

Abstract
X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal disorder caused by mutations in the ABCD1 gene. Accumulation of very long chain fatty acids (VLCFA) that have been attributed to reduced peroxisomal VLCFA β-oxidation activity are the hallmark of the disease. Overexpression of ABCD2 gene, the closest homolog of ABCD1, has been shown to compensate for ABCD1, thus correcting the VLCFA derrangement. The accumulation of VLCFA leads to a neuroinflammatory disease process associated with demyelination of the cerebral white matter. The present study underlines the importance of caffeic acid phenethyl ester (CAPE) in inducing the expression of ABCD2 (ALDRP), and normalizing the peroxisomal β-oxidation as well as the levels of saturated and monounsaturated VLCFAs in cultured human skin fibroblasts of X-ALD patients. The expression of ELOVL1, the single elongase catalyzing the synthesis of both saturated VLCFA (C26:0) and mono-unsaturated VLCFA (C26:1), was also reduced by CAPE treatment. Importantly, CAPE upregulated Abcd2 expression and peroxisomal β-oxidation and lowered the VLCFA levels in Abcd1-deficient U87 astrocytes and B12 oligodendrocytes. In addition, using Abcd1/Abcd2-silenced mouse primary astrocytes we examined the effects of CAPE in VLCFA-induced inflammatory response. CAPE treatment decreased the inflammatory response as the expression of inducible nitric oxide synthase, inflammatory cytokine, and activation of NF-κB in Abcd1/Abcd2-silenced mouse primary astrocytes was reduced. The observations indicate that CAPE corrects both the metabolic disease of VLCFA as well as secondary inflammatory disease; therefore, it may be a potential drug candidate to be tested for X-ALD therapy in humans.
Keywords: X-ALD, Peroxisomes, very long chain fatty acids, glia, nitric oxide, cytokines, ALDP, ABCD2
1. Introduction
X-linked adrenoleukodystrophy (X-ALD) is an inherited demyelinating disease caused by mutation/deletion in ABCD1 gene with clinical heterogeneity varying from presymptomatic individuals to rapidly progressive cerebral ALD forms. The ALD gene (ABCD1), identified by positional cloning [1], encodes ALD protein (ALDP) that is related to the peroxisomal ATP-binding cassette (ABCD) transmembrane transporter proteins [2, 3]. Molecular defects in X-ALD result from ABCD1 gene mutations/deletions. The disease is characterized by increased levels of very long chain fatty acids (VLCFAs) in plasma, adrenal, testicular, and nervous tissues due to decreased peroxisomal β-oxidation as well as increased de novo synthesis of VLCFA by chain elongation enzymes (ELOVL) [4]. Several phenotypes exist without obvious phenotype-genotype relationship. VLCFA, especially hexacosanoic acid (C26:0), has been documented to cause metabolic alterations leading to membrane perturbation, redox imbalance and changes in membrane lipid composition [5–9], as well as the induction of inflammatory response in cultured astrocytes [10].
Recent studies from our laboratory [10] and others [11] have shown the activation of NF-κB and cytokine production in response to VLCFA accumulation. Also, the inflammatory cascade was mediated by activation of lipoxygenase pathway and production of leukotrienes [9]. Inflammatory mediators such as TNF-α and IL-1β have been described to downregulate peroxisomal β-oxidation function [12]. Accordingly, different degrees of VLCFA accumulation were observed in different areas (inflammatory, plaque and non-inflammatory) of X-ALD brain. These observations document that in X-ALD CNS altered activities of ELOVLs and peroxisomal β-oxidation as well as the secondary effects of inflammatory mediators contribute towards the observed pathagnomic levels of VLCFA. Therefore, an effective therapy should be able to correct the metabolic derangements as well as attenuate the inflammatory responses.
