

Abstract
NS3/4A protease is an important emerging target for the cure of hepatitis C. There are many inhibitors of HCV NS3/4A protease that are passing through the clinical improvement indicating momentous reduction in the viral infection rate of patients. In this study molecular docking via MOE-Dock program was used to evaluate binding interactions of ligands with HCV NS3/4A protease. The docking and experimental results were found in good correlation. The best conformations of ligands were analyzed for binding interactions with the residues of binding cavity of NS3/4A protease. The valuable binding interactions and docking scores were observed for compounds 01, 05, 06, 07, 08 and 09.
Keywords: Molecular Docking, HCV NS3/4A protease, inhibitors, Binding interactions
Background
In 1989 it was exposed for the first time that Hepatitis C virus is the main causative agent of hepatitis C [1]. It is a positivestranded RNA virus and classified as a flavivirus [2]. About 200 million individuals are estimated worldwide to be infected with hepatitis C virus [3, 4]. In Pakistan HCV infection was found in about 10 million people [5]. In 50% to 80% of people chronic hepatitis was found due to HCV infection [6]. Eventually some of these patients lead to severe liver disease such as cirrhosis and hepatocellular carcinoma [7].
A polyprotein precursor encoded by HCV RNA genome containing structural proteins capsid [C], membrane [prM], envelope [E] and nonstructural (NS) proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, NS5) [8]. NS3 protease when activated by NS4A causes the cleavage of polyprotein producing the nonstructural proteins 4A, 4B, 5A, and 5B and is therefore very supportive for the replication of virus [9, 10]. NS3 protease required the vital 14-monomer hydrophobic peptide NS4A for its activation [11]. The active site configuration of NS3 protease comprises the residues His-57, Asp-81, and Ser-139 [12]. NS3 protease consists of an N-terminal protease domain and a Cterminal helicase domain [13]. The protease and helicase domains of NS3 have their individual functions i.e. NS3/4A protease causes polyprotein processing and helicase activity is RNA replication. In addition it has also been found that protease increases the helicase activity and the protease activity is enhanced by the helicase [14, 15].
NS3/4A protease is an important emerging target for the cure of hepatitis C [13]. There are many inhibitors of HCV NS3/4A protease that are passing through the clinical improvement indicating momentous reduction in the viral load of patients [16]. Currently, the development of the direct-acting antiviral agents (DDAs) in addition to pegylated interferon and ribavirin have established substantial improvement in the viral treatment rates with reduced treatment period in patients infected with hepatitis C genotype 1 [17]. In 1996 the crystal structures of the NS3 protease of Hepatitis C Virus were published for the first time with and without the cofactor NS4A [18]. Currently Protein Data Bank (PDB) is presented with about 50 structures of NS3/4A protease of HCV both in complexed and free forms [19, 20]. This data provide a valuable source for the development of novel and potential drug against HCV.Molecular docking is a computational method that can be used to explain the interactions of ligands with the receptor. There are a number of docking methods. Among them one is MOE-Dock method [21]. We applied this method to study the best binding interactions of the retrieved ligands with the NS3/4A protease of HCV. The intend of this study was to predict potent ligands that could inhibit NS3 polyprotein processing, replication of hepatitis C virus and to better understand the interactions between the protein binding sites and inhibitor.
Methodology
In this study an effort was made to carry out the docking of the ligands into HCV NS3/4A protein by means of MOE (Molecular Operating Environment) software package. LigPlot implemented in MOE was used to imagine the interactions between HCV NS3/4A protease and Ligands.
Retrieval of ligands:
The inhibitors for NS3/4A Protease included in our study and their IC50 values were all collected from the previous literature [22]. The structures of these inhibitors were constructed using MOE-Builder tool. The 2D structures of these retrieved ligands are shown in (Figure 1). The related 3D structures were also obtained and the energies of the identified molecules were minimized using the default parameters of MOE energy minimization algorithm [gradient: 0.05, Force Field: MMFF94X].
Figure 1.
