. 2003). If the sensitivity of the carotid bodies is too high, such that the withdrawal of capnic stimuli shuts off carotid body excitation too readily causing apnoea, we suggest mild tonic, pharmaceutical stimulation of the carotid body may be all that is required to stave off apnoeas and thereby improve ventilatory stability. To illustrate this, we showed that a bolus of the stress neuropeptide PACAP, when applied through the common carotid artery, is capable of overcoming central apnoeas in the DPP. Consistent with previous studies demonstrating that arterially perfused PACAP strongly excites the carotid body (Ishizuka et al. 1992; Runcie et al. 1995), we demonstrate that the effect of PACAP on overcoming central apnoeas was dependent on intact carotid body afferents; that some activity persisted after transecting the CSN suggests that PACAP excites peripheral targets outside the carotid sinus, the location of which is currently under investigation.
, and if PaCO2 is reduced far enough, apnoea results (i.e. Dempsey et al. 2004; Dempsey, 2005). In support of their doctrine, Smith and Dempsey used an extracorporally perfused carotid body conscious dog model to demonstrate that the withdrawal of capnic stimuli from the carotid body during hyperpnoea likely plays a critical role in causing apnoeas, leading to further respiratory oscillations (Nakayama et al. 2003; Smith et al. 2003). If the sensitivity of the carotid bodies is too high, such that the withdrawal of capnic stimuli shuts off carotid body excitation too readily causing apnoea, we suggest mild tonic, pharmaceutical stimulation of the carotid body may be all that is required to stave off apnoeas and thereby improve ventilatory stability. To illustrate this, we showed that a bolus of the stress neuropeptide PACAP, when applied through the common carotid artery, is capable of overcoming central apnoeas in the DPP. Consistent with previous studies demonstrating that arterially perfused PACAP strongly excites the carotid body (Ishizuka et al. 1992; Runcie et al. 1995), we demonstrate that the effect of PACAP on overcoming central apnoeas was dependent on intact carotid body afferents; that some activity persisted after transecting the CSN suggests that PACAP excites peripheral targets outside the carotid sinus, the location of which is currently under investigation.
Normally, tonic stimulation of the carotid bodies results in hyperventilation; the respiratory system blows off CO2 leading to arterial hypocapnia (Kumar, 2009). If carotid body stimulation has no effect on the central apnoeic threshold, then one would expect that apnoeas would be more likely, not less. However, our results suggest carotid body stimulation reduces the central apnoeic threshold substantially, making apnoeas less likely. Specifically, in the current study using the DPP, with the peripheral chemoreceptors stimulated with hypoxic-hypercapnia, two of nine preparations had no central apnoeic threshold and, in the remainder, the central apnoeic threshold was 3.6 ± 1.4 Torr PCO2. We also showed that this could be raised, in a linear fashion, up to 16.7 ± 0.9 Torr PCO2 by attenuating carotid body output (by increasing PO2 and decreasing PCO2), confirming previous work from our lab (Day & Wilson, 2005). Thus, stimulating the carotid bodies caused a potentially protective hypocapnic shift in the apnoeic threshold of ∼13 Torr PCO2.
In humans with central sleep apnoea, drugs that stimulate respiration tend to stabilize breathing, but this is not the case in patients with obstructive sleep apnoea (OSA). While the carbonic anhydrase inhibitor acetazolamide has a substantial effect on the apnoea/hypopnoea index, most likely through acidification of the blood by action at the kidney (White et al. 1982; Javaheri, 2006) or reductions in the sensitivity of the ventilatory control system (Edwards et al. 2012), drugs that target neuronal components of respiration control have only mild effects on breathing stability in patients with OSA (Saboisky et al. 2009; Kohler & Stradling, 2011). For example, while theophylline stabilizes breathing in central sleep apnoea, in two of three randomized control studies, theophylline is reported to cause only a mild reduction in the apnoea/hypopnoea index in OSA (reviewed in Veasey et al. 2006). Similarly, the effects of naloxone, doxapram and almatrine, all of which are reported to stimulate the carotid body to some degree, have underwhelming effects on the apnoea–hypopnea index in OSA (Krieger et al. 1982; Guilleminault & Hayes, 1983; Atkinson et al. 1985; Suratt et al. 1986; Hackett et al. 1987; Olievier et al. 1987; Yost, 2006). These extensive observations underline the fact that carotid body stimulants are not suitable for the treatment of all types of sleep apnoea, especially when collapse of the airway may be exacerbated by increased inspiratory effort. Therefore, to be effective, pharmaceuticals that target the carotid body will need to be tailored to specific disease phenotypes (White, 2005).
