

Abstract
In silico tools are employed to examine the evolutionary relationship to possible vaccine peptide candidates' development. This perspective sheds light on the proteomic changes affecting the creation of HLA specific T-cell stimulating peptide vaccines for HIV. Full-length sequences of the envelope protein of the HIV subtypes A, B, C and D were obtained through the NCBI Protein database were aligned using CLUSTALW. They were then analyzed using RANKPEP specific to Human Leukocyte Antigen A*02 and B*27. Geneious was used to catalogue the collected gp160 sequences and to construct a phylogenic tree. Mesquite was employed for ancestral state reconstruction to infer the order of amino acid substitutions in the epitopes examined. The results showed that consensus peptide identified SLAEKNITI had changes that indicated predicted escape mutation in strains of HIV responding to pressure exerted by CD8+ cells expressing HLA A*02. The predominating 9-mers IRIGPGQAF of gp120 are significantly less immunogenic toward HLA B*27 than to HLA A*02. The data confirms previous findings on the importance for efficacious binding, of an arginine residue at the 2nd position of the gag SL9 epitope, and extends this principle to other epitopes which interacts with HLA B*27. This study shows that the understanding of viral evolution relating T-cell peptide vaccine design is a development that has much relevance for the creation of personalized therapeutics for HIV treatment.
Keywords: CLUSTALW, Geneious, Human Immunodeficiency Virus type 1, Human Leukocyte Antigen A*02, Human Leukocyte Antigen B*27, Mesquite, ‘personalized therapeutics’, RANKPEP
Background
Human Immunodeficiency Virus type 1 (HIV), the virus which leads to the development of Acquired Immunodeficiency Syndrome (AIDS), currently infects 33 million individuals and is responsible for over 25 million deaths, leading to a global pandemic [1]. Human Leukocyte Antigen A*02 (HLA A*02), a class-I allele, has been extensively studied as it is the most diverse serotype and the most common allele of the human Major Histocompatibility Complex (MHC), particularly within North America and Asia [2]. The expression of HLA A*02 has been linked to strong immune response to HIV infection as well as effects on viral load [3, 4]. This may result from the specificity of conserved epitopes to the alleles associated with the presentation of such amino acid configurations to cytotoxic T lymphocytes (CTL). This is the case with the HLA-A2 restricted, immunodominant SL9 epitope of Gag. Research has shown that mutation in sequences adjacent to the conserved epitope rarely lead to viral escape, which could be possible through interference with recognition of the antigen [5]. Generally, escape occurs via mutation of the presented configuration such that recognition by CTL is reduced or altogether halted. The HLA B*27 has also been studied for its link to long-term nonprogression (LTNP) in HIV-positive individuals [6] as well as to a group of auto-immune diseases collectively known as seronegative spondarthritis [7]. The efficacy of HLA B*27 in responding to the HIV infection is associated with the specificity of the allele for a conserved epitope of the virus' Gag protein. However, mutation in that conserved epitope is linked to viral escape and the onset of AIDS in B*27+ individuals [6, 8].
The envelope (ENV) protein, glycoprotein-160 (gp160), is the precursor to both gp120 and gp41. The two components are separated by the activity of the furin protein of the host cell [9]. The function of gp120, which is secreted on the surface of the envelope, is to bind to CD4 receptors before entry into the host cells, allowing the membranes of the two to fuse [10]. Glycoprotein-120 is of interest in HIV vaccine research because its exposure upon the surface of the envelope means that it is likely to be identified by CTL. An understanding of the evolutionary origins of HIV and how the virus has evolved over the past decades is important in efforts to develop techniques to counter it in the future [11]. Studying the in vivo evolution of the virus also contributes to such efforts. This is supported by a number of studies which have taken the approach of mapping sites of positive selection within the HIV genome in order to identify restricted epitopes under pressure exerted by CTL and anti-viral drugs [12, 13, 14]. The purpose of this study is to examine, using in silico tools, the kinds of changes which have occurred in several ubiquitous immunogenic epitopes of gp120, and how these changes have affected the immunogenicity of the resulting HLA T-cell specific 9-mers.