A successful therapy for X-ALD targeting both the metabolic and neuroinflammatory aspects of the disease remains elusive. Allogenic hematopoietic stem cell transplantation [13] and lentiviral gene therapy [14] can only be achieved in asymptomatic (from screening) or early stage patients. Transgenic or pharmacological overexpression of ABCD2/ABCD3 compensates for ABCD1 deficiency in vitro and in vivo, and therefore, has been postulated to be one of the potential therapeutic strategies for X-ALD [15–17]. Previous studies from our laboratory have shown that lovastatin, and sodium phenylacetate, can enhance VLCFA β-oxidation and reduce VLCFA levels in human skin fibroblasts [18], lymphoblasts [19] and plasma of X-ALD patients [20]. In the setting of X-ALD, most successful strategies to upregulate ABCD2 expression have come from studies with histone deacetylase (HDAC) inhibitors [17, 21, 22]. Non-specific HDAC inhibitors, 4-phenylbutyrate (4-PBA) and valproic acid (VPA), induce the ABCD2 mRNA expression [21, 22]. Despite ABCD2 induction VPA was unable to reduce VLCFA levels [21], while long-term treatment with 4-PBA in Abcd1-KO mouse reduced the drug response and VLCFA levels returned to pre-treatment levels [23]. Additionally, no induction was found in the brain for various reasons. Recent study from our laboratory reported the potential of specific and potent HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) in lowering the VLCFA load in X-ALD fibroblasts and in inhibiting the neuroinflammatory response in Abcd1/2-silenced mouse primary astrocytes [17]. However, the mechanism of SAHA-mediated downregulation of neuroinflammation and its relationship to reduction of VLCFA in X-ALD needs further investigation. Furthermore, the efficacy of SAHA in X-ALD patients is yet to be determined. CAPE, HDAC inhibitor of carboxylic acid class, is a natural flavanoid with potent anti-inflammatory activity. It is a potent antioxidant [24] and completely blocks production of reactive oxygen species (ROS) at a concentration of 10 μM [25]. Moreover, CAPE is a potent and specific inhibitor of NF-κB [26], lipid peroxidation [27], 5- lipoxygenase (5-LOX) activation and leukotriene biosynthesis [25, 28]. CAPE is indicated to cross the blood-brain barrier [29, 30] and we have previously documented the anti-inflammatory potential of CAPE in a rat model of focal cerebral ischemia [30]. In addition, the HDAC inhibitory activity of CAPE was shown to induce SMN2 transcripts and compensate for SMN1 deficiency in human patient skin fibroblasts [31].
CAPE is readily available as the active component of propolis of honeybee hives and is already in human consumption as a folklore medicine [32]. In this study, we investigated the effect of CAPE on β-oxidation activities, elongase expression and VLCFA levels in human skin fibroblasts from X-ALD patients. To determine its efficacy in the setting of X-ALD disease we determined the effect of CAPE on Abcd2 expression and peroxisomal β-oxidation activities in U87 astrocytes and B12 oligodendrocytes stably silenced for Abcd1. Also, the anti-inflammatory potential was investigated in mouse primary astrocytes silenced for Abcd1/2.
2. Materials and Methods
2.1 Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM, 4.5 g/L) was purchased from Invitrogen Life Technologies; fetal bovine serum (FBS) was purchased from BioAbChem Inc. (Ladson, SC) and Hanks’ balanced salt solution (HBSS) was purchased from Gibco (Invitrogen, Carlsbad, CA). ALDP antibody was purchased from Chemicon International Inc. (Temecula, CA). ALDRP antibody was custom-made from ANASPEC against the mouse 20-residue c-terminal sequence: 722 CKILGEDSVLKTIQTPEKTS 741. 5-LOX antibody was purchased from Cayman Chemical (Ann Arbor, Michigan). CAPE was purchased from Sigma (St. Louis, MO). Na+K+ATPase antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ECL and nitrocellulose membranes were purchased from Amersham Biosciences. Fatty acid methyl ester (FAME) standards were obtained from Supelco (Bellefonte, Pennsylvania). [l-14C] Lignoceric acid was prepared as described earlier [33]. [1-14C] Palmitic acid and 125I-labeled protein A were obtained from ICN (Cleveland, Ohio).
