

Abstract
Plasmodium falciparum has limited capacity for de novo amino acid synthesis and rely on degradation of host hemoglobin to maintain protein metabolism and synthesis of proteins. M1 alanine aminopeptidase enzyme of the parasite involved in the terminal degradation of host hemoglobin was subjected to in silico screening with low molecular weight protease inhibitors. The km (avg) of the enzyme M1 alanine aminopeptidase for the substrate DL – Alanine β Napthylamide Hydrochloride was estimated as 322.05µM. The molecular interactions between the enzyme and the substrate and the mechanism of enzyme action were analyzed which paved way for inhibition strategies. Among all the inhibitors screened, Sitagliptin was found to be most potent inhibitor with ki of 0.152 µM in its best orientation whereas the ki(avg) was 2.0055 µM. The ki of Sitagliptin is lower than the km of M1 alanine aminopeptidase for the substrate DL – Alanine β Napthylamide Hydrochloride (322.05 µM) and Ki of the known inhibitor Bestatin. Therefore Sitagliptin may serve as a potent competitive inhibitor of the enzyme M1 alanine aminopeptidase of Plasmodium falciparum.
Keywords: Plasmodium falciparum, M1 alanine aminopeptidase, Computational Docking, DL – Alanine β Napthylamide Hydrochloride, Bestatin, Sitagliptin
Background
Malaria, which is caused by protozoan parasites of the genus Plasmodium, disables and kills more people than any other infectious disease. Malaria due to Plasmodium falciparum is a disease which can involve almost every organ and tissue in the body even though malarial parasites infect only red cells and occasionally platelets. Plasmodium falciparum is the causative agent for the cerebral malaria which includes the features such as loss of consciousness, unresponsiveness to pain, sequestration in cerebral microvasculature, localized hypoglycemia and lactic acidosis, coma and subsequent death [1]. Plasmodium falciparum, being an erythrocytic parasite has limited capacity for de novo amino acid synthesis and rely on degradation of host hemoglobin (Hb) to maintain protein metabolism and synthesis in the erythrocyte. Within the erythrocytes, the malarial parasite consumes as much as 75% of the cellular Hemoglobin [2]. Hemoglobin is initially degraded by the concerted action of cysteine-, aspartyl-, and metalloendoproteases, and a dipeptidase (cathepsin C) within a digestive vacuole (DV) to di- and tripeptide fragments [3]. These fragments are suggested to be exported to the parasite cytoplasm, where further hydrolysis to release free amino acid takes place. The release of amino acid involves metalloexopeptidases such as alanyl aminopeptidase (PfA-M1) regulating the intracellular pool of amino acids required for growth and development inside the red blood cells [4]. These enzymes are essential for parasite viability inside the erythrocyte and are validated therapeutic targets [5].
Although aminopeptidase has been recognized since the 1980s, the three-dimensional structure of this enzyme had been determined only recently [5]. This enzyme is well conserved in a variety of species such as mammals, insects, plants, and bacteria [6]. Mc Gowan et al., 2009 [5] functionally characterized Plasmodium falciparum Aminopeptidase -M1 and validated it as a target with demonstration of the inhibitory activities of Bestatin and PheP[CH2]Phe. They presented the 3D structure of M1 alanine aminopeptidase alone and in complex with both of the inhibitors.
In the present study, M1 alanine aminopeptidase enzyme, the validated drug target of Plasmodium falciparum is subjected to in silico screening using low molecular weight protease inhibitors.
Methodology
The three dimensional structure of the M1 Alanine aminopeptidase (PDB ID = 3EBG) of Plasmodium falciparum was downloaded from the Protein Data Bank. The quality check of the structure is performed through WHAT IF server. The possible molecular interactions of the substrate with M1 alanine aminopeptidase was predicted by docking the known substrate with the enzyme M1 alanine aminopeptidase. Ala- β- naphthylamide (βNA) was used to assay aminopeptidase and to determine Michaelis constant (Km). In the present study, the km of the Enzyme with the substrate, DL – Alanine β Napthylamide Hydrochloride was found out in silico with the Docking server. The low molecular weight protease inhibitors were screened for their efficacy to inhibit the action of M1 alanine aminopeptidase. About 100 low molecular weight protease inhibitors were downloaded from DrugBank, PubChem and MEROPS.