Methodology
Full-length sequences of the ENV protein of the HIV subtypes A, B, C and D were obtained through the NCBI Protein database. These sequences were then queried into NCBI BLAST in order to obtain genetic diversity in the samples to be studied. A total of 25 variants for each of the four subtypes were chosen for the study. These 100 sequences were then aligned using ClustalW on default parameters. ClustalW is a multiplesequence alignment algorithm provided by the European Bioinformatics Institute (EBI) [15]. In addition to performing alignments, the program also generates cladograms and identifies conserved regions in both nucleotide and amino acid sequences. RANKPEP is an immunoinformatics program which performs several functions. Because of the importance of generating a response from both CD8 and CD4 T-cells, RANKPEP uses position specific scoring matrices (PSSMs) to predict peptide binding capability to both sets of T-cells. The algorithm determines a consensus peptide against which the existing sequences are scored. Additionally, the program can determine whether or not the C-terminus of the predicted MHC-I molecule has resulted from proteasomal cleavage [16]. Sequences for gp120 were excised from the aligned gp160 sequences and evaluated by RANKPEP on default parameters to rank epitopes (9-mers) by their binding capability toward HLA A*0201 and HLA B*27 MHC class I alleles. For each of the selected HLAs, one peptide was chosen for its ubiquity among the highest-ranked epitopes. Variants of this ‘archetype’ were identified in all other strains and also scored by RANKPEP. The chosen peptide archetype for the HLA A*02 binding list was SLAEKNITI, and for HLA B*27 it was IRIGPGQAF.
Geneious is an integrated software suite for bioinformatics. It allows convenient access to BLAST and the NCBI database directly through the program and provides ways to easily organize this information. The software also features alignment and tree-building capabilities [17]. In this study, Geneious was used to catalogue the collected gp160 sequences and to construct a phylogeny based upon them. The phylogenetic tree was constructed using the neighbor-joining method, with the Jukes-Cantor genetic distance model and no out-group. The sequences of the variants of each peptide archetype from RANKPEP were placed on the tree with their calculated optimal score (%OPT), alongside their respective accession numbers (Figure 1). This directly relates epitope sequence and immunogenicity to the evolutionary history and relationships of the peptide variants. Mesquite [18] was employed for ancestral state reconstruction (ASR) to infer the order of amino acid substitutions in the epitopes examined. Mesquite serves as a platform for many different modules, which can be loaded and used independently of one another, offering great flexibility in evolutionary analysis.
Figure 1.
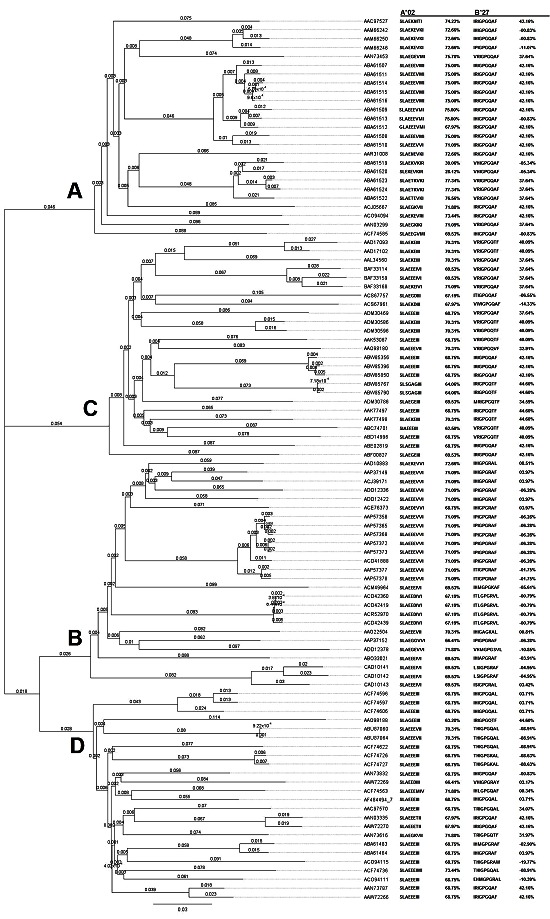
Geneious was used to catalogue the collected gp160 sequences from HIV clade A, B, C, and D in order to construct a phylogenic tree. RANKPEP results consisted of key immunogenic peptides predicted to stimulate to T-cell HLA-A*2 and B*27 clone is presented along with their binding percentages.
