

Abstract
Perivagal application of capsaicin (1% solution) is considered to cause a selective degeneration of vagal afferent C fibres and has been used extensively to examine the site of action of many gastrointestinal (GI) neuropeptides. The actions of both capsaicin and GI neuropeptides may not be restricted to vagal afferent fibres, however, as other non-sensory neurones have displayed sensitivity to capsaicin and brainstem microinjections of these neuropeptides induce GI effects similar to those obtained upon systemic application. The aim of the present study was to test the hypothesis that perivagal capsaicin induces degeneration of vagal efferents controlling GI functions. Experiments were conducted 7–14 days after 30 min unilateral perivagal application of 0.1–1% capsaicin. Immunohistochemical analyses demonstrated that, as following vagotomy, capsaicin induced dendritic degeneration, decreased choline acetyltransferase but increased nitric oxide synthase immunoreactivity in rat dorsal motor nucleus of the vagus (DMV) neurones. Electrophysiological recordings showed a decreased DMV input resistance and excitability due, in part, to the expression of a large conductance calcium-dependent potassium current and the opening of a transient outward potassium window current at resting potential. Furthermore, the number of DMV neurones excited by thyrotrophin-releasing hormone and the gastric motility response to DMV microinjections of TRH were decreased significantly. Our data indicate that perivagal application of capsaicin induced DMV neuronal degeneration and decreased vagal motor responses. Treatment with perivagal capsaicin cannot therefore be considered selective for vagal afferent C fibres and, consequently, care is needed when using perivagal capsaicin to assess the mechanism of action of GI neuropeptides.
Key points
Perivagal application of capsaicin (1% solution) is considered to cause selective degeneration of vagal afferent (sensory) C fibres and has been used extensively to examine the site of action of many gastrointestinal (GI) neuropeptides.
The actions of both capsaicin and GI neuropeptides may not be restricted to vagal afferent fibres, however, as other non-sensory neurones displayed sensitivity to capsaicin and brainstem microinjections of these neuropeptides induce GI effects similar to those obtained upon systemic application.
The present study used immunohistochemical, biophysical and functional approaches to test the hypothesis that perivagal capsaicin induces degeneration of vagal efferents controlling GI functions.
Our data indicate that perivagal application of capsaicin induces degeneration of vagal efferent motoneurones and decreased vagal motor responses. Treatment with perivagal capsaicin cannot therefore be considered selective for vagal afferent C fibres and, consequently, care is needed when using perivagal capsaicin to assess the mechanism of action of GI neuropeptides.
Introduction
Capsaicin (CAP), applied systemically or perivagally, has been used for many years to induce a supposedly selective degeneration of sensory neurones and fibres, including vagal afferents, via actions at TRPV1 receptors (Buck & Burks, 1986; Czaja et al. 2008; Owyang & Heldsinger, 2011). In particular, perivagal application of CAP is considered to cause selective degeneration of vagal afferent C fibres and has been used extensively to examine the site of action of many gastrointestinal (GI) neuropeptides. Using this approach, the ability of several GI neuropeptides and neurohormones to exert vagally mediated effects upon GI functions via paracrine actions on vagal afferent fibres has been explored, namely, loss of effect following CAP administration was indicative of a vagal afferent site and mechanism of action (Holzer, 1986; Raybould & Tache, 1988; South & Ritter, 1988; Lloyd et al. 1993; Holzer et al. 1994; Zittel et al. 1994; Li et al. 1997; Blackshaw et al. 2000; Zafra et al. 2003; Moran, 2006).
One must keep in mind, however, that even a localized, brief perineural application involves using very high concentrations of CAP (usually 1% solution, i.e. ∼33 mm) for 30 min) raising the possibility that the observed effects are due at least in part to non-selective actions on ion channels or receptors other than CAP-sensitive TRPV1 receptors. In fact, it has been shown that systemic administration of CAP induces degeneration of central nervous system neurones and fibres that do not express TRPV1 receptors and even involves neuronal areas that do not receive sensory inputs from the periphery (Holzer, 1991, 1998; Ritter & Dinh, 1992). These effects occur despite the transient nature of CAP effects on ionic conductances (Bevan & Szolcsanyi, 1990; Holzer, 1991; Evans et al. 2003; Jin et al. 2004; Peters et al. 2010; Zsombok et al. 2011). Moreover, even if perivagal CAP abolishes the actions of GI neuropeptides, without definite proof that vagal motoneurones and efferent fibres are unaffected by perivagal CAP, the actions of GI neuropeptides cannot be ascribed exclusively to vagal afferent fibres.
In addition, anatomical evidence argues against the conclusions of the studies that used perivagal CAP to investigate the physiological effects of GI neuropeptides on GI and feeding-related circuits. If the physiological actions of GI neuropeptides were due exclusively to paracrine effects at the level of vagal afferent fibres (which are located between the longitudinal and circular intestinal musculature and within the intestinal mucosa), then the observed effects obtained upon systemic administration of these GI neuropeptides falls short of explaining their mechanism of action. In fact, systemically administered GI neuropeptides are unlikely to cross the lamina propria mucosae to activate vagal afferent terminals at concentrations sufficient to mimic their physiological effects, implying non-paracrine actions (see, for example, Li & Owyang, 1993). Furthermore, one has to consider that the nucleus tractus solitarii (NTS) and dorsal motor nucleus of the vagus (DMV) are circumventricular organs with a leaky blood–brain barrier (BBB), fenestrated capillaries and enlarged perivascular space that allows the passage of large molecules, especially given a recent report of specialized neurones lining the ependymal layer of the central canal and fourth ventricle (Gross et al. 1990; Kastin et al. 2002; Cottrell & Ferguson, 2004; Orts-Del’immagine et al. 2012), raising the possibility that circulating neuropeptides might also reach these neuronal circuits. Additionally, the area postrema lies entirely outside the BBB and has a series of short, communicating vessels that potentially send postremal venous drainage to NTS and DMV (Roth & Yamamoto, 1968). These morphological characteristics raise the possibility that NTS and DMV neuronal activity can be modulated by circulating molecules. This possibility is strengthened by functional evidence demonstrating that GLP-1 and cholecystokinin (CCK), for example, can cross the BBB to activate NTS and DMV neurones directly as well as functional studies demonstrating that, when microinjected into the dorsal vagal complex (DVC; i.e. NTS, DMV and area postrema), GLP-1 and CCK can induce short-term satiety, modulate pancreatic secretions and gastric motility (Hommer, 1985; Blevins, 2000; Zheng et al. 2005; Viard et al. 2007; Holmes et al. 2009a, b; Zhang, 2012).
It is very likely therefore, that the actions of GI neuropeptides are not limited to a paracrine effect on peripheral vagal afferent fibres, but other sites of action, including the second order neurones of the NTS and/or the neurones of the DMV, should be taken into account. In fact, previous studies investigating the effects of CAP on vagally mediated gastric functions have suggested that the efferent vagus nerve may also be damaged by such treatment. Following perivagal CAP, for example, the ability of intracisternal thyrotrophin-releasing hormone (TRH) to stimulate gastric acid secretion and blood flow was reduced significantly (Raybould et al. 1990), while systemic (subcutaneous) CAP reduced the ability of 2-deoxyglucose to increase gastric acid secretion (Evangelista et al. 1989). In brief, major drawbacks exist in the exclusive use of CAP in determining the site and mechanism of action of GI neuropeptides, as the available evidence that perivagal CAP abolishes the effects of systemic application of GI neuropeptides is not sufficient to rule out the possibility that CAP has other actions.
The reader, however, should keep in mind that we do not intend to dispute the powerful and well documented paracrine effects of many GI neuropeptides on peripheral vagal afferent fibres. We prefer, instead, to suggest that perivagal CAP also affects efferent vagal fibres, an action that would impair the identification of alternative sites of action of GI neuropeptides, such as NTS and DMV neurones. The aim of this study was to use immunohistochemical, biophysical and functional approaches to test the hypothesis that perivagal CAP impairs the activity of vagal efferent neurones controlling GI functions.
Methods
Ethical approval
All experiments were carried out under a protocol approved by Penn State University Institutional Animal Care. Sprague–Dawley rats (120–250 g) of either sex were used.
Retrograde labelling
To identify DMV neurones innervating the stomach, injections of the retrograde neuronal tracer cholera toxin B subunit (CTB) were made into the muscular layers of the gastric corpus of rats. Briefly, rats were anaesthetized with isoflurane (2–3%, 600 ml min−1) and, once a deep level of anaesthesia was obtained (abolition of the footpinch withdrawal reflex), an abdominal laparotomy was performed to expose the stomach. Glass micropipettes were used to inject CTB (0.5% solution, 5 μl total volume) in three to five sites along the major curvature of the gastric corpus. The incision was closed in layers using 6/0 vicryl suture and staples and the rats allowed to recover for up to 14 days.
Vagal nerve damage
Rats were divided in three groups: (1) one group received a monolateral subnodosal cervical vagotomy (Ce VgX group); (2) the second group had either their posterior or anterior subdiaphragmatic vagus nerve sectioned (VB VgX group); and (3) rats in the third group were treated with CAP applied unilaterally to the cervical vagus (perivagal CAP group) as described previously (Raybould & Tache, 1988; Li & Owyang, 1993; Baptista et al. 2007). (NB Unilateral, rather than bilateral, CAP application as described in the latter two references was used in the present study to allow direct comparison between control and CAP treated DMV neurones within any one brainstem slice.) We have demonstrated previously that this protocol induces damage to the afferent vagus nerve (Baptista et al. 2007). Briefly, rats were anaesthetized with a mixture of ketamine/xyalzine/acepromazine (80/1.6/5 mg ml−1 kg−1 i.p.). Once a deep level of anaesthesia was obtained, a ventral midline incision on the neck exposed approximately 4 mm of the left cervical vagal trunk, which was either sectioned (Ce VgX) or isolated over a strip of Parafilm® before a cotton pellet soaked in 1% CAP solution (8:1:1 saline/DMSO/ethanol) was applied to the nerve for 30 min; the Parafilm® was removed and the area was dried (perivagal CAP group). The contralateral vagus was isolated and treated similarly with a cotton pellet soaked with vehicle solution only. The incision was closed in layers using 6/0 vicryl and the rats allowed to recover for 7–14 days. In rats that underwent posterior or anterior subdiaphragmatic vagus nerve section (VB VgX group), an abdominal laparotomy was used to expose the subdiaphragmatic vagus nerves along the oesophagus. Under microscopic guidance, the anterior or posterior gastric branches were identified and sectioned. The incision was closed in layers as above. Further groups of naïve rats, those that were not injected with CTB, received a monolateral application of perivagal CAP and were used for electrophysiological recording or in vivo measurement of gastric motility (see below).