Docking server offers a web-based easy to use interface that handles all aspects of molecular docking from ligand and protein setup. The active site of the enzyme was defined in the server prior to docking. The knowledge about the active site was obtained from the structural data provided by Mc Gowan et al., 2009 [5]. The Docking server was used to further identify the inhibitors of the active site for M1 alanine aminopeptidase. Computational docking was carried out using DockingServer. Gasteiger partial charges were added to the ligand atoms. Non polar hydrogen atoms were merged, and rotatable bonds were defined. Docking calculations were carried out on the protein – ligand interaction models. Essential hydrogen atoms, Kollman united atom type charges, and salvation parameters were added with the aid of AutoDock tools [7]. Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method [8]. Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was derived from 10 different runs that were set to terminate after a maximum of 2,50,000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied and the docking were performed.
Results & Discussion
km Estimation and proposed Mechanism of Action:
In the present study, km (av) for the enzyme M1 alanine aminopeptidase for the substrate DL – Alanine β Napthylamide Hydrochloride was found as 322.05µM which is the average of km obtained due to binding of substrate in all possible configurations. Ito et al., (2006) [9] has reported km for E.Coli aminopeptidase with the substrate Alanyl β Napthylamide as 304 ± 10 µM. In the current studies, computed km (avg) (322.05µM) is closely correlating experimental data [9].
The active site incorporate the zinc-binding motif H496EYFHX17KE519 and the well-conserved G490AMEN motif involved in substrate recognition [9, 10, 11]. The substrate Alanyl β Napthylamide is observed to be oriented antiparallel to the β strand defined by residues of the G460AMEN motif, with the N-terminus anchored by the zinc moiety and Glu526. The free amine group of the substrate donates one hydrogen bond to the carboxyl group of Glu526. The catalytic zinc ion is coordinated by N2 atoms of His496 and His500, the carboxyl O atom of Glu519, and a water molecule that acts as the nucleophile that attacks the carbonyl carbon of the substrate [9]. This water molecule forms a slightly longer metallo bond with the zinc ion and is also coordinated by Glu497 and Glu463 [5]. Ito et al., 2006 [9], proposed that amide carbonyl oxygen of the substrate Ala-p-NA is bound to the Zn2 ion, and the carbonyl carbon is at a suitable position for attack from a water molecule activated by Glu296 in E. Coli aminopeptidase. Correspondingly, the carbonyl oxygen of the scissile bond interacts with the zinc ion [9] and forms a polar interaction between the NH2 atom of the Arg 489 and hydroxyl group of Tyr580.
This positions the scissile peptide bond optimally for catalysis, presenting the carbonyl carbon to both Glu497 and the nucleophilic water. Together with Tyr580 and the zinc ion polarize the carbonyl bond of the scissile peptide bond. This increases the electropositivity of the carbonyl carbon, facilitating the nucleophilic attack of a water molecule. Presumably, the bound peptide displaces the water molecule, and the absence of a charged Glu497 prevents a new water molecule from binding when substrate is present. The possible mechanism of enzyme action is depicted in Figure 1 (A – D). The molecular interactions between the amino acids and the substrate are shown in Figure 2 (A).
Figure 1.