In conclusion, the current data from the decerebrate, vagotomized in situ rat preparation suggest that the peripheral respiratory chemoreceptors are demigod chemoreceptors capable of sustaining the frequency of the respiratory rhythm generator in the absence of vagal feedback or substantive central respiratory chemoreceptor activity. With the respiratory rhythm sustained by input from the peripheral chemoreceptors, the role of the central chemoreceptor is one of determining tidal volume. Given the Smith–Dempsey doctrine that predicts removal of capnic stimuli from the carotid body is the primary cause of apnoea, and assuming the demonstration herein that carotid body stimulation is capable of shifting the apnoeic threshold to very low levels of PCO2 is reflective of the role of the carotid body in intact sleeping humans, we surmise that pharmacological treatments that selectively stimulate the carotid bodies may be an effective treatment for some forms of sleep apnoea.
Acknowledgments
This study was funded by the Canadian Institutes of Health Research. Salary support for R.J.A.W. was provided by Alberta Innovates-Health Solutions (AIHS). M.-N.F. was supported in part by a grant ‘Legs Poix’ of the Chancellerie des Universités de Paris, France, and by a grant from the Bourse de voyage de la Société de Pneumologie de Langue Française (Boehringer-Ingelheim). I.Z. was funded by the AIHS (both HYRS program and summer studentship). The authors thank Sherry Moore and Dr Trevor Day for technical assistance.
Glossary
- CSN
carotid sinus nerve
- DPP
dual-perfused preparation
- OSA
obstructive sleep apnoea
- PACAP
pituitary adenylate cyclase-activating peptide
- REM
rapid eye movement
- RTN
retrotrapezoid nucleus
Author contributions
This study was performed at the University of Calgary. All authors contributed to this study: M.-N.F., I.Z. and R.J.A.W. developed the concept, performed initial experiments, analysed data and wrote the first draft of the manuscript; E.T.O.C., A.R. and R.J.A.W. designed substantive additional experiments; these additional experiments were performed and analysed by E.T.O.C. and A.R.; E.T.O.C. and R.J.A.W. wrote the final draft of the manuscript. All authors approved the final version to be published.
References
- Atkinson RL, Suratt PM, Wilhoit SC, Recant L. Naloxone improves sleep apnea in obese humans. Int J Obes. 1985;9:233–239. [PubMed] [Google Scholar]
- Berkenbosch A, van Beek JH, Olievier CN, De Goede J, Quanjer PH. Central respiratory CO2 sensitivity at extreme hypocapnia. Respir Physiol. 1984;55:95–102. doi: 10.1016/0034-5687(84)90119-1. [DOI] [PubMed] [Google Scholar]
- Bisgard GE, Forster HV, Orr JA, Buss DD, Rawlings CA, Rasmussen B. Hypoventilation in ponies after carotid body denervation. J Appl Physiol. 1976;40:184–190. doi: 10.1152/jappl.1976.40.2.184. [DOI] [PubMed] [Google Scholar]
- Boden AG, Harris MC, Parkes MJ. Apneic threshold for CO2 in the anesthetized rat: fundamental properties under steady-state conditions. J Appl Physiol. 1998;85:898–907. doi: 10.1152/jappl.1998.85.3.898. [DOI] [PubMed] [Google Scholar]
- Carroll JL, Canet E, Bureau MA. Dynamic ventilatory responses to CO2 in the awake lamb: role of the carotid chemoreceptors. J Appl Physiol. 1991;71:2198–2205. doi: 10.1152/jappl.1991.71.6.2198. [DOI] [PubMed] [Google Scholar]
- Cherniack NS. Sleep apnea and its causes. J Clin Invest. 1984;73:1501–1506. doi: 10.1172/JCI111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran AE, Hodges MR, Wu Y, Wang W, Wylie CJ, Deneris ES, Richerson GB. Medullary serotonin neurons and central CO2 chemoreception. Resp Physiol Neurobiol. 2009;168:49–58. doi: 10.1016/j.resp.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté A, Porras H, Meehan B. Age-dependent vulnerability to carotid chemodenervation in piglets. J Appl Physiol. 1996;80:323–331. doi: 10.1152/jappl.1996.80.1.323. [DOI] [PubMed] [Google Scholar]