Unilateral vagal deafferentation
To assess whether the perivagal CAP-induced changes in brainstem neuronal properties were due to loss of monosynaptic vagal afferent inputs on to DMV neurones, an additional subgroup of rats (n = 10) received surgical vagal deafferentation 3–7 days before removal of the brainstem for electrophysiological recordings. Surgical deafferentation was achieved as described previously (Baptista et al. 2007). Briefly, rats were anaesthetized as above and, once a deep level of anaesthesia was obtained, were placed in a stereotaxic frame. A dorsolateral incision at the occipital bone followed by blunt dissection was used to expose the area between the occipital bone and right first cervical vertebra. The occipital bone was trimmed with a no. 6 dental drill to expose the vagal afferent rootlets, located ∼1 mm medial to the occipital condyle. Under microscopic guidance, the afferent rootlets were severed using a 27-gauge needle. The cervical incision was closed using 4/0 suture and the rats allowed to recover.
Immunohistochemistry
Immunohistochemical analyses were conducted on rats 7 days following Ce VgX or either VB VgX or 7, 10, or 14 days following perivagal CAP treatments. Rats were anaesthetized (Inactin®, 125–150 mg kg−1 i.p.) before being perfused transcardially with heparinized saline followed by formaldehyde fixative (4% in PBS; see below for composition). Brainstems were removed and post-fixed for 3–4 days at room temperature before being transferred to PBS containing 20% sucrose at 4°C overnight. The entire rostrocaudal extent of the DVC was cut, using a cryostat, into four series of 50 μm transverse sections. After sectioning, one of every four brainstem sections were washed 3 × 20 min in Tris-phosphate-buffered saline (TPBS; see below for composition) containing 0.3% Triton X-100 and then blocked in 10% normal horse serum (NHS) in TPBS-Triton.
Free-floating brainstem slices were incubated with antibodies on a shaking platform at room temperature. After each incubation period, sections were rinsed in TPBS 3 × 20 min. We used goat-α-CTB (1:100,000; List Biologicals, Campbell, CA, USA), rabbit-α-choline acetyltransferase (ChAT, 1:5000; Chemicon, Temecula, CA, USA) or mouse-α-NOS (1:3000; Sigma, Chemical Co. St Louis, MO, USA) primary antibodies and donkey-α-goat, rabbit or mouse (1:500; Jackson ImmunoResearch, West Grove, PA, USA) secondary antibodies. Tissues were incubated in primary antibodies diluted in TPBS containing 10% NHS for 2–3 days and in secondary antibodies diluted in TPBS containing 1% NHS for 24 h and for 4–6 h in ExtrAvidin-HRP. After ExtrAvidin exposure, an imidazole-intensified DAB reaction was used to stain CTB-labelled or nitric oxide synthase (NOS)-immunoreactive (IR) neurones; this reaction gives the neurones a brown reaction product. Two-colour immunoperoxidase labelling was used to localize ChAT (blue–grey reaction product using a Vector Laboratories (Burlingame, CA, USA) SG kit with peroxide generated by glucose oxidase) with CTB-IR (brown reaction product). Immunostained sections were mounted on gelatin-subbed slides, dehydrated through a series of alcohols and xylene before being coverslipped using Cytoseal® (Thermo Fisher Scientific, Waltham, MA, USA). Images were captured with a SPOT RT colour camera mounted on a Nikon E400 microscope and digitally enhanced with Adobe Photoshop or Picasa® software.
NOS-IR neurones were counted every fourth brainstem slice by two independent investigators unaware of the treatment. If the cell count differed by more than 10%, a third investigator then analysed the brainstem slices. The final cell count was the mathematical average of the independent cell counts. To minimize errors in counting, we counted only those neurones in which the nucleus was clearly visible. Despite this precaution, we have to consider cell counts as best proportional estimates only, rather than absolute values, when comparing labelled subpopulations. Cell count values are given as mean +/− s.e.m. neurones/section.
Electrophysiology
Electrophysiological recordings were made from brainstem slices (300 μm thick) prepared as described previously (Browning et al. 1999, 2011; Browning & Travagli, 2007; Holmes et al. 2009a). In brief, rats were anaesthetized with isoflurane, the brainstem was removed and placed in ice-cold, oxygenated Krebs’ solution. Four to six coronal slices spanning the entire rostrocaudal extent of the DVC were cut using a vibratome and incubated in oxygenated Krebs’ solution (35 ± 1°C) for at least 90 min before electrophysiological recording. A single brainstem slice was then placed in a custom-made perfusion chamber (volume 0.5 ml) on the stage of a Nikon E600FN microscope and held in place with a nylon mesh. Slices were perfused continuously with warmed, oxygenated Krebs’ solution and maintained at 32 ± 1°C. Electrophysiological recordings were made under brightfield illumination, using differential interference contrast optics, at a final magnification of ×400.
Whole cell patch clamp recordings were made using micropipettes of resistance 2–4 MΩ when filled with a potassium gluconate solution (see below for composition). A single electrode voltage clamp amplifier (Axoclamp 1D, Molecular Devices, Union City, CA, USA) was used to capture data, which were filtered at 2 kHz, digitized via a Digidata 1320 interface and analysed using pClamp 10 software (Molecular Devices). Recordings were accepted only if the series resistance was <15 MΩ. Recordings were made from neurones located in the medial DMV, which, as demonstrated previously, contains gastric-projecting neurones (Fox & Powley, 1985; Norgren & Smith, 1988; Browning et al. 1999).
Basic membrane properties
The input resistance (Rin) was calculated by measuring the instantaneous current displacement (at the end of the stimulus artifact) obtained by stepping the membrane from −50 mV to −60 mV. The membrane capacitance was calculated by applying repetitive square wave voltage-command pulses (10 mV) to the cell and fitting the resulting transient to Cm = Qt/ΔV; where Cm = membrane capacitance, Qt = total charge under the transient response to the square wave voltage command and ΔV = the change in voltage across the membrane.
Action potential characteristics
To examine action potential and afterhyperpolarization (AHP) properties, depolarizing current pulses (15–30 ms duration) of intensity sufficient to evoke a single action potential at current pulse offset were injected into DMV neurones current clamped at −60 mV. The action potential duration at threshold was measured, as was the amplitude of the AHP. The duration of the AHP (constant of decay, τ) was fitted to a single exponential equation.
To measure the firing frequency of DMV neurones, depolarizing current pulses (400 ms duration) of increasing intensity (30–270 pA) were injected into DMV neurones current clamped at −60 mV. The number of action potentials fired during the depolarizing current pulse was noted and expressed as pulses per second (p.p.s.).
Voltage-dependent potassium currents
All voltage-dependent potassium currents were measured in the presence of tetrodotoxin (0.3–1 μm) to block sodium currents. The inactivation curve (H∞) for the transient outward potassium current, IA, was constructed using hyperpolarizing steps (400 ms duration) from a holding potential of −50 mV in 10 mV decrements to −120 mV and repolarization to −50 mV. The resultant current values were normalized (Imax = 100), averaged and plotted (Fig. 2A).
Figure 2. CAP pretreatment increases NOS but decreases choline acetyltransferase IR in the dorsal vagal complex.
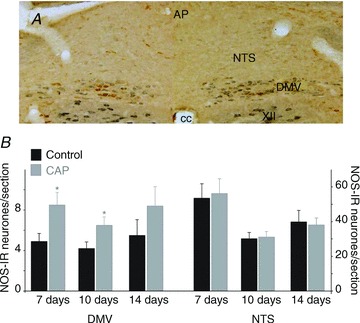
A, NOS (brown) and choline acetyltransferase (blue) expression 7 days after perivagal CAP treatment to the left cervical vagus. The contralateral (right) cervical vagus was treated with vehicle only and is used as control. Note the increased NOS expression and decrease in choline acetyltransferase IR in the DMV area of the CAP treated side. B, graphical representation of the number of NOS-IR neurones per brainstem slice in the DMV (left) and NTS (right) following perivagal CAP (grey bars) as compared to the contralateral control (black) brainstem. Note the increase in NOS-IR in DMV neurones, but not in NTS neurones following perivagal CAP. *P < 0.05. AP, area postrema; CAP, capsaicin; cc, central canal; DMV, dorsal motor nucleus of the vagus; IR, immunoreactivity; NOS, nitric oxide synthase; NTS, nucleus tractus solitarii; XII, hypoglossus nucleus.
The activation curve for IA was constructed by first hyperpolarizing the DMV membrane from a holding potential of −50 mV to −120 mV (400 ms duration) to remove IA inactivation. The membrane was then depolarized (400 ms duration) in steps from +20 mV to −70 mV in 10 mV decrements. To isolate IA from the other currents activated upon membrane depolarization, the brainstem slice was then perfused with 4-aminopyridine (5 mm); the activation protocol was repeated and the resulting current trace subtracted from the initial activation protocol to allow measurement of the 4-aminopyridine-sensitive IA current. Another protocol to isolate IA consisted of digitally subtracting the IKV trace from the current ensemble (see Fig. 2B). There was no difference in the results, which were therefore pooled. These currents were measured, normalized (Imax = 100) averaged and plotted (See Fig. 2C).
The delayed rectifier current, IKV, was assessed by passing depolarizing current pulses (400 ms duration, holding potential = −50 mV) from +20 mV to −70 mV in 10 mV decrements. The resulting current was measured isochronically at the end of the depolarizing current step, averaged and plotted (Fig. 2D).