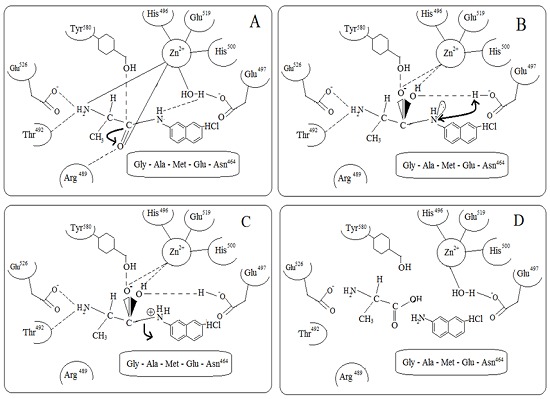
(A –D): Possible Reaction mechanism for hydrolysis of the substrate Alanine β Napthylamide Hydrochloride by M1 alanine aminopeptidase (A) The Substrate displaces the zinc associated water molecule and the absence of a charged Glu497 prevents a new water molecule from binding when substrate is present .The substrate chelates the zinc ion by its free amine group. Together with Tyr580, the zinc ion polarizes the carbonyl bond of the scissile peptide bond (Represented by an arrow mark). This results in the increase in the electro positivity of the carbonyl carbon facilitating the nucleophilic attack; (B) The polalized carbonyl carbon is prone to nucleophilic attack. Glu497 acts as a base for the nucleophilic attack. Pentahedral zinc coordination is required for the transition state of the enzyme that exists after the nucleophilic attack at the carbonyl carbon of the substrate. There is initiation of proton shift from the Glu497 to the amino terminal of the leaving group which is indicated by double headed arrow; (C) Upon the formation of the reaction intermediate the substrate becomes slightly shifted, leading to the exchange of zinc coordinating groups and strengthened H bonds to nearby residues. The amino moiety H bonds tighter to Glu497 and the oxyanion which is bound to the carbonyl carbon forms two strong bonds, a co ordinate bond with zinc ion and low barrier hydrogen bond to Tyr580. As a result of previous base catalysis, a proton resides on the carboxylate oxygen of Glu497. To create a good leaving group, allowing the peptide to break apart, the leaving amine acquires an additional proton which resides on the carboxylate oxygen of Glu497 by direct shuffling. As a result, the scissile peptide bond is prone to hydrolysis which is indicated by an arrow mark; (D) The products along with the amino acids participating in the enzyme catalysis. Also, the tetrahedral geometry of the Zinc ion is seen which is coordinated with N2 atoms of His496 and His500, the carboxyl O2 of Glu519, and a water molecule which forms a slightly longer metallo bond also coordinated by Glu497.
Figure 2.
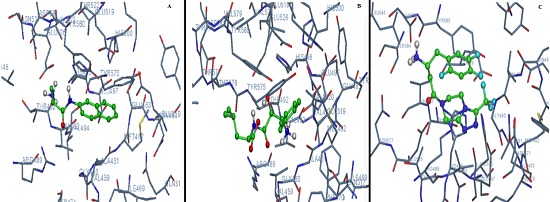
A) Molecular interactions of M1 alanine aminopeptidase with DL – Alanine β Napthylamide Hydrochloride; B) Bestatin and C) Sitagliptin.
Role of Glu526:
The N-terminal specificity of aminopeptidases suggest the presence in these enzymes of an anionic binding site which involves a negatively charged carboxylate side chain, expected to be located at the end of the S1 subsite, allowing recognition of the free amino group of substrates and inhibitors. The negative charge of the glutamate side-chain carboxylate may constitute the anionic binding site. Glu526 is ideally positioned in the structure to bind the N terminus of the substrate DL – Alanine β Napthylamide Hydrochloride. In addition, this residue, or any equivalent functionality, is absent in the structure of thermolysin, a classical zinc endopeptidase that accommodates peptide substrates of any length [12]. Mc Gowan et al., (2009) [5] observed that Glu526 side chain that moved away from the active site, removing what would otherwise form a close contact with P1 position of inhibitors. The position of Glu526 in M1 alanine Aminopeptidase is within hydrogen bonding distance (2.87 Å) to the terminal amino group of the substrate. This data imply that Glu526 acts as an N-terminal recognition site for peptide substrates.