- Cummings KJ, Wilson RJA. Time-dependent modulation of carotid body afferent activity during and after intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1571–R1580. doi: 10.1152/ajpregu.00788.2004. [DOI] [PubMed] [Google Scholar]
- Cunningham DJ. Studies on arterial chemoreceptors in man. J Physiol. 1987;384:1–26. doi: 10.1113/jphysiol.1987.sp016440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva GSF, Giusti H, Benedetti M, Dias MB, Gargaglioni LH, Branco LGS, Glass ML. Serotonergic neurons in the nucleus raphe obscurus contribute to interaction between central and peripheral ventilatory responses to hypercapnia. Pflugers Arch. 2011;462:407–418. doi: 10.1007/s00424-011-0990-x. [DOI] [PubMed] [Google Scholar]
- Daristotle L, Berssenbrugge AD, Engwall MJ, Bisgard GE. The effects of carotid body hypocapnia on ventilation in goats. Respir Physiol. 1990;79:123–135. doi: 10.1016/0034-5687(90)90012-n. [DOI] [PubMed] [Google Scholar]
- Day TA, Wilson RJA. Specific carotid body chemostimulation is sufficient to elicit phrenic poststimulus frequency decline in a novel in situ dual-perfused rat preparation. Am J Physiol Regul Integr Comp Physiol. 2005;289:R532–R544. doi: 10.1152/ajpregu.00812.2004. [DOI] [PubMed] [Google Scholar]
- Day TA, Wilson RJA. Brainstem PCO2 modulates phrenic responses to specific carotid body hypoxia in an in situ dual perfused rat preparation. J Physiol. 2007;578(Pt 3):843–857. doi: 10.1113/jphysiol.2006.119594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TA, Wilson RJA. A negative interaction between brainstem and peripheral respiratory chemoreceptors modulates peripheral chemoreflex magnitude. J Physiol. 2009;587(Pt 4):883–896. doi: 10.1113/jphysiol.2008.160689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H, Skatrud JB. The ventilatory responsiveness to CO(2) below eupnoea as a determinant of ventilatory stability in sleep. J Physiol. 2004;560(Pt 1):1–11. doi: 10.1113/jphysiol.2004.072371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey J, Skatrud J. A sleep-induced apneic threshold and its consequences. Am Rev Respir Dis. 1986;133:1163–1170. doi: 10.1164/arrd.1986.133.6.1163. [DOI] [PubMed] [Google Scholar]
- Dubreuil V, Ramanantsoa N, Trochet D, Vaubourg V, Amiel J, Gallego J, Brunet JF, Goridis C. A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnea, and specific loss of parafacial neurons. Proc Natl Acad Sci U S A. 2008;105:1067–1072. doi: 10.1073/pnas.0709115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin J. The role of the central chemoreceptors: a modeling perspective. Resp Physiol Neurobiol. 2010;173:230–243. doi: 10.1016/j.resp.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Dutschmann M, Wilson RJ, Paton JF. Respiratory activity in neonatal rats. Auton Neurosci: Basic. 2000;84:19–29. doi: 10.1016/S1566-0702(00)00177-6. [DOI] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, Malhotra A, Wellman A. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590(Pt 5):1199–211. doi: 10.1113/jphysiol.2011.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham MMJ, Li Q, Goodchild AK, Pilowsky PM. PACAP is expressed in sympathoexcitatory bulbospinal C1 neurons of the brain stem and increases sympathetic nerve activity in vivo. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1304–R1311. doi: 10.1152/ajpregu.00753.2007. [DOI] [PubMed] [Google Scholar]