Morphological properties
To assess the morphological properties of the recorded DMV neurones, Neurobiotin® (2.5%) was included in the recording pipette. At the conclusion of the experiment, Neurobiotin® was injected into the neurone by passing subthreshold direct current pulses (400 ms on, 600 ms off for 20 min) through the recording pipette. Following removal of the pipette, the neuronal membrane was allowed to reseal for 10–20 min before the brainstem slice was fixed in Zamboni's fixative (see below for composition) at 4°C. As described previously (Browning et al. 1999, 2005; Martinez-Pena y Valenzuela et al. 2004), brainstem slices were cleared of fixative by repeated washes in PBS containing Triton X-100 (PBS-TX; see below for composition) before incubation with avidin-D-HRP solution for 2 h. After repeated rinsing in PBS-TX, brainstem slices were incubated in a PBS solution containing DAB, cobalt chloride and nickel sulphate (see below for composition) and reacted in the presence of 3% H2O2 for a time sufficient to allow adequate visualization of the Neurobiotin-filled neurone. Brainstem slices were mounted on gelatin-subbed slices, air dried and dehydrated via a graded series of alcohols and xylene before being mounted in Cytoseal® (Thermo Fisher Scientific).
Three-dimensional reconstructions of individual Neurobiotin-filled neurones were made at a final magnification of ×400 using the Neurolucida® software system (Microbrightfield, Williston, VT, USA) connected to a Nikon E400 microscope. The optical and physical compression of the slice that may have occurred during fixation and processing was corrected by a subroutine of the software, which rescaled the section to 300 μm, the original thickness of the slice at the time of sectioning.
Several morphological features were assessed, including soma size (in both the x- and y-axes) and soma area, form factor (a measure of soma circularity where 1 = perfect circle and 0 = a straight line), dendritic branching (number of segments, branching order). To be accepted, the neuronal reconstructions had to have a mediolateral and rostrocaudal branch extension of at least 200 μm, no major branches would have been severed during the initial sectioning of the brainstem slice and the neuronal soma could not have been damaged during pipette retrieval.
Drug application and statistical analysis
Drugs were applied to DMV neurones via perfusion through a series of manually operated valves, at concentrations demonstrated previously to be effective. TRH was applied for a time sufficient for the response to reach plateau, or a minimum of 3 min if the neurone was unresponsive. Ion channel blockers were applied for at least 5 min. Each neurone served as its own control, i.e. the response of any neurone was assessed before and after drug application using the paired Students’t test. Results are expressed as mean ± s.e.m. with significance defined as P < 0.05.
In vivo studies: surgical preparations and thyrotrophin-releasing hormone applications
Male Sprague–Dawley rats were fasted overnight (water ad libitum) before being anaesthetized with thiobutabarbital (Inactin®; 120–150 mg kg−1 i.p.). An adequate depth of anaesthesia was assessed (absence of foot pinch withdrawal reflex) throughout the experimental period. Body temperature was monitored by a rectal thermometer and maintained at 37 ± 1°C with a heating pad.
Rats were intubated with a tracheal catheter and, following a laparotomy, an encapsulated miniature strain gauge (RB Products, Minneapolis, MN, USA) was sutured to the anterior gastric corpus in alignment with the circular smooth muscle fibres. The strain gauge signal was amplified (QuantaMetrics EXP CLSG-2, Newton, PA, USA), low pass filtered (Fc = 0.5 Hz) and recorded on a polygraph (Grass model 79, Quincy, MA, USA) and on a computer using Axotape® software (Axon Instruments, Union City, CA, USA). Following surgical instrumentation, animals were placed in a stereotaxic frame with the head of the animal slightly tilted (−7 mm) to expose the fourth ventricle upon midline incision and removal of the overlying neck musculature. Following 1 h of stabilization, baseline values of gastric motility were determined as the mean value of the 5 min period immediately preceding drug application.
TRH was dissolved in isotonic PBS and microinjected (100 pmol in 60 nl) at (mm from calamus scriptorius): +0.2–0.3 rostrocaudal, 0.1–0.3 mediolateral and −0.5 dorsoventral.
Signals of baseline motility and tone were acquired for 30 min after drug application. The drug-induced gastric effects were measured as the average of the 30 s period centred around the peak effect. The basal strain gauge output was monitored for any changes for 10 min following drug infusion.
Animals were separated into three groups: (1) control; (2) pretreatment with a perivagal solution of 0.1% CAP; and (3) pretreatment with a perivagal solution of 1% CAP. The perivagal CAP pretreatment occurred 7–8 days before the gastric motility recordings.
In vivo data analysis and statistics
The strain gauges were calibrated with a 1 g weight before and after the experimental procedures. Gastric motility was calculated using the following formula:
Where N equals the number of peaks in a particular force range. Therefore, presuming that a 0 mV signal is indicative of no gastric motility, the grouping of peak to peak sinusoidal signals reflected 25–50 mg, 60–100 mg, 110–200 mg and signals greater than 210 mg for N1 through N4, respectively.
The TRH-induced effects on gastric motility were measured against the averaged value of gastric motility before microinjection. Data were evaluated by comparing the change in response between pre- and post-treatment values within each group by ANOVA or paired t test (SPSS Inc., Chicago, IL, USA). In all instances, significance was set at P < 0.05.
Drugs and solutions
Krebs’ solution (mm): 126 NaCl; 25 NaHCO3; 2.5 KCl; 1.2 MgCl2; 2.4 CaCl2; 1.2 NaH2PO4; and 11 dextrose, maintained at pH 7.4 by bubbling with 95/5% O2/CO2. Potassium gluconate intracellular solution (mm): 128 potassium gluconate; 10 KCl, 0.3 CaCl2; 1 MgCl2; 10 Hepes, 1 EGTA, 2 Na2-ATP; 0.25 Na-GTP, adjusted to pH 7.36 with KOH. PBS-TX (mm): 115 NaCl; 75 Na2HPO4; 7.5 KH2PO4 and 0.15% Triton X-100. Zamboni's fixative (mm): 1.6% paraformaldehyde, 19 KH2PO4 and 100 Na2PO4 in 240 ml saturated picric acid and 1600 ml water, adjusted to pH 7.4 with NaOH. Avidin-D-HRP solution: 0.002% avidin D-HRP in PBS containing 1% Triton X-100. DAB solution:0.05% DAB in PBS supplemented with 0.025% CoCl2 and 0.02% NiNH4SO4.
Tetrodotoxin was purchased from Alomone Labs Ltd, (Jerusalem, Israel), Neurobiotin and avidin D-HRP were purchased from Vector Laboratories; all other chemicals were purchased from Sigma Chemical Co.
Results
Immunohistochemistry
Control CTB-IR neurones in the DMV contralateral to the injured vagus showed a consistent pattern of labelling irrespective of injury type. In the contralateral DMV, dendrites of corpus-projecting neurones were densely packed and retrogradely labelled somata showed homogeneous immunostaining for CTB with clearly defined nuclei that lacked immunoreactivity. In contrast, ipsilateral to a Ce VgX, signs of degeneration and death of vagal motoneurones were present 7 days after injury. At 7 days postsurgical injury, there was a consistent reduction in the number of CTB-IR dendrites ipsilateral to the vagal injury and a few CTB-IR neurones displayed vacuoles and/or dark, shrunken CTB-IR nuclei (pyknosis). The small number of CTB-IR axons that exited the ventral medulla from labelled DMV neurones showed numerous swellings along the length of the vagal bundle.
Seven days after VB VgX, CTB-labelled corpus-projecting DMV neurones also displayed vacuoles, pyknotic nuclei and a decreased number of dendrites although the proportion of CTB-labelled DMV neurones showing damage was lower than in rats with Ce VgX (data not shown).
These results suggest that vagotomy, both cervical as well as subdiaphragmatic, induces damage in DMV neurones, including neurones projecting to the stomach.
CTB-labelled corpus-projecting DMV neurones also showed signs of damage 7 days after perivagal CAP. While retrogradely labelled CTB-IR cell bodies showed no clear signs of degeneration, the density of CTB-IR dendrites was significantly decreased. In sections double stained for CTB- and ChAT-IR, there was also a noticeable decrease in the number and staining intensity of ChAT-positive neurones, as compared to the contralateral (intact) DMV (Fig. 1).
Figure 1. Capsaicin (CAP) pretreatment induces neuronal degeneration of the dorsal motor nucleus of the vagus.
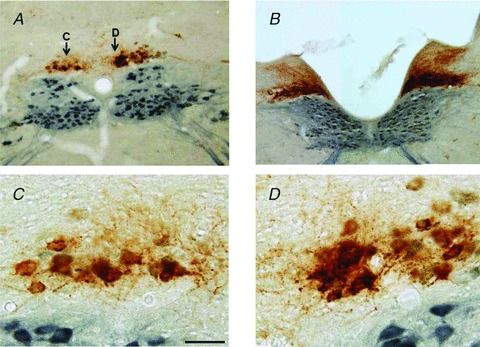
A and B, low magnification photomicrograph of a representative brainstem at a level caudal (A) or rostral (B) to area postrema taken from a rat that received injections of choleratoxin B along the major curvature of the gastric corpus and, 3 days later, apposition of perivagal CAP on the left cervical vagus. Rats were fixed 7 days after the perivagal CAP treatment. Note the decrease in choleratoxin B immunoreactivity (brown) as well as the decreased choline acetyltransferase immunoreactivity (blue) and reduced dendritic arborization on the left side of the brainstem, i.e. in CAP-treated neurones in the dorsal motor nucleus of the vagus. C and D, high magnification photomicrograph of the areas labelled (C) and (D) in A. CAP-treated neurones of the dorsal motor nucleus of the vagus (C) show clear signs of degeneration (consistent reduction in the number of choleratoxin B immunoreactivity dendrites, vacuolization and pyknotic nuclei). Bar is 300 μm in A and B; 50 μm in C and D.
These results suggest that perivagal CAP induces damage in DMV neurones, including neurones projecting to the stomach, and that qualitatively the neuronal damage was similar to that observed following vagotomy.
NOS-IR neurones were distributed within the boundaries of the DMV (as determined by ChAT-IR) as well as in the commissuralis, centralis, dorsalis and medialis subnuclei of the NTS. In the portion of the DVC contralateral to the treatment (controls), there were 5.3 ± 0.44 NOS-IR neurones per section (n = 43) in the DMV and 42.1 ± 3.16 NOS-IR neurones per section (n = 25) in the NTS, the majority of which were located in the subnucleus centralis.