Role of Glu497:
Hydrophobic environment around Glu298 is supposed to be important for the activation of the E.coli Aminopeptidase [5] which is equivalent to Glu497 in M1 alanine aminopeptidase. Presumably, the bound peptide displaces the water molecule, and the absence of a charged Glu497 prevents a new water molecule from binding when substrate is present. To create a good leaving group, allowing the peptide to break apart, the leaving amine must acquire an additional proton. Based on mutagenesis, Tyr383 in leucotriene hydrolase was previously proposed to act as an acid catalyst for this purpose [13]. However, Tholander et al., (2008) [14] found that Tyr383 is too far from the amine nitrogen in leucotriene hydrolase. They proposed that the most obvious acid catalyst is Glu298 which is equivalent to Glu497 in M1 alanine aminopeptidase, protonated as a consequence of the previous catalytic step. In this way, the newly formed glutamic acid shuffles a proton to the leaving amine. This is equivalent to a proposed function for the corresponding Glu residue as a catalytic base in thermolysin [15].
Role of His496, Glu497, His500 and Glu519:
Zinc coordination geometry appears to be critical for ligand binding and is basically maintained throughout the reaction. Thus, bound substrate, inhibitor, or reaction intermediates must provide the fourth zinc binding ligand at a distance close to 2Å and a fifth, slightly more distant ligand. Pentahedral zinc coordination, rather than the tetrahedral geometry observed in the unbound structure, is required for the transition state of the enzyme that exists after the nucleophilic attack at the carbonyl carbon of the substrate [5].
The catalytic zinc ion is coordinated by N2 atoms of His496 and His500, the carboxyl O2 of Glu519, and a water molecule that acts as the nucleophile that attacks the carbonyl carbon of the substrate [9]. This water molecule forms a slightly longer metallo bond with the zinc ion and is also coordinated by Glu497 and Glu463 [5] which are required for the transition state of the enzyme.
Role of Tyr580:
Site directed mutagenesis of Tyr383 in LTA4 hydrolase corresponding to Tyr580 in M1 alanine aminopeptidase resulted in inactive mutants towards peptidase activity [13]. Tyr471 in Aminopeptidase A seems to stabilize the transition state of the catalytic process acting as an electrophilic catalyst through interaction of the tyrosine hydroxy group with the oxyanion [16]. Tyr 580 is well conserved in peptidase family M1; the corresponding residues are Tyr381 in E.coli aminopeptidase [9] and Tyr383 in leukotriene A4 hydrolase [6].
Role of Gly460, Ala461, Met462, Glu463 and Asn464:
The GAMEN motif is a substrate recognition motif in M1 alanine aminopeptidase. For all ligands, the peptide backbone binds as an extended β strand antiparallel to the β strand defined by the GXMEN motif, which is conserved among M1 Aminopeptidases [5]. In the present study the GAMEN motif is found between Gly460 to Asn464.
Role of Thr492:
The substrate specificity of mono zinc aminopeptidases depends not only on interactions occurring in the Michaelis complex between the residues of the enzyme and the side chain of the substrate but also on the optimal positioning of the substrate during catalysis, thereby optimizing the hydrolysis of the substrate scissile peptide bond.
Nishiyama et al., (1991) [17] performed random mutagenesis of malate dehydrogenase from a thermophilic bacterium, Thermus flavus AT-62 and revealed that the replacement of Thr190 with Ile replacement near the essential catalytic residue His187 caused marked modulation of the catalytic properties.
Claperon et al., (2009) [18] postulated that in Aminopeptidase A, Thr348 adjusts the position of the substrate in the APA active site, strengthening, together with Glu215 and Glu352, the polarization of the catalytic water molecule to optimize the nucleophilic attack on the scissile peptide bond of acidic substrates.
In the present study, proximity of Thr492 to the catalytic residues such as His496, Glu497, His500 and Glu519 suggests that similar role can be played by Thr492. The amino terminal of the substrate binds with Thr492 along with Glu526 enabling the optimal positioning of the substrate during catalysis and may contribute substrate specificity.