2.2 Cell Culture
2.2.1 Fibroblasts
Human skin fibroblasts derived from normal (control; GM03348), X-ALD (GM04932, GM04934), and AMN (GM07531) patients were obtained from the NIGMS Human Genetic Mutant Cell Repository at the Coriell Institute for Medical Research (ccr.coriell.org/). The fibroblasts were cultured in DMEM containing 10% FBS and antibiotic/antimicotic. X-ALD fibroblast cell line GM04932 was derived from a patient with positive family history of X-ALD (10-year-old male whose cultured fibroblasts presented elevated C26:0 fatty acids). Fibroblasts GM04934 were from a 7 year old boy that presented elevated very long chain (C26:0) fatty acids levels. Fibroblast GM04904 was from a 11 year old male with elevated C26:0/C22:0 ratio in fibroblasts; adrenal insufficiency; progressive white matter disease; progressively deteriorating intellectual function; with a family history of X-ALD. Fibroblast GM07531 was from a 19 year old male characterized as AMN with adrenal insufficiency since age 11; unsteady gait since age 13; slight weakness and hyperreflexia since age 19; brother has mild spastic paraparesis; mother had progressive paraparesis; elevated level of C26 fatty acid in plasma and fibroblasts (ccr.coriell.org/). Normal (control) fibroblasts (GM03348) were from an apparently healthy 10-year-old male.
2.2.2 Mouse primary mixed glia and astrocytes
C57BL6 mouse breeding pairs were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained at the Medical University of South Carolina (MUSC) animal facility. All animal procedures were approved by the MUSC Animal Review Committee and all animals received humane care in compliance with the MUSC experimental guidelines and the National Research Council’s criteria for humane care (Guide for Care and Use of Laboratory Animals)
Primary astrocyte-enriched cultures were prepared from the whole cortex of 1-day-old C57BL/6 mice as described earlier [34]. Briefly, the cortex was rapidly dissected in ice-cold calcium/magnesium-free HBSS at pH 7.4 as described previously [35]. The tissue was minced, incubated in HBSS containing trypsin (2 mg/ml) for 20 min, and washed twice in plating medium containing 10% FBS and 10 μg/ml gentamicin and then disrupted by triturating through a Pasteur pipette, after which cells were seeded in 75-cm2 culture flasks (Falcon, Franklin, NJ). After incubation at 37 °C in 5% CO2 for 1 day, the medium was completely changed to the culture medium (Dulbecco’s modified Eagle’s medium containing 10% FBS and 10 μg/ml gentamicin). The cultures received half exchanges with fresh medium twice a week. After 10 days, the cells were shaken for at least 30 min on an orbital shaker to remove the microglia, and flasks were incubated for 1 day after which they were shaken again for 8 h to remove the oligodendrocytes. The remaining population was used as astrocytes culture. All cultured cells were maintained at 37 °C in 5% CO2.
2.3 siRNA interference of Abcd1 and/or Abcd2 in mouse primary astrocytes
The Silencer siRNA (Ambion, Austin, TX, USA) was used for Abcd1 and Abcd2 silencing in primary mouse astrocytes as described previously [10]. Briefly, mice astrocytes cultured in DMEM with 10% serum and in the presence of antibiotic were transfected with siRNA for Abcd1 and Abcd2 using siPORT NeoFX transfection agent (Ambion, Austin, TX). Three siRNA for Abcd1 and Abcd2 (Ambion, Austin, TX) each were used (Abcd1 siRNA1, ID 162218; 5′-CCUCUACAACCUAAUUUAUtt-3′; 5′-AUAAAUUAGGUUGUAGAGGtg-3′, siRNA2, ID60153; 5′-GGUAUUUGAAGAUGUCAAAtt-3′; 5′-UUUGACAUCUUCAAAUACCtg-3′, siRNA3, ID 60064; 5′-GGAAAUUGCCUUCUACGGGtt-3′; 5′-CCCGUAGAAGGCAAUUUCCtc-3′; Abcd2 siRNA1, ID 188185; 5′-GGCUUUAGCUUACCAGAUGtt-3′; 5′-CAUCUGGUAAGCUAAAGCCtt-3′, siRNA2, ID 214996; 5′-GGUAAAUGUCUAGAAAUGGtt-3′; 5′-CCAUUUCUAGACAUUUACCtg-3′, siRNA3, ID 214997, 5′-GCUGUAGAGAUCAAUAGAGtt-3′; 5′-CUCUAUUGAUCUCUACAGCtc-3′). The siRNAs were mixed and diluted in OPTI-MEM1 medium to a final concentration of 30 nM/well. siRNA/transfection agent was dispensed into culture plates as directed by the manufacturer. A negative control with sequence similarity to no known human, mouse, or rat gene was included. Cells were maintained in DMEM with reduced serum (2%). Silencing was observed with Western blot and mRNA quantification. 48h after silencing for Abcd1/2, cells were treated with CAPE and harvested 72h later for protein and RNA analysis. For protein analysis of the transfected cells, three wells per plate were lysed and used for protein measurements and protein levels (Western blot). Cells were maintained for 6 days in DMEM with 2% FBS before harvesting for the analysis.