Seven days after cervical vagotomy (n = 4), posterior gastric branch vagotomy (n = 6) or anterior gastric branch vagotomy (n = 4), the number of NOS-IR neurones in the DMV was increased significantly (6.3 ± 0.70 and 13.8 ± 1.61 NOS-IR neurones per section, in controls and following vagotomy, respectively; P < 0.05). Conversely, the number of NOS-IR neurones in the NTS was unaffected (46.0 ± 3.72 and 46.7 ± 4.11 NOS-IR neurones per slice, in control and following vagotomy, respectively; P > 0.05). Normalized data are detailed in Table 1.
Table 1.
Vagotomy and perivagal capsaicin (CAP) increase nitric oxide synthase immunoreactivity in neurones of the dorsal motor nucleus of the vagus (normalized data)
| Control | Cervical vagotomy | Anterior gastric branch vagotomy | Posterior gastric branch vagotomy |
| 100 | 298 ± 126* | 523 ± 190* | 149 ± 22* |
| (n = 14) | (n = 4) | (n = 4) | (n = 6) |
| Control | Perivagal CAP: 7days | Perivagal CAP: 10 days | Perivagal CAP: 14 days |
| 100 | 201 ± 33* | 178 ± 30* | 173 ± 24* |
| (n = 11) | (n = 10) | (n = 8) |
P < 0.05 vs. control.
These data indicate that severing the axons of DMV neurones induces upregulation of NOS-IR, whereas a subnodosal vagotomy is not sufficient to induce upregulation of NOS-IR in NTS neurones.
As 7 and 10 days, but not 14 days, after a 30 min long perivagal application of CAP, we observed an increase in NOS-IR DMV neurones from 4.8 ± 0.79 to 8.5 ± 1.2 NOS-IR neurones per section, in controls and 7 days after perivagal CAP, respectively (n = 11; P < 0.05); from 4.2 ± 0.65 to 6.5 ± 0.82 NOS-IR neurones per section, in controls and 10 days after perivagal CAP, respectively (n = 10; P < 0.05); and, from 5.5 ± 1.54 to 8.4 ± 1.91 NOS-IR neurones per section, in controls and 14 days after perivagal CAP, respectively (n = 8; P > 0.05). Conversely, the number of NOS-IR neurones in the NTS was unaffected at all the time points (40.7 ± 4.14 and 41.4 ± 4.04 NOS-IR neurones per section, in control and following perivagal CAP, respectively; combined data n = 18; P > 0.05) (Fig. 2). Normalized data are detailed in Table 1.
These data indicate that, similar to vagotomy, perivagal CAP induces an upregulation of NOS-IR in DMV neurones, which appears to recover 2 weeks after treatment. As in vagotomized animals, perivagal CAP does not induce upregulation of NOS-IR in NTS neurones.
Electrophysiological recordings
Whole cell patch clamp recordings were made from 97 DMV neurones from 24 rats. Twelve of those rats underwent unilateral application of CAP to the left cervical vagus, 7 days before recording; 10 rats underwent unilateral surgical deafferentation 3–7 days before recording and the remaining two rats were naïve controls. The properties of DMV neurones contralateral to the site of CAP application were not significantly different from those recorded from naïve rats; the results from these recordings were therefore pooled and designated ‘control DMV’ neurones. Recordings from DMV neurones ipsilateral to the site of CAP application were designated ‘CAP DMV’ neurones while DMV neurones ipsilateral to the unilateral deafferentation were designated ‘deafferentation DMV’ neurones.
Note that patch clamp recordings from CAP DMV neurones were conducted on cells that had no obvious signs of damage and, consequently, the variations in basic and pharmacological properties reported below may represent an underestimation of the CAP-induced effects.
Basic membrane properties
The basic membrane properties of DMV neurones were altered by perivagal CAP treatment. In fact, following perivagal CAP, the input resistance of DMV neurones was decreased (from 440 ± 25 MΩ to 325 ± 20 MΩ; P < 0.05) while the membrane capacitance was increased (from 74 ± 4.2 pF to 92 ± 7.5 pF; P < 0.05; n = 33 and 35 for control and CAP DMV neurones, respectively). In contrast, unilateral deafferentation had no effect on the input resistance of DMV neurones (439 ± 37 MΩ, P > 0.05 vs. controls; P < 0.05 vs. CAP; n = 29).
These results suggest that perivagal CAP, but not unilateral deafferentation, increases the resting conductance(s) of the DMV neuronal membrane.
Action potential characteristics
Following perivagal CAP, the action potential AHP was increased in DMV neurones (from 14.2 ± 0.86 mV to 16.9 ± 0.65 mV; P < 0.05; n = 33 and 35 for control and CAP DMV neurones, respectively), although neither the duration of the action potential (2.6 ± 0.1 and 2.8 ± 0.1 ms for control and CAP DMV neurones, respectively) nor duration of AHP (126 ± 19.7 and 126 ± 18.2 ms, for control and CAP DMV neurones, respectively) were altered (Fig. 3). The action potential duration (2.7 ± 0.1 ms) and AHP amplitude (14.3 ± 0.7 mV) and duration (131 ± 14.4 ms) in unilateral deafferentation neurones were similar to control (n = 29; P > 0.05 vs. control neurones for all). We, and others, have demonstrated previously that an apamin-sensitive (SK), but not a charybdotoxin-sensitive (BK), calcium-dependent potassium currents contribute to the AHP in rat DMV neurones (Sah & McLachlan, 1992; Travagli et al. 1992; Browning et al. 1999). In light of the larger amplitude, AHP in DMV neurones the contribution of calcium-dependent potassium currents following perivagal CAP was reinvestigated.
Figure 3. CAP pretreatment alters the basic membrane properties of DMV neurones.
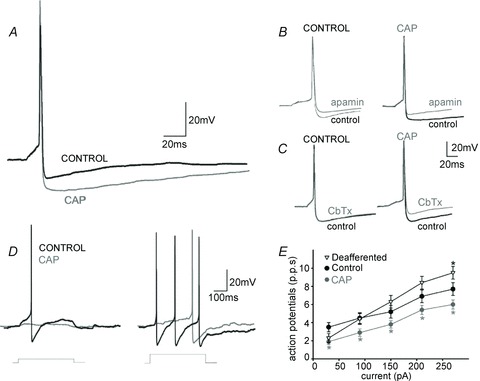
A, representative trace of single action potentials evoked in DMV neurones current clamped at −60 mV before injection of a 16 ms long direct current pulse of intensity sufficient to evoke an action potential at its offset. Note that the afterhyperpolarization from CAP DMV neurones (CAP) has a larger amplitude than that obtained in neurones from control rats. B, representative trace of single action potentials evoked in DMV neurones treated with the small conductance calcium-dependent potassium current antagonist apamin (100 nm). Note that perfusion with apamin decreased the amplitude and duration of the afterhyperpolarization to a similar degree in both controls and CAP DMV neurones. C, representative trace of single action potentials evoked in DMV neurones treated with the large conductance calcium-dependent potassium current antagonist CbTx (40 nm). Note that perfusion with CbTx decreased the amplitude and duration of afterhyperpolarization in CAP DMV neurones only. D, representative trace of trains of action potentials evoked in DMV neurones current clamped at −60 mV before injection of 400 ms long direct current pulses of increasing intensity. Note the faster frequency of action potential firing in control neurones compared with CAP DMV neurones. F, frequency–response curves for control, deafferented and CAP DMV neurones. Note that CAP DMV neurones fire fewer action potentials than neurones obtained from control or deafferented animals. *P < 0.05 vs. control neurones. CAP, capsaicin; CbTx, charibdotoxin; DMV, dorsal motor nucleus of the vagus.
Perfusion with the SK calcium-dependent potassium channel antagonist, apamin (100 nm) decreased the amplitude and duration of the AHP to a similar degree in both control and CAP DMV neurones (29.8 ± 5.9%vs. 20 ± 3.3% and 41 ± 8.7%vs. 66 ± 7.4% decrease in control and CAP DMV neuronal amplitude and duration, respectively; n = 6 for both, P > 0.05 for each; Fig. 3).
In contrast, perfusion with the BK calcium-dependent potassium channel antagonist, charybdotoxin (40 nm) had no effect upon the control DMV AHP amplitude or duration (100.9 ± 2.1 and 128 ± 26.0% of AHP amplitude and duration in the absence of charybdotoxin; n = 6; P > 0.05). Conversely, in CAP-DMV neurones, charybdotoxin decreased AHP amplitude and duration in five of seven neurones tested (27 ± 6.1% and 63 ± 11.2% decrease in amplitude and duration, respectively; P < 0.05; Fig. 3).
These results suggest that, following perivagal CAP, a charybdotoxin-sensitive (BK) calcium-dependent potassium channel is activated in DMV neurones. This BK current may be responsible, at least in part, for the larger action potential AHP observed in these neurones.
To examine whether the larger AHP in CAP-DMV neurones exerts any effect upon the frequency of action potential firing, the number of action potentials fired in response to injection of direct current pulses of increasing amplitude (400 ms duration; 30–270 pA) were examined. Following perivagal CAP treatment, CAP-DMV neurones fired fewer action potentials than control DMV neurones at all intensity of current injection examined (Fig. 3). Unilateral vagal deafferentation, however, had no effect upon action potential firing frequency, apart from the largest amplitude of current injected (270 pA; 9.5 ± 0.7 p.p.s. vs. 7.7 ± 0.7 p.p.s., P < 0.05; Fig. 3). In contrast, the firing frequency of unilateral deafferented neurones was significantly faster than CAP DMV neurones at all except the lowest current amplitude injected (2.3 ± 0.5 p.p.s. vs. 1.9 ± 0.3 p.p.s. P < 0.05; P > 0.05 for all other amplitudes).
These results suggest that, following perivagal CAP, DMV neurones are less excitable and fire fewer action potentials in response to membrane depolarization. In contrast, unilateral vagal deafferentation has no effect upon the excitability of DMV neurones suggesting that changes in DMV neuronal excitability are not due to loss of vagal afferent input.
Voltage-dependent potassium currents
The delayed rectifier current, IKV, was examined in 18 control and 16 CAP DMV neurones. As seen in Fig. 4, the magnitude of the outward current was decreased in CAP DMV neurones at potentials above 0 mV. Physiologically, IKV is activated during the action potential and contributes to action potential repolarization in rat DMV neurones (Sah & McLachlan, 1992); the present results would suggest that following perivagal CAP, action potential repolarization is delayed.
Figure 4. Capsaicin (CAP) pretreatment alters the activation of IA and IKV.