Discussion
The phylogenetic tree used in this paper was resampled using the bootstrap method with 100 iterations (results not shown). The placement of the subtypes received strong (100) support, with subtype C as the progenitor of A, B and D. Some relationships among finer branches received significantly weaker support. However, discrepancy in these ambiguous relationships would not have compromised any of the conclusions of the present research. ASR performed on Mesquite found SLAEEEIII as the most parsimonious ancestral sequence of all sampled strains of HIV for the peptide archetype analyzed for HLA A*02. IRIGPGQAF and IHIGPGQAF were found to be equally parsimonious ancestral states of the archetype tested for binding to HLA B*27. However, it seems unreasonable to suppose that the ancestral state of the pandemic HIV envelope protein should be a recognized potential escape mutant [6, 8]. In addition, histidine at the 2nd position is just one of many different potential escape mutants shown in the data. Thus, it may be concluded that the IRIGPGQAF sequence used as the archetype is most likely the ancestral sequence of this epitope in HIV (Figure 1).
Nearly all of the variants of the SLAEKNITI peptide archetype examined for HLA A*02 have a high %OPT score, typically ranging from approximately 64 – 77 percent. The most immunogenic variant is SLAETKVKI; with a %OPT of 77.34 percent. This variant is present in two strains, accessioned as ABA61523 and ABA61524. These are sisters to the two leastimmunogenic variants. One of these variants, SLAEKVKIR, differs from SLAETKVKI by the deletion of the threonine at the 5th position. This deletion alone brings the peptide's %OPT score down from 77.34 percent to 39.06 percent, suggesting that the deletion may have been of adaptive significance to the particular strain of HIV. The other variant with a low binding score is SLEKEVKIR, which incurred an independent deletion of alanine at the 3rd position. This variant is scored at 28.12 percent. Both of these deletions have the effect of shifting arginine into the binding frame at the 9th position (Figure 2). It is noteworthy that these two closely related strains are the only ones which display deletion mutations within the targeted epitope. Single residues were systematically changed, and the resulting 9-mers were tested for predicted immunogenicity again using the RANKPEP algorithm. This analysis revealed that while the specific deletions of the alanine and threonine residues themselves had negative effects on the peptides' immunogenicity, the brunt of the reduction in binding score was due to the resulting replacement of isoleucine with arginine as the residue in the 9th position. This data may identify such a change as a candidate escape mutation in strains of HIV responding to pressure exerted by CTL expressing HLA A*02 (Figure 1). The phylogenetic proximity of the two rare mutations suggests that they may represent such a case, as both sequences were submitted to Genbank as part of the same study [19].
Figure 2.
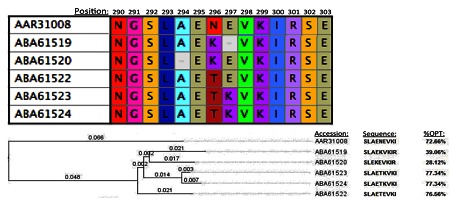
Based on the alignment and ancestral sequence reconstruction, (ASR), there appear to have been two independent deletions.