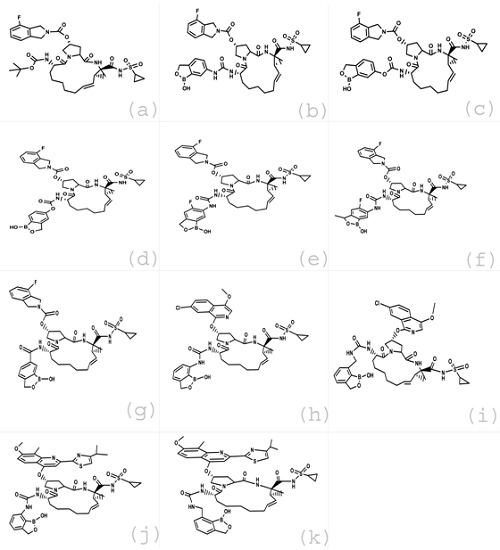
2D Structures of retrieved ligands, (a) compound 01,(b) compound 02, (c) compound 03, (d) compound 04, (e) compound 05, (f) compound 06, (g) compound 07, (h) compound 08, (i) compound 09, (j) compound 10, (k) compound 11
Preparation of Receptor Protein:
The protein molecule included in our study, HCV NS3/4A protease was obtained from Protein Data Bank [PDB Code 4a92]. Water molecules were removed and the 3D protonation of the protein molecule was carried out. The energy of the retrieved protein molecule was minimized using the default parameters of MOE energy minimization algorithm [gradient: 0.05, Force Field: MMFF94X].
Molecular Docking:
The default parmeters of MOE-Dock program were used for the molecular docking of the ligands. To find the correct conformations of the ligands and to obtain minimum energy structures, ligands were allowed to be flexible. At the end of docking, the best conformations of the ligands were analyzed for their binding interactions.
Results & Discussion
Validation of the docking procedure:
In order to assess the accuracy of MOE-Dock parogram the cocrystallized ligand was removed from the active site and redocked within the inhibitor binding cavity of NS3/4A protease. In this study, RMSD value (Figure 2) was found as 1.8299 Å, showing that our docking method is valid for the studied inhibitors [23] and MOE-Dock method, therefore, is reliable for docking of these inhibitors.
Figure 2.
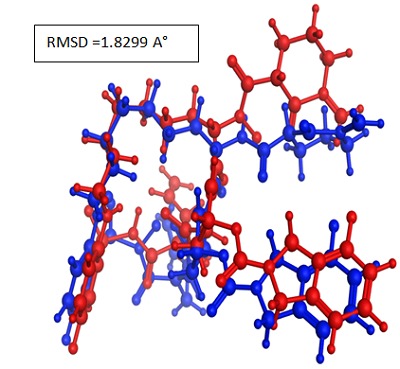
Blue Native co-crystallized ligand and red docked ligand.
Docking Analysis:
Correlation between docking score and IC50 value:
From the docking studies it was observed that the most active compound 05 (IC50; <0.2 nM) was ranked as second Table 1 (see supplementary material) indicating good agreement between the docking and experimental results. The results show that the molecular docking approach is reliable and produces a good correlation coefficient (r² = 0.519) between pdocking score and IC50 values of the ligands after the removal of two compounds (04 & 11) as shown in (Figure 3).
Figure 3.
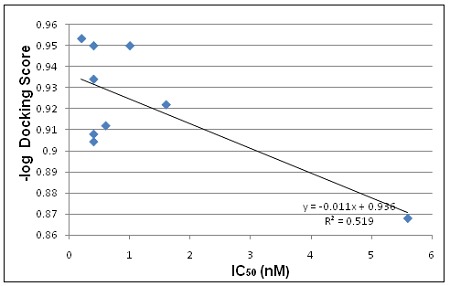
A correlation graph for docking predicted activity and IC50 values.
Binding interactions of ligands and Protein:
The most active compound 05 is clear from (Figure 4a) that this compound was bound deeply into the binding cavity of NS3 protease making interactions with the residues Gly137, Ser139, His57, Ala157 and Gln 526. Gly137 interacts with the oxygen double bonded to S on one side while Ser139 and His57 were found in interaction with the oxygen double bonded to the same S on the other side. The interaction of Ala157 was found with the oxygen of amide functional group bonded to pyrrolidine ring. Gln 526 was in a polar interaction with the N of urea linker of the ligand.
Figure 4.