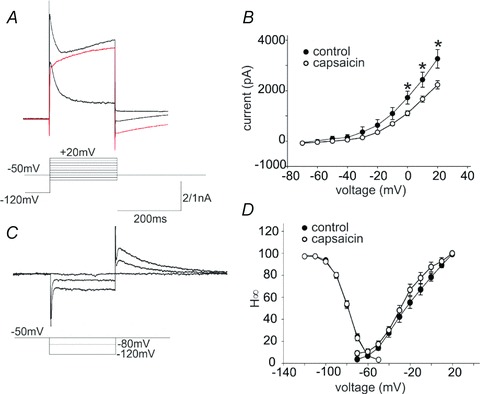
A, representative traces showing activation of the ensemble of potassium and calcium currents (upper trace) or IKV (middle trace, red) upon stepping to +20 mV from −120 mV (ensemble currents) or from −50 mV (IKV). The lower trace is the current obtained upon subtraction of the IKV from the ensemble current. The peak current obtained comprises IA mainly. For the purposes of clarity, only traces from −120 and −50 mV are shown. Similar results were obtained upon perfusion of the ensemble currents with 5 mm 4-aminopyridine (not shown). B, activation curve of IKV in neurones of the dorsal motor nucleus of the vagus from untreated (control) and perivagal CAP-treated (capsaicin) rats. Note that at potentials positive to 0 mV, the peak IKV from CAP neurones is significantly reduced compared to control. C, representative traces showing inactivation of IA obtained upon hyperpolarization of the membrane from −50 mV to −120 mV and repolarization to −50 mV. For clarity, only traces from −50, −80 and −120 mV are shown. D, inactivation (H∞) and activation curves of IA in DMV neurones from untreated (control) and perivagal CAP treated (capsaicin) rats. Note that pretreatment with perivagal CAP did not affect the voltage dependency of IA inactivation, but shifted the voltage dependence of IA activation leftwards, opening up a window current; this suggests a larger IA is activated at resting potential.
The inactivation curve (H∞), either Imax or conductance, of the fast transient IA was not altered following perivagal CAP (Fig. 4). In contrast, the IA activation curve was significantly different between control and CAP DMV neurones, in particular CAP DMV neurones showed the presence of a window current between −70 and −45 mV.
These data suggest that at membrane potentials close to resting, IA is active and may delay further the repolarization phase between action potentials thus slowing DMV neuronal firing. Furthermore, the interspike depolarization phase is further delayed by the earlier activation of IA in CAP vs. control DMV neurones.
Response of neurones of the dorsal motor nucleus of the vagus to thyrotrophin-releasing hormone
We, and others, have demonstrated previously that TRH activates DMV neurones directly to increase vagal efferent outflow (Stephens et al. 1988; Travagli et al. 1992; Tache & Yang, 1993). To determine whether the responsiveness and excitability of DMV neurones was altered by perivagal CAP treatment, DMV neurones were current clamped to a membrane potential that elicited a spontaneous firing rate of approximately 1 p.p.s. before superfusion with TRH (100 nm). The number of action potentials fired was counted over a 30 s period both before, during the peak response and after TRH application and expressed as a percentage of the control firing rate. TRH was applied to eight control DMV neurones; seven of these neurones responded to TRH with an increase in action potential firing rate (259 ± 33% of control firing frequency; P < 0.05; Fig. 5). In contrast, only three of 11 CAP DMV neurones responded to TRH application with an increase in action potential firing rate (295 ± 75% of control firing frequency; P < 0.05; P < 0.05 vs. control DMV neurones).
Figure 5. Perfusion with thyrotrophin-releasing hormone (TRH) increases the firing rate of a smaller subpopulation of neurones in the dorsal motor nucleus of the vagus from rats pretreated with perivagal capsaicin (CAP).
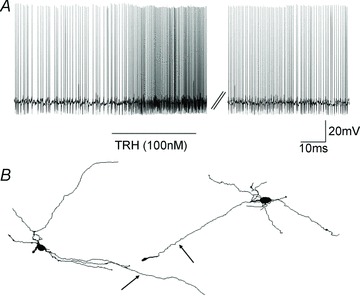
A, representative trace showing the increase in action potential firing frequency upon perfusion with 100 nm TRH. In control neurones of the dorsal motor nucleus of the vagus, TRH increased the action potential firing frequency in seven of eight neurones, whereas following CAP, only three of 11 neurones responded to TRH (P < 0.05). B, brightfield micrograph at low magnification of a coronal brain stem slice at approximately ± X mm from obex. C, computer-aided reconstruction of the neurone depicted in B. Note that the cell has a multipolar dendritic arbor. The axonal terminal has been indicated by an arrow. There were no significant morphological differences between neurones of the dorsal motor nucleus of the vagus from control and CAP pretreated rats.
These results suggest that, following perivagal CAP treatment, the ability of DMV neurones to be excited by TRH is reduced.
Morphological properties of dorsal motor nucleus of the vagus neurones
Complete morphological properties were assessed in 18 control and 14 CAP DMV neurones and are summarized in Table 2. Perivagal CAP treatment did not alter any of the morphological features of the recorded DMV neurones (Fig. 5).
Table 2.
Morphological properties of neurones of the dorsal motor nucleus of the vagus
| Control (n = 18) | Perivagal capsaicin (n = 14) | |
|---|---|---|
| x-Axis (μm) | 398 ± 84 | 391 ± 52 |
| y-Axis (μm) | 291 ± 66 | 281 ± 28 |
| Soma area | 228 ± 15 | 243 ± 21 |
| Soma diameter (μm) | 22.8 ± 0.97 | 21.9 ± 0.96 |
| Form factor | 0.56 ± 0.07 | 0.57 ± 0.08 |
| Segment ends | 9.4 ± 1.12 | 9.7 ± 0.77 |
| Segment length | 207 ± 17.4 | 210 ± 15.2 |
| Branch order | 3.7 ± 0.35 | 3.9 ± 0.34 |
Measurement of gastric motility
Baseline motility index values were not significantly different between groups and were thus combined. The preinjection motility index value was 63 ± 11, microinjection of PBS in the DVC did not induce any significant effect on gastric motility (n = 15; P > 0.05).
In five naive rats, microinjection of 100 pmol of TRH in the left DVC induced an increase in gastric motility index values from 96 ± 18 to 349 ± 18 (P < 0.05 vs. baseline) that returned to baseline values within 16.9 ± 1.61 min. Three of these rats also received a microinjection of 100 pmol of TRH in the right DVC, which induced an increase in gastric motility index values from 55 ± 10 to 239 ± 23 (P < 0.05 vs. baseline) that returned to baseline values within 12 ± 0.73 min (Fig. 6).
Figure 6. Perivagal CAP pretreatment reduces the gastric motility response to DVC microinjection of TRH.
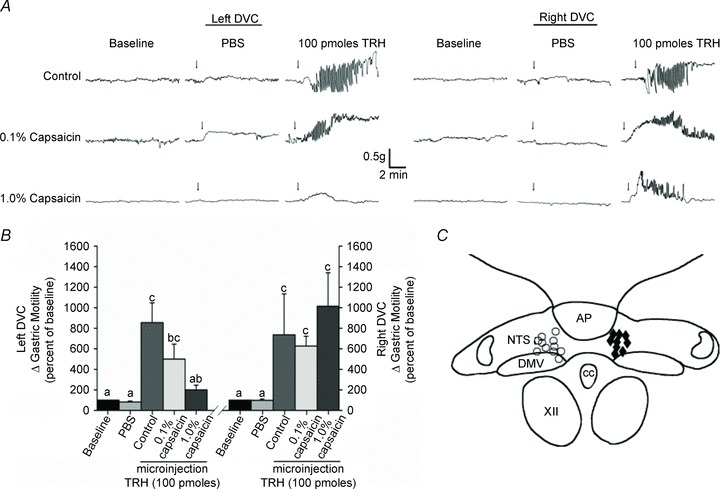
A, representative traces of gastric motility in three animals following microinjection of vehicle (PBS) or 100 pmol of TRH into the left or right DVC. Perivagal CAP was applied to the left cervical vagus only. Note that microinjection of TRH in the left DVC reveals that unilateral perivagal CAP attenuates gastric responsiveness. Conversely, microinjection into the contralateral (right) DVC elicited a robust increase in gastric motility even in CAP-treated animals. B, graphic summarizing the responses to microinjection of TRH in the left and right DVC. Bars that do not share the same letters are significantly different (P < 0.05). C, summary diagram illustrating the cumulative plots of successful microinjection sites into the DVC. CAP, capsaicin; DMV, dorsal motor nucleus of the vagus; DVC, dorsal vagal complex; PBS, phosphate-buffered saline; TRH, thyrotrophin-releasing hormone.
In five rats, which 9–10 days earlier received pretreatment with 0.1% perivagal CAP, microinjection of 100 pmol of TRH in the left DVC increased the gastric motility index values from 46 ± 7 to 197 ± 30 (P < 0.05 vs. baseline; P > 0.05 vs. naïve rats) that returned to baseline values within 13.9 ± 3.36 min. Three of these rats also received a microinjection of 100 pmol of TRH in the right DVC, which increased the gastric motility index values from 39 ± 5 to 235 ± 14 (P < 0.05 vs. baseline; P > 0.05 vs. naïve rats) that returned to baseline values within 17.3 ± 0.31 min (Fig. 6).
In five rats, which 9–10 days earlier received pretreatment with 1% perivagal CAP, microinjection of 100 pmol of TRH in the left DVC induced a non-significant increase in gastric motility index values from 46 ± 10 to 109 ± 38 (P > 0.05 vs. baseline; P < 0.05 vs. TRH in naïve rats) that returned to baseline values within 8.7 ± 2.26 min. Four of these rats received also a microinjection of 100 pmol of TRH in the right DVC, which induced an increase in gastric motility index values from 27 ± 5 to 241 ± 44 (P < 0.05 vs. baseline; P > 0.05 vs. naïve rats) that returned to baseline values within 12.4 ± 2.4 min (Fig. 6).
These results suggest that, following perivagal CAP, the ability of TRH to activate DMV neurones and to increase gastric motility is impaired significantly.