The question arises, however, of why, in both of these cases, a deletion occurred to move arginine into the 9th position rather than a simple substitution of isoleucine for arginine. One possible explanation would be that isoleucine or an amino acid with similar chemical properties is important in some other aspect of protein structure, and so its deletion or replacement would be deleterious. An alternative hypothesis is that, as two non-synonymous nucleotide substitutions are required to reach most codons associated with arginine from a codon associated with isoleucine, the intermediate residue could be deleterious. RANKPEP analysis shows this to be the case if leucine is the transitional residue between isoleucine and arginine. Leucine at the 9th position yields approximately the same predicted immunogenicity as isoleucine. However, serine can also be used as a ‘bridge’ to arginine, and it yields nearly as low a binding score as arginine (data unpublished). It is possible that serine might disrupt the structure of the envelope protein in the same way as the loss of isoleucine might, but it is unclear whether that is the case. Further research into the physical and chemical importance of those residues might be fruitful. It was found that 9-mers of gp120 are significantly less immunogenic overall toward HLA B*27 than they are toward HLA A*02. Many of the IRIGPGQAF archetype variants examined with respect to HLA B*27 were placed among the most immunogenic epitopes by RANKPEP, but none of these were assigned a %OPT score above approximately 45 percent. There appears to be a great diversity of B*27 binding scores present among the variants of this peptide archetype, which range from -19.77% to 44.60%. However, all of these variants fall into two discrete classes with respect to immunogenicity. These classes are differentiated solely on the basis of the presence or absence of arginine at the 2nd position. It is known from the study of escape mutations in HLA B*27-restricted epitopes in the gag protein that arginine at this position is critical for efficacious binding, and is thus a common escape mutant from HLA B*27 [6, 8].
Based upon that knowledge, this common substitution could be supposed from the data to be adaptive. However, this is probably not the case for two reasons: First, it is unlikely that any variant of this peptide archetype, or for that matter, any epitope on gp120 will be immunodominant toward HLA B*27 given the comparatively low binding score given by the RANKPEP algorithm. Thus, it will probably face relatively little selective pressure from CTL in the case of an immune response. The second reason why the substitution is not necessarily adaptive is that all of the sampled amino acid variants (e.g. tryptonphan and histidine) at that position can be reached by a single nucleotide substitution from one of the codons associated with arginine. This means that the replacement of arginine in this context may simply be an evolutionary accident.
Application
Amino acid replacements that can occur with HIV expressed proteins could lead to the identification of conserved regions that are vaccine targets. There have been several attempts to develop a prophylaxis vaccine for HIV infection over the past few decades. However, the hurdles for developing any type of vaccine for this pathology have been daunting [20, 21, 22]. This is also true for peptide derived vaccines. Peptide vaccines may never become the basis of a universal prophylaxis regiment for HIV. There are several reasons for this. The lack of success for peptide vaccine development may include the dependence on uninterrupted vaccine candidate epitopes, the over confidence in antibodies specificity, the monoclonal antibodies that are used to characterize the epitopes are created with operational bias, and then there is the challenge of associated with the underestimation of what construes the differences between antigenicity and immunogenicity. Van Regenmortel et al. [23] noted that these misconceptions need to be overcome if there is ever to be a peptide vaccine for HIV. Therefore, this in silico study which looked at the evolution of HIV in vivo could be extrapolated to propose a novel methodology that could revolutionize how peptide vaccines are designed for viral infections. Using the concepts purported by this study in silico tools may be utilized for the development of therapeutics vaccines that could lead for a cure from HIV infection within a single individual. The process of creating an individual treatment paradigm could include the use of dendritic cells in conjunction to algorithmic derived HLA specific T-cell based peptide vaccine design.
Ultra-deep pyrosequencing is one technique in which HIV nucleotides could be translated into sequences of amino acids utilizing a correction algorithm. Rozera et al. [24] showed that cell membranes that are virally associated and are acquired by HIV can provide information dealing with the circulating virions cellular sources. In this case, the monocytic and lymphocytic markers CD26 were targeted for identification from HIV patients which had their HAART therapy interrupted. The study created phylogenetic trees representing the third variable region (V3) associated with HIV gp120 envelope (env) glycoprotein sequences from individual patients (Figure 3) [24]. HIV-1 gp120 env glycoprotein V3 region possesses features necessary for coreceptor binding and is immunodominant [25]. HIV V3 protein sequences have been considered a prime vaccine target [26]. HIV quasispecies analysis that leads to the identification of targeted proviral sequences that has been archived or is in the actual stage of viron replication does have therapeutic relevance as being proposed as an application of this study [24]. Quasispecies identification gives an evolutionary snapshot of HIV viron proteins in vivo and therefore can lend itself to the next stage of analysis in order to identify peptide vaccine candidates.
Figure 3.