- Fewell JE. Protective responses of the newborn to hypoxia. Resp Physiol Neurobiol. 2005;149:243–255. doi: 10.1016/j.resp.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Forster HV, Martino P, Hodges M, Krause K, Bonis J, Davis S, Pan L. The carotid chemoreceptors are a major determinant of ventilatory CO2 sensitivity and of PaCO2 during eupneic breathing. Adv Exp Med Biol. 2008;605:322–326. doi: 10.1007/978-0-387-73693-8_56. [DOI] [PubMed] [Google Scholar]
- Gargaglioni LH, Hartzler LK, Putnam RW. The locus coeruleus and central chemosensitivity. Resp Physiol Neurobiol. 2010;173:264–273. doi: 10.1016/j.resp.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Bévengut M, Dutschmann M. The dual role of the orexin/hypocretin system in modulating wakefulness and respiratory drive. Curr Opin Pulm Med. 2008;14:512–518. doi: 10.1097/MCP.0b013e32831311d3. [DOI] [PubMed] [Google Scholar]
- Guilleminault C, Hayes B. Naloxone, theophylline, bromocriptine, and obstructive sleep apnea. Negative results. Bull Eur Physiopathol Respir. 1983;19:632–634. [PubMed] [Google Scholar]
- Guyenet PG. The 2008 Carl Ludwig Lecture: retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol. 2008;105:404–416. doi: 10.1152/japplphysiol.90452.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bayliss DA. Central respiratory chemoreception. J Comp Neurol. 2010;518:3883–3906. doi: 10.1002/cne.22435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett PH, Roach RC, Harrison GL, Schoene RB, Mills WJ., Jr Respiratory stimulants and sleep periodic breathing at high altitude. Almitrine versus acetazolamide. Am Rev Respir Dis. 1987;135:896–898. doi: 10.1164/arrd.1987.135.4.896. [DOI] [PubMed] [Google Scholar]
- Inglott MA, Farnham MMJ, Pilowsky PM. Intrathecal PACAP-38 causes prolonged widespread sympathoexcitation via a spinally mediated mechanism and increases in basal metabolic rate in anesthetized rat. Am J Physiol Heart Circ Physiol. 2011;300:H2300–H2307. doi: 10.1152/ajpheart.01052.2010. [DOI] [PubMed] [Google Scholar]
- Ishizuka Y, Kashimoto K, Mochizuki T, Sato K, Ohshima K, Yanaihara N. Cardiovascular and respiratory actions of pituitary adenylate cyclase-activating polypeptides. Regul Pept. 1992;40:29–39. doi: 10.1016/0167-0115(92)90081-5. [DOI] [PubMed] [Google Scholar]
- Iturriaga R. Carotid body chemoreception: the importance of CO2-HCO3- and carbonic anhydrase (review) Biol Res. 1993;26:319–329. [PubMed] [Google Scholar]
- Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind, prospective study. Am J Respir Crit Care Med. 2006;173:234–237. doi: 10.1164/rccm.200507-1035OC. [DOI] [PubMed] [Google Scholar]
- Khoo MC. Determinants of ventilatory instability and variability. Respir Physiol. 2000;122:167–182. doi: 10.1016/s0034-5687(00)00157-2. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Filmyer WG, Mitchell GS, Milsom WK. Vagal input enhances responsiveness of respiratory discharge to central changes in pH/CO2 in bullfrogs. J Appl Physiol. 1994;77:2048–2051. doi: 10.1152/jappl.1994.77.4.2048. [DOI] [PubMed] [Google Scholar]
- Kiwull-Schöne H, Teppema L, Wiemann M, Kalhoff H, Kiwull P. Pharmacological impact on loop gain properties to prevent irregular breathing. J Physiol Pharmacol. 2008;59:37–45. [PubMed] [Google Scholar]
- Kohler M, Stradling JR. Pitfalls of clinical trials on pharmacological treatment for obstructive sleep apnoea: future directions. Expert Opin Invest Drugs. 2011;20:1033–1037. doi: 10.1517/13543784.2011.590473. [DOI] [PubMed] [Google Scholar]
- Krieger J, Mangin P, Kurtz D. Almitrine and sleep apnoea. Lancet. 1982;320:210. doi: 10.1016/s0140-6736(82)91050-9. [DOI] [PubMed] [Google Scholar]