Enzyme inhibition:
The inhibitor Bestatin was docked with the M1 alanine aminopeptidase and molecular interactions are shown in (Figure 2B). The ki of M1 alanine aminopeptidase for Bestatin as an inhibitor in its best orientation is found to be 98.81µM. The ki were 1830µM and 6860 µM in other two orientations and so the ki (avg) is 1005.4 µM.
According to Mc Gowan et al., 2009, [5] the Bestatin interacts with the catalytic zinc ion. The carbonyl carbon (O3) of the Bestatin form hydrogen bonds with the side chain of Tyr580, stabilizing this reaction intermediate. A cis-peptide (Glu319- Ala320) allows the side chain of Glu319 to extend into the active site, where it forms a hydrogen bond with the N2 atom of bestatin. The GAMEN recognition motif residues also contribute hydrogen bonds to ligand binding with the side chain of Glu463 and main-chain amide of Gly460 was also found to interact with bestatin. It also forms a hydrogen bond with the main chain amide of Ala461.
According to the docking results in the present study, Bestatin interacts with Glu 319, Ala461, Met462, Glu 463, Arg 489, His 496, Glu497, His 500 and Tyr 580. The amino acids His 496, Glu 497, His 500 form a part of Zn recognition motif. The inhibitor forms hydrogen and polar interactions with Oε2 of Glu497 and forms hydrophobic interaction with CD2 of His 496. The amino acid Ala461, Met462, Glu 463 contributes a part of GAMEN substrate recognition motif. The inhibitor Bestatin interacts with the backbone carbon of Ala461, Carboxyl group of Glu 463 and forms hydrophobic interaction with sulfur in Met462. The inhibitor forms many interactions with Tyr 580 which is a stabilizing amino acid in the reaction intermediate.
In the present study, ki of M1 alanine aminopeptidase for Bestatin as an inhibitor in its best orientation (Shown in Figure 2B) is found to be 98.81µM and the ki avg is 1005.4 µM. The km in the best orientation of the substrate and the km (avg) for the enzyme M1 alanine aminopeptidase for the substrate DL – Alanine β Napthylamide Hydrochloride are found as 278.6µM and 322.05µM respectively. Though the km is higher than the ki in the best orientation of the ligands, the undesirable aspect is the much lower value of km (avg) than ki (avg). Thus, there is a need for the inhibitors with desired properties.
In the docking studies, the ki of M1 alanine aminopeptidase for inhibitors such as Sitagliptin , Chloridoxipoxide, Alprazolam, Ergotamine, Dihydroergotamine, 4,7 dimethyl 1,10 phenanthroline, 9,4 hydroxy phenyl phenanthroline and Camptothecin are found to be lower than Bestatin in their best orientations. The values are tabulated in Table 1 (see supplementary material).
To cite specifically, the ki of M1 Alanine aminopeptidase for Sitagliptin in its best orientation is 0.152 µM which is much lower than the ki of the enzyme for bestatin (98.81µM ) and km (278.60 µM). The ki (avg) for sitagliptin is 2.0055µM which is much lower than the ki (avg) of the enzyme for bestatin (1005.4 µM) as well as kmavg (322.05µM) for the substrate DL – Alanine β Napthylamide Hydrochloride. The molecular interactions between Sitagliptin and the enzyme Alanine aminopeptidase are shown in Figure 2C.
Sitagliptin phosphate was approved by the US FDA for the treatment of type 2 diabetes mellitus in October 2006. It is the first in a new class of drugs that inhibit the proteolytic activity of dipeptidyl peptidase-4, thereby potentiating the action of endogenous glucoregulatory peptides, known as incretins. It reduces blood glucose levels without significant increases in hypoglycaemia.