Rozera et al. developed individual phylogentic trees of the V3 protein sequences from patients analyzed after longterm suppression of viremia. The CD26-provirus is indicated in red and represents monocytes that harbor proviral quasispecies. The green circles represent the CD26-provirus proviral quasispecies harbored in T-lymphocytes. The nucleotide sequences obtained for this patient was translated in amino acid sequences and then filtered.
Many studies have shown that predictive algorithms are useful in predicting virally important immunogenic epitopes. Algorithms have been developed to predict immunogenic peptides restricted to both MHC Class I and II. MHC Class I and II in silico tools include ProPred, MHC2PRED, RANKPEP, SVMHC, MHCPred, and MHC-BPS, SYFPEITHI, BIMAS, IEDB_ANN, EpiJen, Rankpep, HLApred, NetCTL and Multipred, among others. These algorithms have been shown to be useful in identifying both MHC Class I and/or MHC Class II restricted epitopes [27, 28, 29, 30]. Online in silico tools, though advantageous, may be overwhelmed by the amount of data which has to be entered manually by the data generated from quasispecies techniques. EpiMatrix is a tool kit of algorithms that has been designed to identify potential immunogenic MHC Class I and II peptides. It has been successfully applied to the identification of potential HIV T-cell restricted epitopes [31, 32]. These in vivo evolutionary selected and conserved peptides can be pulsed to dendritic cells. Dendritic cells have been used in therapy that is truly personalized. Tumor derived medicine have been found to be a possibility in treating cancer. These cells can be primed with tumor specific T-lymphocytic immunogens and early studies have been shown to be promising. This type of approach has been combined with chemotherapy [33]. Dendritic cells have also been utilized in the possible development of a HIV vaccine. Data showed that there was an increase immune response to dendritic cells pulsed with HIV Gag and p24 proteins in nonhuman primates [34]. Using a personalized medicine approach in the treatment of individuals with HIV infection could lead to individualize cures (Figure 4). However, though this proposed methodology could be successful in AIDS patients undergoing the HAART regiment, for example, the cost per treatment would probably be tremendous. Thus, ethical questions would need to be evaluated on how the treatment would be rendered.
Figure 4.

The development of a HIV vaccine has been elusive. Using a personalized approach may be helpful in a treatment that is designed to the individual patient. The possible protocol could include: Massively parallel pyrosequencing; Use of in silico tools predictive programs (MHC Class I and/or Class II); Dendritic cell pulse with selected in vivo evolutionary conserved peptide; Personalized HIV vaccine; Weaning of the patient from conventional HIV drug therapy.
Conclusion
The present paper combines the use of several in silico tools to represent evolutionary changes in HIV together with their precise immunological significance. From this study, it may be concluded that vaccines based upon the envelope glycoprotein of HIV are unlikely to be effective in eliciting an immune response in vivo in HLA B*27+ individuals. A gp120-based vaccine may, however, be effective in individuals expressing HLA A*02. This work identifies potential target peptides for such vaccines, and important antigenic residues within them. In particular, isoleucine at the 9th position of an antigen is important for its recognition by HLA A*02+ T-cells, and thus its replacement at that site by certain other amino acids could lead to immune escape. Further, the data confirms previous findings on the importance for efficacious binding, of an arginine residue at the 2nd position of the gag SL9 epitope, and extends this principle to other epitopes which interact with HLA B*27. Such information may be useful in developing individual treatments for rapidly evolving viral infections. Though much more work is necessary in validating the proposed methodology of treating HIV infected patients, the hypothesis lends itself to tremendous possibilities of treating many infectious viral diseases that currently does not have a vaccine, such as, Dengue Fever, Epstien Barr Virus, Hepatitic C Virus, among others. It is certain that the understanding the evolution of HIV in vivo and applying massively parallel pyrosequencing that highlights HIV env quasispecies protein variants and in silico peptide design could lead to a personalized medical approached given therapeutic relief for HIV infected individuals.
Competing Interests
The authors declared that no competing interests relating to the publication exists.
Acknowledgments
Authors would like to thank Dr. Yonnette Semple-Dormer of Semple Professional Enterprises for their editing services. In addition, authors desire to thank Dormer-Owusu BioInstitute, Inc. for their support of the research conducted for this publication.