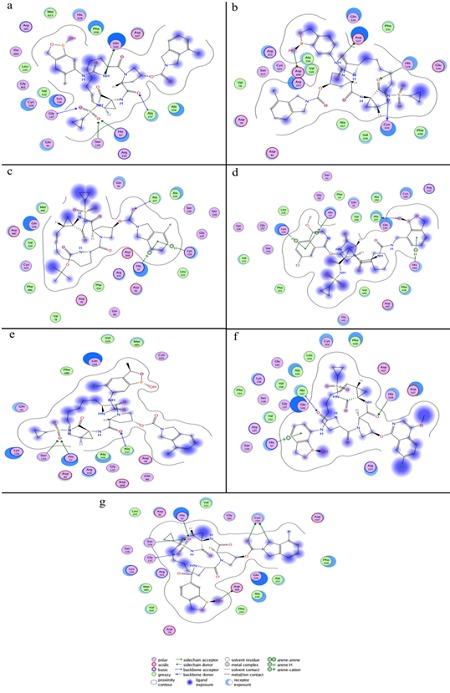
The 2D pictures of the docked conformations of most active compounds, (a) compound 05, (b) compound 02, (c) compound 01, (d) compound 09, (e) compound 06, (f) compound 08, (g) compound 07.
Compounds 1, 2, 6 and 9 are the second most active compounds (IC50; 0.4 nM) among the compounds included in this study. Among these four compounds, compound 02 and 09 showed four interactions while each one of the other two compounds (01, 06) was found making three interactions with the residues of the binding cavity of protein.
There were some four prominent interactions observed between compound 02 and the binding site residues Cys159, His528, Asp527 and Asp168 (Figure 4b). Cys159 showed interaction with the N and His528 with the Oxygen of amide functional group bonded to S. A hydrogen bonding interaction was found between the Asp168 and the hydroxyl group of benzoxaborole ring. The fourth interaction was found between Asp527 and the N of urea linker of benzoxaborole moiety.
The structural difference between compound 02 and most active compound 05 that may lead to the difference in activity and protein ligand interactions is the presence of Fluorine on the benzoxaborole ring of compound 05. It may be suggested that the polarity of compound 05 increases due to the presence of Fluorine on the benzoxaborole ring which enhances its activity and interactions with the protein.
The equipotent compounds 01 and 09 showed almost similar interactions with the protein. Compound 01 showed (Figure 4c) three interactions with the residues His57, Lys136 and Ala157. His57 and Lys136 were found in arene-cation and arenehydrogen interactions respectively with the benzene ring of indoline group. Ala157 made polar interaction with the double bonded oxygen of carboxylate group between indoline rings and pyrrolidine.
Some similar interactions were also found in compound 09- protein docked complex (Figure 4d). Compound 09 has an isoquinoline ring instead of indoline as were in compounds 01, 02 and 05. Here His57 and Lys136 showed the same arenecation and arene-hydrogen interactions respectively with the benzene ring of isoquinoline moity as were shown with indoline in compound 01. Ala157 was found making interaction with the hydroxyl group of benzoxaborole ring. Benzoxaborole ring was absent in compound 01 where Ala157 made polar interaction with the double bonded oxygen of carboxylate group between indoline rings and pyrrolidine. In addition to these three interactions His528 was involved in an arenehydrogen interaction with the benzoxaborole ring of the compound.
The isoquinoline moiety of compound 09 and indoline group of compound 01 showed similar behavior regarding interactions. However the substitution of the benzoxaborole ring in compound 09 improved number of interactions with the target protein. The main structural difference found between compound 09 and the most active compound 05 was Fluorine group bonded to benzoxaborole ring in compound 05 that might be the cause of slightly high activity of compound 05.
Compound 06 is also equipotent to compound 01, 02 and 09 (IC50; 0.4nM) although it came up with low ranking on docking score. Here three interactions were found between the ligand and target protein (Figure 4e). Ser139 and His57 showed interactions with the oxygen double bonded to S. The third interaction was shown by Ala157 with the oxygen of amide functional group bonded to pyrrolidine ring.
The structural difference found between compound 06 and most active compound 05 was the methyl group substituted on benzoxaborole ring in compound 06. The indoline and benzoxaborole rings showed no interaction in this conformation and may be a cause of low ranking of this compound on docking score. This may suggest that further optimization especially in this compound should be possible.
The third most active compound 08 (IC50; 0.6 nM) was found making three interactions with the residues His57, Ala157 and His528 (Figure 4f). His57 showed arene-cation interaction with the benzoxaborole ring of the ligand. Ala157 made hydrogen bond with the oxygen of urea linker of the side chain of compound. The third interaction was made by His528 with the oxygen of amide functional group bonded to pyrrolidine ring. The presence of isoquinoline ring and lack of Fluorine group on the benzoxaborole ring in compound 08 might be a cause of somewhat low enzymatic potency of compound 08 as compared to compound 05.