- Kumar P. Systemic effects resulting from carotid body stimulation-invited article. Adv Exp Med Biol. 2009;648:223–233. doi: 10.1007/978-90-481-2259-2_26. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Stornetta RL, Bayliss DA, Guyenet PG. Orexin A activates retrotrapezoid neurons in mice. Resp Physiol Neurobiol. 2011;175:283–287. doi: 10.1016/j.resp.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton RA, Band DM, McLoughlin P. Adaptation of carotid chemoreceptors to step increases in PaCO2 in anesthetized cats. J Neurophysiol. 1995;74:2707–2712. doi: 10.1152/jn.1995.74.6.2707. [DOI] [PubMed] [Google Scholar]
- Longobardo GS, Gothe B, Goldman MD, Cherniack NS. Sleep apnea considered as a control system instability. Respir Physiol. 1982;50:311–333. doi: 10.1016/0034-5687(82)90026-3. [DOI] [PubMed] [Google Scholar]
- Lowry TF, Forster HV, Pan LG, Serra A, Wenninger J, Nash R, Sheridan D, Franciosi RA. Effects on breathing of carotid body denervation in neonatal piglets. J Appl Physiol. 1999;87:2128–2135. doi: 10.1152/jappl.1999.87.6.2128. [DOI] [PubMed] [Google Scholar]
- Lugliani R, Whipp BJ, Seard C, Wasserman K. Effect of bilateral carotid-body resection on ventilatory control at rest and during exercise in man. N Engl J Med. 1971;285:1105–1111. doi: 10.1056/NEJM197111112852002. [DOI] [PubMed] [Google Scholar]
- Martino PF, Davis S, Opansky C, Krause K, Bonis JM, Pan LG, Qian B, Forster HV. The cerebellar fastigial nucleus contributes to CO2-H+ ventilatory sensitivity in awake goats. Resp Physiol Neurobiol. 2007;157:242–251. doi: 10.1016/j.resp.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouradian GC, Forster HV, Hodges MR. Acute and chronic effects of carotid body denervation (CBD) on ventilation and chemoreflexes in three rat strains. J Physiol. 2012;590:3335–3347. doi: 10.1113/jphysiol.2012.234658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Carotid body denervation eliminates apnea in response to transient hypocapnia. J Appl Physiol. 2003;94:155–164. doi: 10.1152/japplphysiol.00722.2002. [DOI] [PubMed] [Google Scholar]
- Nattie E. Julius H. Comroe, Jr, Distinguished Lecture: central chemoreception: then … and now. J Appl Physiol. 2011;110:1–8. doi: 10.1152/japplphysiol.01061.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E, Li A. Central chemoreception is a complex system function that involves multiple brain stem sites. J Appl Physiol. 2009;106:1464–1466. doi: 10.1152/japplphysiol.00112.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Regan RG, Majcherczyk S. Role of peripheral chemoreceptors and central chemosensitivity in the regulation of respiration and circulation. J Exp Biol. 1982;100:23–40. doi: 10.1242/jeb.100.1.23. [DOI] [PubMed] [Google Scholar]
- Olievier CN, Berkenbosch A, Degoede J, Kruyt EW. Almitrine bismesylate and the central and peripheral ventilatory response to CO2. J Appl Physiol. 1987;63:66–74. doi: 10.1152/jappl.1987.63.1.66. [DOI] [PubMed] [Google Scholar]
- Olson EB, Jr, Vidruk EH, Dempsey JA. Carotid body excision significantly changes ventilatory control in awake rats. J Appl Physiol. 1988;64:666–671. doi: 10.1152/jappl.1988.64.2.666. [DOI] [PubMed] [Google Scholar]
- Pan LG, Forster HV, Martino P, Strecker PJ, Beales J, Serra A, Lowry TF, Forster MM, Forster AL. Important role of carotid afferents in control of breathing. J Appl Physiol. 1998;85:1299–1306. doi: 10.1152/jappl.1998.85.4.1299. [DOI] [PubMed] [Google Scholar]
- Paton JF. A working heart-brainstem preparation of the mouse. J Neurosci Meth. 1996;65:63–68. doi: 10.1016/0165-0270(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Peña F. PACAP modulates the respiratory rhythm generated in the brainstem slice preparation. Adv Exp Med Biol. 2010;669:119–122. doi: 10.1007/978-1-4419-5692-7_24. [DOI] [PubMed] [Google Scholar]