Discussion
Results of the present study suggest that, following perivagal CAP, vagal efferent motoneurones innervating the GI tract exhibit immunohistochemical, biophysical and functional signs of degeneration and damage, including: (1) dendritic degeneration; decrease in ChAT-IR and an increase in NOS-IR; (2) decreased neuronal excitability (decreased membrane input resistance, increased capacitance, decreased action potential firing frequency) due, at least in part, to the expression of a charybdotoxin-sensitive (BK) calcium-dependent potassium current, and the opening of the transient outward potassium (IA) window current at resting potentials; (3) decreased response to neurotransmitter (TRH)-induced excitation; and (4) decreased gastric motility response to brainstem microinjection of TRH. These results demonstrate, using immunohistochemical, electrophysiological and functional approaches, that perivagal CAP damages and induces degeneration of vagal efferent motoneurones. In contrast, unilateral vagal deafferentation had no effect on DMV neuronal excitability implying that the changes observed following perivagal CAP were due to vagal efferent motoneurone damage rather than loss of monosynaptic vagal afferent inputs. Treatment with perivagal CAP cannot therefore be considered selective for vagal afferent C fibres and care must be taken when using perivagal CAP to interpret the site and mechanism of action of GI neuropeptides.
The TRPV1 receptor agonist, CAP, applied systemically or perivagally, has been used over the years to induce degeneration of sensory neurones and fibres, including vagal afferents, via supposedly receptor-mediated actions at TRPV1 receptors (Buck & Burks, 1986; Czaja et al. 2008; Owyang & Heldsinger, 2011). Given the loss of GI effects observed as a consequence of CAP treatment, the site of action of many GI neuropeptides was described as almost exclusively via paracrine, namely the neuropeptide would have activated vagal terminals within the GI tract muscle layers to induce sensory vagal fibre activation (Holzer, 1986; Raybould & Tache, 1988; South & Ritter, 1988; Lloyd et al. 1993; Holzer et al. 1994; Zittel et al. 1994; Li et al. 1997; Blackshaw et al. 2000; Zafra et al. 2003; Moran, 2006; and many others). CAP-induced damage to vagal efferent nerve function has been described in a few previous studies, however, particularly with respect to gastric acid secretion and blood flow (Evangelista et al. 1989; Raybould et al. 1990). Thus, CAP-induced neurodegeneration does not appear to be selective for afferent (sensory) neurones and TRPV1 receptors expressed on neurones and glial may contribute to synaptic integration, processing and plasticity as well as glial reactivity and cytokine release within the central nervous system.
Electrophysiological studies, for example, have demonstrated that CAP acts presynaptically within the DVC to enhance glutamate release on to inhibitory GABAergic nerve terminals (Derbenev et al. 2006) as well as enhance glutamate release from vagal afferent terminals on to NTS neurones (Peters et al. 2010, 2011; Shoudai et al. 2010) or inhibit synaptic transmission on to DMV neurones (Evans et al. 2003). Activation of TRPV1 has also been found to increase synaptic glutamate release within the locus coeruleus and substantia nigra (Marinelli et al. 2002; Kim et al. 2005), indeed, microinjection of CAP into the substantia nigra has been used to induce degeneration of mesencephalic dopaminergic neurones (Kim et al. 2005). Within the hypothalamus, systemic (subcutaneous) CAP desensitizes thermosensors within the preoptic area of the anterior hypothalamus (Jancso-Gabor et al. 1970; Hajos et al. 1985) and, when applied acutely, increases glutamatergic synaptic transmission (Sasamura et al. 1998; Karlsson et al. 2005).
Over 40 years ago, it was first demonstrated that systemic CAP may induce neuronal degeneration in the central nervous system; subcutaneous CAP was shown to induce mitochondrial swelling and perikarya impairment in 1971 (Szolcsanyi et al. 1971), suggesting that the actions of CAP are not restricted to small diameter sensory fibres. Similar degeneration of terminals and neurones, in both neonatal and adult rats, was shown throughout the central nervous system following intraperitoneal CAP (Ritter & Dinh, 1988, 1990).
While we do not wish to suggest that perivagal application of CAP activates TRPV1 receptors in the DVC, these studies demonstrate clearly that TRPV1 are not restricted to sensory afferent fibres but, rather, they have a widespread distribution within the peripheral and central nervous systems and may participate in diverse integrative functions.
Despite this significant body of evidence indicating that the actions of CAP are not selective for sensory neurones, high doses of CAP (1% perivagal CAP, i.e. ∼33 mm), have been used to investigate the site of action of many GI neuropeptides, while dismissing the possibility that high doses of CAP used in these studies may have induced non-TRPV1-mediated effects. Based on this flawed approach, the site of action of many GI peptides has been attributed exclusively to a paracrine site, i.e. terminal vagal afferents in the visceral organ under consideration, because actions of these GI neuropeptides were abolished by pretreatment with perivagal CAP (Holzer, 1986; Raybould & Tache, 1988; South & Ritter, 1988; Li & Owyang, 1993, 1994; Lloyd et al. 1993; Holzer et al. 1994; Zittel et al. 1994; Li et al. 1997; Blackshaw et al. 2000; Zafra et al. 2003; Moran, 2006; and many others). Furthermore, data indicating that one of the most studied GI neuropeptides, CCK, induces effects that are not mediated exclusively by activation of vagal afferent fibres have been overlooked. In fact, many studies have shown that, in rats with intact vagal afferents, CCK-8s can increase glutamatergic excitatory inputs to NTS neurones (Appleyard et al. 2005; Baptista et al. 2005); CCK-8 excites DMV neurones directly (Branchereau et al. 1993; Zheng et al. 2005; Wan et al. 2007); CCK-8 or its segretagogue casein increase pancreatic exocrine secretion even following surgical removal of vagal afferent fibres (Viard et al. 2007); CCK-8 microinjection in the DVC decreases gastric tone (Holmes et al. 2009b); CCK-8 increases c-Fos in DVC neurones in surgically deafferented as well as in CAP-treated rats (Sayegh & Ritter, 2000; van de Wall et al. 2005; Baptista et al. 2007) and CCK-8 depolarizes similar proportions of A- and C-type neurones of the nodose ganglion (Simasko & Ritter, 2003).
In the current study, the degeneration of gastric-projecting vagal efferent motoneurones was examined following perivagal CAP application and compared to neurodegeneration resulting from surgical resection, either cervical or subdiaphragmatic, of the vagus nerve. Previous studies have described damage to central vagal nuclei following vagotomy, including a decrease in DMV neuronal number (Ji et al. 2002), a decrease in ChAT-IR DMV neurones (Saito et al. 2009) and an increase in inflammatory markers and activated microglia (Ji et al. 2002, 2005; Gallaher et al. 2012) occurring as early as 1 day post-vagotomy. In the present study, ipsilateral to vagal nerve section, gastric-projecting DMV neurones displayed clear signs of degeneration, including somatic vacuoles, dark, shrunken (pyknotic) nuclei, a decrease and eventual loss of dendrites, swollen, disconnected axons and a decrease in number and intensity of ChAT-IR neurones 7 days after vagotomy, i.e. the earliest time point in the present study. While there were no overt signs of somatic degeneration in gastric-projecting DMV neurones following perivagal CAP, there was a clear decrease in the number of proximal dendrites and a decrease in number of ChAT-IR neurones as well as intensity of ChAT-IR staining, similar to that observed following vagal nerve section.
Alterations in the number of NOS-IR neurones were observed ipsilateral to perivagal CAP as well as cervical or subdiaphragmatic vagotomy. An upregulation of NOS-IR has frequently been observed following neuronal injury and insults, including DVC neurones (Lin et al. 1997; Briski, 1999).
In the present study, perivagal CAP did not alter the morphological properties of the recorded DMV neurones, despite the neurodegeneration observed immunohistochemically. This discrepancy almost certainly occurs due to the inability to make electrophysiological recordings and morphological reconstructions from damaged or degenerating neurones. Despite selecting apparently undamaged neurones to record from, however, the present study uncovered alterations in the biophysical properties of even these ‘healthy’ DMV neurones in response to perivagal CAP. Neurones ipsilateral to perivagal CAP were less excitable, had a decreased membrane input resistance and increased membrane capacitance suggesting opening of membrane conductance(s) at resting potential. Perivagal CAP also appeared to induce the expression of a previously unrecorded, in rat DMV neurones, charybdotoxin-sensitive (BK) calcium-dependent potassium current, which contributed to the larger action potential AHP and slower action potential firing frequency observed in these neurones. Charybodotoxin-sensitive (BK) channel activity has previously been observed following ischaemic injury and has been described as determining the extent of neuronal degeneration following hypoxic and glucoprivic conditions (Gong et al. 2000; Runden-Pran et al. 2002; Bentzen et al. 2009; Liao et al. 2010). The appearance of BK in DMV neurones following perivagal CAP may be a response to vagal injury and a means of limiting neuronal damage and preserving neuronal survival.
In addition to altering the membrane properties of DMV neurones, perivagal CAP also decreased the responsiveness of these neurones to neurotransmitters. TRH has been described previously as acting directly upon DMV neurones to increase neuronal excitability, increase vagal efferent output and, by consequence, increase gastric motility and tone (Stephens et al. 1988; Travagli et al. 1992; Tache & Yang, 1993). In the present study, the majority (∼87%) of control DMV neurones responded to superfusion with TRH with a membrane depolarization and increase in action potential firing rate. In contrast, following perivagal CAP, less than 30% of DMV neurones were excited by TRH.
The functional consequence of this decreased responsiveness to TRH was assessed by examining the effects of the fourth ventricular microinjection of TRH on gastric motility. Perivagal CAP decreased the TRH-induced increase in gastric motility and tone dramatically, demonstrating clearly the vagal efferent dysfunction induced by perivagal CAP.
Conclusions
The present study provides multiple lines of immunohistochemical, biophysical and functional evidence to demonstrate that CAP is not selective for vagal afferent fibres but, rather, also induces neurodegeneration of non-sensory fibres, including vagal efferent motoneurones regulating GI functions. Previous studies have demonstrated clearly that CAP induces neurodegeneration of vagal afferent sensory neurones and fibres, and several GI neuropeptides and neurohormones unmistakably exert vagally mediated actions via paracrine effects upon vagal afferent neurones and fibres. CAP-induced damage to non-sensory afferent fibres, however, will result in underestimation of neuropeptide and neurohormone effects at brainstem, vagal efferent and GI sites of action. Treatment with perivagal CAP cannot therefore be considered selective for vagal afferent C fibres and, consequently, care is needed when using perivagal CAP to assess the mechanism of action of GI neurohormones.