Footnotes
Citation:Hecht & Dormer, Bioinformation 9(6): 315-320 (2013)
References
- 1.Pandit A, Sinha S. BMC Bioinformatics. 2010;11:S26. doi: 10.1186/1471-2105-11-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Middleton D, et al. Tissue Antigens. 2003;61:403. doi: 10.1034/j.1399-0039.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 3.Grene E, et al. J Infect Dis. 2001;183:409. doi: 10.1086/318085. [DOI] [PubMed] [Google Scholar]
- 4.Tang J, et al. J Virol. 2002;76:8276. doi: 10.1128/JVI.76.16.8276-8284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brander C, et al. J Virol. 1999;73:10191. doi: 10.1128/jvi.73.12.10191-10198.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ammaranond P, et al. AIDS Res Hum Retroviruses. 2005;21:395. doi: 10.1089/aid.2005.21.395. [DOI] [PubMed] [Google Scholar]
- 7.Shankarkumar U, et al. Br J Biomed Sci. 2002;59:38. doi: 10.1080/09674845.2002.11783633. [DOI] [PubMed] [Google Scholar]
- 8.Schneidewind A, et al. J Virol. 2007;81:12382. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyatt R, Sodroski J. Science. 1998;280:1884. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 10.Kwong PD, et al. Structure. 2000;8:1329. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 11.Rambaut A, et al. Nat Rev Genet. 2004;5:52. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- 12.Brander C, et al. J Clin Invest. 1998;101:2559. doi: 10.1172/JCI2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bazykin GA, et al. Proc Natl Acad Sci U S A. 2006;103:19396. doi: 10.1073/pnas.0609484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Oliveira T, et al. Genetics. 2004;167:1047. doi: 10.1534/genetics.103.018135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chenna R, et al. Nucleic Acids Res. 2003;31:3497. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reche PA, et al. Immunogenetics. 2004;56:405. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 17.Drummond AJ, et al. Geneious. 2009;5:17. [Google Scholar]
- 18. http://mesquiteproject.org/mesquite/mesquite.html.
- 19.Wu X, et al. J Virol. 2006;80:835. doi: 10.1128/JVI.80.2.835-844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munier CM, et al. Drugs. 2011;71:387. doi: 10.2165/11585400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 21.Kim JH, et al. Curr Opin HIV AIDS. 2010;5:428. doi: 10.1097/COH.0b013e32833d17ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Regenmortel MHV. The Open Vaccine Journal. 2009;2:33. [Google Scholar]
- 23.Van Regenmortel MH. J Mol Recognit. 2011;24:741. doi: 10.1002/jmr.1116. [DOI] [PubMed] [Google Scholar]
- 24.Rozera G, et al. Retrovirology. 2009;12:6. doi: 10.1186/1742-4690-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang CC, et al. Science. 2005;310:1025. [Google Scholar]
- 26.Mester B, et al. Biochemistry. 2009;48:7867. doi: 10.1021/bi900146g. [DOI] [PubMed] [Google Scholar]
- 27.Seyed N, et al. PLoS Negl Trop Dis. 2011;5:e1295. doi: 10.1371/journal.pntd.0001295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, et al. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2010;27:1146. [PubMed] [Google Scholar]
- 29.Xu W, et al. Tumour Biol. 2011;32:63. doi: 10.1007/s13277-010-0098-5. [DOI] [PubMed] [Google Scholar]
- 30.Warren RL, Holt RA. Hum Immunol. 2010;71:245. doi: 10.1016/j.humimm.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 31.De Groot AS, et al. Vaccine. 2005;23:2136. doi: 10.1016/j.vaccine.2005.01.097. [DOI] [PubMed] [Google Scholar]
- 32.De Groot AS, et al. Vaccine. 2008;26:3059. doi: 10.1016/j.vaccine.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain KK. Expert Opin Biol Ther. 2010;10:1637. doi: 10.1517/14712598.2010.531256. [DOI] [PubMed] [Google Scholar]
- 34.Flynn BJ, et al. Proc Natl Acad Sci U S A. 2011;108:7131. [Google Scholar]