- Praud JP, Canet E, Bureau MA. Chemoreceptor and vagal influences on thyroarytenoid muscle activity in awake lambs during hypoxia. J Appl Physiol. 1992;72:962–969. doi: 10.1152/jappl.1992.72.3.962. [DOI] [PubMed] [Google Scholar]
- Ramanantsoa N, Hirsch MR, Thoby-Brisson M, Dubreuil V, Bouvier J, Ruffault PL, Matrot B, Fortin G, Brunet JF, Gallego J, Goridis C. Breathing without CO2 chemosensitivity in conditional Phox2b mutants. J Neurosci. 2011;31:12880–12888. doi: 10.1523/JNEUROSCI.1721-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runcie MJ, Ulman LG, Potter EK. Effects of pituitary adenylate cyclase-activating polypeptide on cardiovascular and respiratory responses in anaesthetised dogs. Regul Pept. 1995;60:193–200. doi: 10.1016/0167-0115(95)00131-x. [DOI] [PubMed] [Google Scholar]
- Saboisky JP, Chamberlin NL, Malhotra A. Potential therapeutic targets in obstructive sleep apnoea. Expert Opin Ther Targets. 2009;13:795–809. doi: 10.1517/14728220903005608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Nakayama H, Dempsey JA. The essential role of carotid body chemoreceptors in sleep apnea. Can J Physiol Pharmacol. 2003;81:774–779. doi: 10.1139/y03-056. [DOI] [PubMed] [Google Scholar]
- Spengler CM, Gozal D, Shea SA. Chemoreceptive mechanisms elucidated by studies of congenital central hypoventilation syndrome. Respir Physiol. 2001;129:247–255. doi: 10.1016/s0034-5687(01)00294-8. [DOI] [PubMed] [Google Scholar]
- Spyer KM, Gourine AV. Chemosensory pathways in the brainstem controlling cardiorespiratory activity. Phil Trans R Soc B. 2009;364:2603–2610. doi: 10.1098/rstb.2009.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suratt PM, Wilhoit SC, Brown ED, Findley LJ. Effect of doxapram on obstructive sleep apnea. Bull Eur Physiopathol Respir. 1986;22:127–131. [PubMed] [Google Scholar]
- Timmers HJLM, Wieling W, Karemaker JM, Lenders JWM. Denervation of carotid baro- and chemoreceptors in humans. J Physiol. 2003;553(Pt 1):3–11. doi: 10.1113/jphysiol.2003.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veasey SC, Guilleminault C, Strohl KP, Sanders MH, Ballard RD, Magalang UJ. Medical therapy for obstructive sleep apnea: a review by the Medical Therapy for Obstructive Sleep Apnea Task Force of the Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep. 2006;29:1036–1044. doi: 10.1093/sleep/29.8.1036. [DOI] [PubMed] [Google Scholar]
- Wade JG, Larson CP, Jr, Hickey RF, Ehrenfeld WK, Severinghaus JW. Effect of carotid endarterectomy on carotid chemoreceptor and baroreceptor function in man. N Engl J Med. 1970;282:823–829. doi: 10.1056/NEJM197004092821501. [DOI] [PubMed] [Google Scholar]
- White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370. doi: 10.1164/rccm.200412-1631SO. [DOI] [PubMed] [Google Scholar]
- White DP, Zwillich CW, Pickett CK, Douglas NJ, Findley LJ, Weil JV. Central sleep apnea. Improvement with acetazolamide therapy. Arch Intern Med. 1982;142:1816–1819. [PubMed] [Google Scholar]
- Williams RH, Jensen LT, Verkhratsky A, Fugger L, Burdakov D. Control of hypothalamic orexin neurons by acid and CO2. Proc Natl Acad Sci U S A. 2007;104:10685–10690. doi: 10.1073/pnas.0702676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RJ, Remmers JE, Paton JF. Brain stem PO(2) and pH of the working heart-brain stem preparation during vascular perfusion with aqueous medium. Am J Physiol Regul Integr Comp Physiol. 2001;281:R528–R538. doi: 10.1152/ajpregu.2001.281.2.R528. [DOI] [PubMed] [Google Scholar]
- Yost CS. A new look at the respiratory stimulant doxapram. CNS Drug Rev. 2006;12:236–249. doi: 10.1111/j.1527-3458.2006.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]