Acknowledgments
Funds provided by NIH (DK 55530) and NSF (1049618). The authors would like to thank Cesare M. and Zoraide Travagli and W. Nairn Browning for support and encouragement.
Glossary
- AHP
Afterhyperpolarization
- ANOVA
Analysis of variance
- BBB
Blood brain barrier
- BK
Large conductance (charybdotoxin sensitive) calcium-dependent potassium current
- CAP
Capsaicin
- CbTX
Charybdotoxin
- CC
Central canal
- Ce VgX
Cervical vagotomy
- CCK; CCK-8
Cholecystokinin; cholecystokinin octapeptide
- ChAT
Choline acetyltransferase
- Cm
Membrane capacitance
- CTB
Cholera toxin B subunit
- DMV
Dorsal motor nucleus of the vagus
- DVC
Dorsal vagal complex
- GI
gastrointestinal
- GLP-1
Glucagon-like peptide 1
- HRP
Horseradish peroxidase
- IA
Transient outward potassium A-current
- IKV
Delayed rectifier potassium current
- -IR
-immunoreactive
- NHS
Normal horse serum
- NOS
Nitric oxide synthase
- NTS
Nucleus of the tractus solitarius
- PBS
Phosphate buffered saline
- P.P.S.
Pulses per second
- Rin
Input resistance
- SK
Small conductance (apamin-sensitive) calcum-dependent potassium current
- TRH
Thyrotropin releasing hormone
- TPBS
Tris-phosphate buffered saline
- TRPV1
Transient receptor potential vanilloid type 1
- TX
Triton-X 100
- VB VgX
Vagal branch vagotomy
- XII
Hypoglossus nucleus
Author contributions
K.N.B. and T.B. conducted the electrophysiology experiments. G.M.H. and E.S. conducted the in vivo experiments. T.B. and R.A.T. conducted the immunocytochemistry experiments. All authors contributed to the planning and interpretation of the experiments and to the writing of the manuscript.
References
- Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci. 2005;25:3578–3585. doi: 10.1523/JNEUROSCI.4177-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista V, Zheng Z, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol. 2005;94:2763–2771. doi: 10.1152/jn.00351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista V, Browning KN, Travagli RA. Effects of cholecystokinin-8s in the nucleus tractus solitarius of vagally deafferented rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1092–R1100. doi: 10.1152/ajpregu.00517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen BH, Osadchii O, Jespersen T, Hansen RS, Olesen SP, Grunnet M. Activation of big conductance Ca(2+)-activated K (+) channels (BK) protects the heart against ischemia- reperfusion injury. Pflugers Arch. 2009;457:979–988. doi: 10.1007/s00424-008-0583-5. [DOI] [PubMed] [Google Scholar]
- Bevan S, Szolcsanyi J. Sensory neuron-specific actions of capsaicin: mechanisms and applications. TIPS. 1990;11:330–333. doi: 10.1016/0165-6147(90)90237-3. [DOI] [PubMed] [Google Scholar]
- Blackshaw LA, Page AJ, Partosoedarso ER. Acute effects of capsaicin on gastrointestinal vagal afferents. Neuroscience. 2000;96:407–416. doi: 10.1016/s0306-4522(99)00547-3. [DOI] [PubMed] [Google Scholar]
- Blevins JE, Stanley BG, Reidelberger RD. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res. 2000;860:1–10. doi: 10.1016/s0006-8993(99)02477-4. [DOI] [PubMed] [Google Scholar]
- Branchereau P, Champagnat J, Denavit-Saubie M. Cholecystokinin-gated currents in neurons of the rat solitary complex in vitro. J Neurophysiol. 1993;70:2584–2595. doi: 10.1152/jn.1993.70.6.2584. [DOI] [PubMed] [Google Scholar]
- Briski KP( Induction of Fos immunoreactivity by acute glucose deprivation in the rat caudal brainstem: relation to NADPH diaphorase localization. Histochem Cell Biol. 1999;111:233. doi: 10.1007/s004180050352. [DOI] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Functional organization of presynaptic metabotropic glutamate receptors in vagal brainstem circuits. J Neurosci. 2007;27:8979–8988. doi: 10.1523/JNEUROSCI.1105-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Renehan WE, Travagli RA. Electrophysiological and morphological heterogeneity of rat dorsal vagal neurones which project to specific areas of the gastrointestinal tract. J Physiol. 1999;517:521–532. doi: 10.1111/j.1469-7793.1999.0521t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Coleman FH, Travagli RA. Characterization of pancreas-projecting rat dorsal motor nucleus of the vagus neurons. Am J Physiol Gastrointest Liver Physiol. 2005;288:G950–G955. doi: 10.1152/ajpgi.00549.2004. [DOI] [PubMed] [Google Scholar]
- Browning KN, Wan S, Baptista V, Travagli RA. Vanilloid, purinergic, and CCK receptors activate glutamate release on single neurons of the nucleus tractus solitarius centralis. Am J Physiol Regul Integr Comp Physiol. 2011;301:R394–R401. doi: 10.1152/ajpregu.00054.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck SH, Burks TF. The neuropharmacology of capsaicin: review of some recent observations. Pharmacol Rev. 1986;38:179–226. [PubMed] [Google Scholar]
- Cottrell GT, Ferguson AV. Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul Pept. 2004;117:11–23. doi: 10.1016/j.regpep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Czaja K, Burns GA, Ritter RC. Capsaicin-induced neuronal death and proliferation of the primary sensory neurons located in the nodose ganglia of adult rats. Neurosci. 2008;154:621–630. doi: 10.1016/j.neuroscience.2008.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbenev AV, Monroe MJ, Glatzer NR, Smith BN. Vanilloid-mediated heterosynaptic facilitation of inhibitory synaptic input to neurons of the rat dorsal motor nucleus of the vagus. J Neurosci. 2006;26:9666–9672. doi: 10.1523/JNEUROSCI.1591-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista S, Santicioli P, Maggi CA, Meli A. Increase in gastric secretion induced by 2-deoxy-D-glucose is impaired in capsaicin pretreated rats. Br J Pharmacol. 1989;98:35–37. doi: 10.1111/j.1476-5381.1989.tb16858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C, Baxi S, Neff R, Venkatesan P, Mendelowitz D. Synaptic activation of cardiac vagal neurons by capsaicin sensitive and insensitive sensory neurons. Brain Res. 2003;979:210–215. doi: 10.1016/s0006-8993(03)02937-8. [DOI] [PubMed] [Google Scholar]
- Fox EA, Powley TL. Longitudinal columnar organization within the dorsal motor nucleus represents separate branches of the abdominal vagus. Brain Res. 1985;341:269–282. doi: 10.1016/0006-8993(85)91066-2. [DOI] [PubMed] [Google Scholar]
- Gallaher ZR, Ryu V, Herzog T, Ritter RC, Czaja K. Changes in microglial activation within the hindbrain, nodose ganglia, and the spinal cord following subdiaphragmatic vagotomy. Neurosci Lett. 2012;513:31–36. doi: 10.1016/j.neulet.2012.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Gao TM, Li X, Huang H, Tong Z. Enhancement in activities of large conductance calcium-activated potassium channels in CA1 pyramidal neurons of rat hippocampus after transient forebrain ischemia. Brain Res. 2000;884:147–154. doi: 10.1016/s0006-8993(00)02923-1. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol. 1990;259:R1131–1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Hajos M, Obal F, Jr, Jancso G, Obal F. Capsaicin impairs preoptic serotonin-sensitive structures mediating hypothermia in rats. Neurosci Lett. 1985;54:97–102. doi: 10.1016/s0304-3940(85)80124-5. [DOI] [PubMed] [Google Scholar]
- Holmes GM, Browning KN, Tong M, Qualls-Creekmore E, Travagli RA. Vagally mediated effects of glucagon-like peptide 1: in vitro and in vivo gastric actions. J Physiol. 2009a;587:4749–4759. doi: 10.1113/jphysiol.2009.175067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GM, Tong M, Travagli RA. Effects of brainstem cholecystokinin-8 s on gastric tone and esophageal-gastric reflex. Am J Physiol Gastrointest Liver Physiol. 2009b;296:G621–G631. doi: 10.1152/ajpgi.90567.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer HH, Turkelson CM, Solomon TE, Raybould HE. Intestinal lipid inhibits gastric emptying via CCK and a vagal capsaicin-sensitive afferent pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1994;267:G625–G629. doi: 10.1152/ajpgi.1994.267.4.G625. [DOI] [PubMed] [Google Scholar]
- Holzer P. Capsaicin-sensitive afferent neurones and gastrointestinal propulsion in the rat. Arch Pharmacol. 1986;332:62–65. doi: 10.1007/BF00633198. [DOI] [PubMed] [Google Scholar]
- Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- Holzer P. Neural injury, repair, and adaptation in the GI tract. II. The elusive action of capsaicin on the vagus nerve. Am J Physiol Gastrointest Liver Physiol. 1998;275:G8–G13. doi: 10.1152/ajpgi.1998.275.1.G8. [DOI] [PubMed] [Google Scholar]
- Hommer DW, Palkovits M, Crawley JN, Paul SM, Skirboll LR. Cholecystokinin-induced excitation in the substantia nigra: evidence for peripheral and central components. J Neurosci. 1985;5:1387–1392. doi: 10.1523/JNEUROSCI.05-06-01387.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancso-Gabor A, Szolcsanyi J, Jancso N. Stimulation and desensitization of the hypothalamic heat-sensitive structures by capsaicin in rats. J Physiol. 1970;208:449–459. doi: 10.1113/jphysiol.1970.sp009130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Dheen ST, Wah Tay SS. Molecular analysis of the vagal motoneuronal degeneration after right vagotomy. J Neurosci Res. 2002;69:406–417. doi: 10.1002/jnr.10300. [DOI] [PubMed] [Google Scholar]