Compound 07 is the fourth active compound (IC50; 1.0 nm) and here six important interactions were observed with the residues Asp168, Gly137, Ser139, His57 and Cys159 (Figure 4g). A hydrogen bonding interaction was found between the Asp168 and the hydroxyl group of benzoxaborole ring of the ligand. Gly137 was found making interaction with oxygen double bonded to S on one side while Ser139 and His57 made interactions with the oxygen double bonded to S on the other side. Cys159 interestingly showed two interactions, one with the oxygen of carboxylate group and second with the C of indoline ring.
The structural difference between compound 07 and the most active compound 05 is the amide linker with benzoxaborole ring in compound 07 which is urea linker in compound 05. This amide linker in compound 07 may provide enzymatic potency slightly inferior to compound 05.
Conclusion
The docking analysis resulted in the detection of important ligand interactions with respect to binding site of targeted protein. As a result of this study we concluded that the inhibitors computationally studied here have shown good relationship among IC50 values, docking score and binding interactions. The compounds 01, 05, 06, 07, 08 and 09 distinctly showed interactions with one or two reported active site residues [12] of the target protein NS3/4A protease. So these compounds may be potent drug candidates and their potency may be increased against HCV NS3/4A protease with relatively simple structural changes.
Supplementary material
Footnotes
Citation:Wadood et al, Bioinformation 9(5): 309-314 (2013)
References
- 1.Choo QL, et al. Science. 1989;244:359. [Google Scholar]
- 2.Ismail NSM, Hattori M. Bioorganic and Medicinal Chemistry Letters. 2011;19:374. doi: 10.1016/j.bmc.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 3.Wands JR, et al. N Engl J Med. 2004;351:1567. doi: 10.1056/NEJMe048237. [DOI] [PubMed] [Google Scholar]
- 4. http://www.debiopharm.com/press-releases/debio- 025/clinical-update-debio-025-in-hepatitis-c.html.
- 5.Hamid S, et al. J Pak Med Assoc. 2004;54:146. [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. MMWR Recomm Rep. 1998;47:1. [Google Scholar]
- 7.Liverton NJ, et al. Chem Soc. 2008;130:4607. doi: 10.1021/ja711120r. [DOI] [PubMed] [Google Scholar]
- 8.Assenberg R, et al. J Virol. 2009;83:12895. doi: 10.1128/JVI.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartenschlager R, et al. J Virol. 1995;69:7519. doi: 10.1128/jvi.69.12.7519-7528.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brass V, et al. Proc Natl Acad sci USA. 2008;105:14545. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taremi SS, et al. Protein Sci. 1998;7:2143. doi: 10.1002/pro.5560071011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love RA, et al. Cell. 1996;87:331. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 13.Schiering N, et al. PNAS. 2011;108:21052. doi: 10.1073/pnas.1110534108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beran RK, et al. J Biol Chem. 2007;282:34913. doi: 10.1074/jbc.M707165200. [DOI] [PubMed] [Google Scholar]
- 15.Beran RK, Pyle AM. J Biol Chem. 2008;283:29929. doi: 10.1074/jbc.M804065200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melnikova I. Nat Rev Drug Discovery. 2011;10:93. doi: 10.1038/nrd3361. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann WP, Zeuzem S. Nat Rev Gastroenterol Hepatol. 2011;8:257. doi: 10.1038/nrgastro.2011.49. [DOI] [PubMed] [Google Scholar]
- 18.Kim JL, et al. Cell. 1996;87:343. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 19.Cummings MD, et al. Angew Chem Int Ed Engl. 2010;49:1652. doi: 10.1002/anie.200906696. [DOI] [PubMed] [Google Scholar]
- 20.Romano KP, et al. Proc Natl Acad Sci USA. 2010;107:20986. doi: 10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. http://www.chemcomp.com/
- 22.Ding CZ, et al. Bioorganic and Medicinal Chemistry Letters. 2010;20:7317. doi: 10.1016/j.bmcl.2010.10.071. [DOI] [PubMed] [Google Scholar]
- 23.Bostrom J, et al. J Mol Graph Model. 2003;21:449. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.