The present study reveals that Sitagliptin interacts with the aminoacids such as His496, Glu497 and Glu526 (which are the components of the essential Zn binding motif) by forming hydrogen bonds, polar interactions or hydrophobic interactions. The halogen bond interaction of sitagliptin with Ala461 which is the amino acid present in substrate recognizing GA461 MEN motif, may inhibit the function of the motif. The hydroxyl group of Tyr580 forms halogen bond with F1 of Sitagliptin besides its hydrophobic and pi-pi interactions thus, stabilizing this reaction intermediate. Sitagliptin binds with the amino acids such as Asp581, Val493, Val523, Val459, Arg489, Thr492 and Leu546 which are present at the vicinity of the active site. The enzyme activity not only depends on interactions occurring in the Michaelis complex between the residues of the enzyme and the side chain of the substrate but also on the optimal positioning of the substrate during catalysis, thereby optimizing the hydrolysis of the substrate scissile peptide bond.
Conclusion
Sitagliptin shows higher binding affinity towards the active site of M1 alanine aminopeptidase than its substrate Ala- β- naphthylamide (βNA) and any inhibitors that has been used in the study. Hence, sitagliptin competes with the substrate for the binding at the active site. Thus, sitagliptin may serve as a potent competitive inhibitor for the enzyme M1 alanine aminopeptidase of Plasmodium falciparum, thus may serve as a potent drug candidate. Further studies are required in the wet lab to confirm these results which are predicted. Also, target specific drug delivery system has to be developed that the infected erythrocytes are specifically recognised and destroyed on the release of the drug.
Supplementary material
Footnotes
Citation:Mohana & Achary, Bioinformation 9(6): 293-298 (2013)
References
- 1.Clark IA, Cowden WB. Pharmacol Ther. 2003;99:221. doi: 10.1016/s0163-7258(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg DE. Curr Top Microbiol Immunol. 2005;295:275. doi: 10.1007/3-540-29088-5_11. [DOI] [PubMed] [Google Scholar]
- 3.Klemba M, et al. J Biol Chem. 2004;279:43000. doi: 10.1074/jbc.M408123200. [DOI] [PubMed] [Google Scholar]
- 4.Gavigan CS, et al. Mol Biochem Parasitol. 2001;117:37. doi: 10.1016/s0166-6851(01)00327-9. [DOI] [PubMed] [Google Scholar]
- 5.Gowan Mc, et al. Proc Natl Acad Sci U S A. 2009;106:2537. [Google Scholar]
- 6.Thunnissen MM, et al. Nat Struct Biol. 2001;8:131. doi: 10.1038/84117. [DOI] [PubMed] [Google Scholar]
- 7.Morris GM, Goodsell DS. J comp chem. 1996;19:1639. [Google Scholar]
- 8.Solis FJ, Wets RJB. Minimization of Operations Research. 1981;6:19. [Google Scholar]
- 9.Ito K, et al. J Biol Chem. 2006;281:33664. doi: 10.1074/jbc.M605203200. [DOI] [PubMed] [Google Scholar]
- 10.Addlagatta A, et al. Proc Natl Acad Sci U S A. 2006;103:13339. doi: 10.1073/pnas.0606167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardiner DL, et al. J Biol Chem. 2006;281:1741. doi: 10.1074/jbc.M508955200. [DOI] [PubMed] [Google Scholar]
- 12.Holmes MA, Matthews BW. J Mol Biol. 1982;160:623. doi: 10.1016/0022-2836(82)90319-9. [DOI] [PubMed] [Google Scholar]
- 13.Blomster M, et al. Eur J Biochem. 1995;231:528. doi: 10.1111/j.1432-1033.1995.0528d.x. [DOI] [PubMed] [Google Scholar]
- 14.Tholander F, et al. Chem Biol. 2008;15:920. doi: 10.1016/j.chembiol.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 15.Matthews BW. Acc Chem Res. 1988;21:333. [Google Scholar]
- 16.Vazeux G, et al. Biochem J. 1997;327:883. doi: 10.1042/bj3270883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishiyama SM, et al. J Biol Chem. 1991;266:14294. [PubMed] [Google Scholar]
- 18.Claperon C, et al. J Biol Chem. 2009;284:10618. doi: 10.1074/jbc.M806783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.