- Ji JF, Dheen ST, Kumar SD, He BP, Tay SS. Expressions of cytokines and chemokines in the dorsal motor nucleus of the vagus nerve after right vagotomy. Brain Res Mol Brain Res. 2005;142:47–57. doi: 10.1016/j.molbrainres.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson U, Sundgren-Andersson AK, Johansson S, Krupp JJ. Capsaicin augments synaptic transmission in the rat medial preoptic nucleus. Brain Res. 2005;1043:1–11. doi: 10.1016/j.brainres.2004.10.064. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18:7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- Kim SR, Lee DY, Chung ES, Oh UT, Kim SU, Jin BK. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J Neurosci. 2005;25:662–671. doi: 10.1523/JNEUROSCI.4166-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Owyang C. Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest. 1993;92:418–424. doi: 10.1172/JCI116583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Owyang C. Endogenous cholecystokinin stimulates pancreatic enzyme secretion via vagal afferent pathway in rats. Gastroenterology. 1994;107:525–531. doi: 10.1016/0016-5085(94)90180-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Hao Y, Owyang C. High-affinity CCK-A receptors on the vagus nerve mediate CCK-stimulated pancreatic secretion in rats. Am J Physiol Gastrointest Liver Physiol. 1997;273:G679–G685. doi: 10.1152/ajpgi.1997.273.3.G679. [DOI] [PubMed] [Google Scholar]
- Liao Y, Kristiansen AM, Oksvold CP, Tuvnes FA, Gu N, Runden-Pran E, Ruth P, Sausbier M, Storm JF. Neuronal Ca2+-activated K+ channels limit brain infarction and promote survival. PLoS One. 2010;5:e15601. doi: 10.1371/journal.pone.0015601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L-H, Sandra A, Boutelle S, Talman WT. Up-regulation of nitric oxide synthase and its mRNA in vagal motor nuclei following axotomy in rat. Neurosci Lett. 1997;221:97–100. doi: 10.1016/s0304-3940(96)13287-0. [DOI] [PubMed] [Google Scholar]
- Lloyd KCK, Holzer HH, Zittel TT, Raybould HE. Duodenal lipid inhibits gastric acid secretion by vagal, capsaicin-sensitive afferent pathways in rats. Am J Physiol. 1993;264:G659–G663. doi: 10.1152/ajpgi.1993.264.4.G659. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Christie MJ, Connor M. Capsaicin activation of glutamatergic synaptic transmission in the rat locus coeruleus in vitro. J Physiol. 2002;543:531–540. doi: 10.1113/jphysiol.2002.022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela IM, Browning KN, Travagli RA. Morphological differences between planes of section do not influence the electrophysiological properties of identified rat dorsal motor nucleus of the vagus neurons. Brain Res. 2004;1003:54–60. doi: 10.1016/j.brainres.2003.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran TH. Gut peptide signaling in the controls of food intake. Obesity (Silver Spring) 2006;14:250S–253S. doi: 10.1038/oby.2006.318. [DOI] [PubMed] [Google Scholar]
- Norgren R, Smith GP. Central distribution of subdiaphragmatic vagal branches in the rat. J Comp Neurol. 1988;273:207–223. doi: 10.1002/cne.902730206. [DOI] [PubMed] [Google Scholar]
- Orts-Del’immagine A, Wanaverbecq N, Tardivel C, Tillement V, Dallaporta M, Trouslard J. Properties of subependymal cerebrospinal fluid contacting neurones in the dorsal vagal complex of the mouse brainstem. J Physiol. 2012;590:3719–3741. doi: 10.1113/jphysiol.2012.227959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyang C, Heldsinger A. Vagal control of satiety and hormonal regulation of appetite. J Neurogastroenterol Motil. 2011;17:338–348. doi: 10.5056/jnm.2011.17.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC. Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron. 2010;65:657–669. doi: 10.1016/j.neuron.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Andresen MC. TRPV1 marks synaptic segregation of multiple convergent afferents at the rat medial solitary tract nucleus. PLoS One. 2011;6:e25015. doi: 10.1371/journal.pone.0025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE, Tache Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- Raybould HE, Holzer P, Reddy SN, Yang H, Tache Y. Capsaicin-sensitive vagal afferents contribute to gastric acid and vascular responses to intracisternal TRH analog. Peptides. 1990;11:789–795. doi: 10.1016/0196-9781(90)90196-c. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT. Capsaicin-induced neuronal degeneration: silver impregnation of cell bodies, axons, and terminals in the central nervous system of the adult rat. J Comp Neurol. 1988;271:79–90. doi: 10.1002/cne.902710109. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT. Capsaicin-induced neuronal degeneration in the brain and retina of preweanling rats. J Comp Neurol. 1990;296:447–461. doi: 10.1002/cne.902960310. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT. Age-related changes in capsaicin-induced degeneration in rat brain. J Comp Neurol. 1992;318:103–116. doi: 10.1002/cne.903180108. [DOI] [PubMed] [Google Scholar]
- Roth GI, Yamamoto WS. The microcirculation of the area postrema of the rat. J Comp Neurol. 1968;133:329–340. doi: 10.1002/cne.901330304. [DOI] [PubMed] [Google Scholar]
- Runden-Pran E, Haug FM, Storm JF, Ottersen OP. BK channel activity determines the extent of cell degeneration after oxygen and glucose deprivation: a study in organotypical hippocampal slice cultures. Neurosci. 2002;112:277–288. doi: 10.1016/s0306-4522(02)00092-1. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J Neurophysiol. 1992;68:1834–1841. doi: 10.1152/jn.1992.68.5.1834. [DOI] [PubMed] [Google Scholar]
- Saito A, Sato T, Okano H, Toyoda KI, Bamba H, Kimura S, Bellier JP, Matsuo A, Kimura H, Hisa Y, Tooyama I. Axotomy alters alternative splicing of choline acetyltransferase in the rat dorsal motor nucleus of the vagus nerve. J Comp Neurol. 2009;513:237–248. doi: 10.1002/cne.21959. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Sasaki M, Tohda C, Kuraishi Y. Existence of capsaicin-sensitive glutamatergic terminals in rat hypothalamus. NeuroReport. 1998;9:2045–2048. doi: 10.1097/00001756-199806220-00025. [DOI] [PubMed] [Google Scholar]
- Sayegh AI, Ritter RC. Vagus nerve participates in CCK-induced Fos expression in hind brain but not myenteric plexus. Brain Res. 2000;878:155–162. doi: 10.1016/s0006-8993(00)02731-1. [DOI] [PubMed] [Google Scholar]
- Shoudai K, Peters JH, McDougall SJ, Fawley JA, Andresen MC. Thermally active TRPV1 tonically drives central spontaneous glutamate release. J Neurosci. 2010;30:14470–14475. doi: 10.1523/JNEUROSCI.2557-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simasko SM, Ritter RC. Cholecystokinin activates both A- and C-type vagal afferent neurons. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1204–G1213. doi: 10.1152/ajpgi.00132.2003. [DOI] [PubMed] [Google Scholar]
- South EH, Ritter RC. Capsaicin application to central or peripheral vagal fibers attenuates CCK satiety. Peptides. 1988;9:601–612. doi: 10.1016/0196-9781(88)90171-4. [DOI] [PubMed] [Google Scholar]
- Stephens RL, Jr, Ishikawa T, Winer H, Novin D, Tache Y. TRH analogue, RX 77368, injected into dorsal vagal complex stimulates gastric secretion in rats. Am J Physiol Gastrointest Liver Physiol. 1988;254:G639–G643. doi: 10.1152/ajpgi.1988.254.5.G639. [DOI] [PubMed] [Google Scholar]
- Szolcsanyi J, Joo F, Jancso-Gabor A. Mitochondrial changes in preoptic neurons after capsaicin desensitization of the hypothalamic thermodetectors in rats. Nature. 1971;229:116–117. doi: 10.1038/229116a0. [DOI] [PubMed] [Google Scholar]
- Tache Y, Yang H. Role of medullary TRH in the vagal regulation of gastric function. In: Tache Y, Wingate DL, Burks TF, editors. Innvervation of the Gut: Pathophysiological Implications. Boca Raton: CRC Press; 1993. pp. 67–80. [Google Scholar]
- Travagli RA, Gillis RA, Vicini S. Effects of thyrotropin-releasing hormone on neurons in rat dorsal motor nucleus of the vagus, in vitro. Am J Physiol Gastrointest Liver Physiol. 1992;263:G508–G517. doi: 10.1152/ajpgi.1992.263.4.G508. [DOI] [PubMed] [Google Scholar]
- van de Wall EH, Duffy P, Ritter RC. CCK enhances response to gastric distension by acting on capsaicin-insensitive vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;289:R695–R703. doi: 10.1152/ajpregu.00809.2004. [DOI] [PubMed] [Google Scholar]
- Viard E, Zheng Z, Wan S, Travagli RA. Vagally-mediated, non paracrine effects of cholecystokinin-8s on rat pancreatic exocrine secretion. Am J Physiol Gastrointest Liver Physiol. 2007;293:G494–500. doi: 10.1152/ajpgi.00118.2007. [DOI] [PubMed] [Google Scholar]
- Wan S, Coleman FH, Travagli RA. Cholecystokinin-8s excites identified rat pancreatic-projecting vagal motoneurons. Am J Physiol Gastrointest Liver Physiol. 2007;293:G484–492. doi: 10.1152/ajpgi.00116.2007. [DOI] [PubMed] [Google Scholar]
- Zafra MA, Molina F, Puerto A. Effects of perivagal administration of capsaicin on post-surgical food intake. Auton Neurosci. 2003;107:37–44. doi: 10.1016/S1566-0702(03)00128-0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ritter RC. Circulating GLP-1 and CCK-8 reduce food intake by capsaicin-insensitive, nonvagal mechanisms. Am J Physiol Regul Integr Comp Physiol. 2012;302:R264–R273. doi: 10.1152/ajpregu.00114.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Lewis MW, Travagli RA. In vitro analysis of the effects of cholecystokinin (CCK) on rat brainstem motorneurons. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1066–G1073. doi: 10.1152/ajpgi.00497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zittel TT, Rothenhofer I, Meyer JH, Raybould HE. Small intestinal capsaicin-sensitive afferents mediate feedback inhibition of gastric emptying in rats. Am J Physiol Gastrointest Liver Physiol. 1994;267:G1142–G1145. doi: 10.1152/ajpgi.1994.267.6.G1142. [DOI] [PubMed] [Google Scholar]
- Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN. Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin-treated hyperglycemic mice. J Neurosci. 2011;31:14024–14031. doi: 10.1523/JNEUROSCI.2081-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]