2.4 Lentiviral stable silencing of Abcd1 in human U87 astrocytoma and rat B12 oligodendrocytes
A set of 3 human (SK-009605-00-10) and rat (SK-098142-00-10) specific SMART vector 2.0 lentiviral shRNA particles (108 TU/ml) for ABCD1 were purchased from Thermo Fisher Scientific Dharmacon (CO, USA). The vector had an hCMV promoter, a TurboGFP reporter gene and a puromycin selection gene. SMARTvector 2.0 non-targeting shRNA control particles (NT) (108 TU/ml, Thermo Fisher Scientific Dharmacon, CO, USA) designed so that no known gene in human, mouse or rat will be targeted were used as negative controls.
Human U87 astrocytes and rat B12 oligodendrocytes were cultured in DMEM with 10% FBS in the presence of antibiotic, and viral particles (ABCD1 and non-targeting control) were added with a multiplicity of infection (MOI) of 2.5 and 3.0 respectively for U87 astrocytes and B12 oligodendrocytes. TurboGFP-expression was analysed using microscopy and GFP-positive cells were selected using puromycin (3.0μg/ml for U87 astrocytes and 2.0μg/ml for B12 oligodendrocytes). The selected cells were maintained in culture media containing puromycin at 0.5μg/ml until further use.
2.5 Fatty Acid β-Oxidation
The peroxisomal oxidation of fatty acids in control, X-ALD fibroblasts and CAPE-treated X-ALD fibroblasts was determined in 6-well plates, as described previously [10]. β-oxidation of fatty acids to acetate (water soluble product) was determined using [1-14C]-labeled fatty acids as substrate (C24:0, lignoceric acid or C16:0, palmitic acid (ARC, St. Louis, MO); 150,000 dpm suspended in 0.25 mg of α-cyclodextrin/assay) [36]. Briefly, plates were washed 3 times with serum-free medium and the substrate in 0.250 ml of serum-free media was added to each well. The plates were incubated 1 h with lignoceric acid or for 30 min with palmitic acid, at 37 °C. Blank wells were included in each set of plates. The reaction mixture was stopped with 1 M KOH (0.625 ml) in methanol and transferred to capped tubes. The methanolic solution was then incubated at 60 °C for 1 h, neutralized with of 6 N HCl (0.125 ml) and partitioned with 1.25 ml of chloroform [36]. The amount of radioactivity in the upper phase represents the amount of [1-14C]-labeled fatty acid oxidized to acetate. Cells grown in parallel in same plate were used to determine the protein present in the assays. Experiments were performed in triplicate.
2.6 Lipid Extraction and Fatty Acid Analysis
Total lipids were extracted from control and treated cells as described previously [37]. The fatty acid methyl esters (FAME) were analyzed by GC (Shimadzu chromatograph GC-17A) using a fused silica capillary column (25 M 007 series methyl silicone, 0.25-mm internal diameter, 0.25-μm film thickness) from Quadrex Corporation (New Haven, CT). The column temperature was programmed at 125 °C for 5 min, raised to 280 °C at the rate of 5 °C per min, and then to 295 °C, at the rate of 25 °C per min. This temperature was held constant at 295 °C for 5 min. The injection block was set at 200 °C and the detector was set at 320 °C. The separated components were identified by comparison with retention times of standard FAME. The area under the peaks of identified fatty acids was set as 100%. The individual fatty acids were measured as area percent.
2.7 Preparation of carbonate membranes
Cells were harvested with sucrose buffer (0.25 M sucrose, 1 mM EDTA, 3 mM imidazole buffer, pH 7.4), and subjected to sonication (10 sec at 8–9 watts output power). The homogenate was centrifuged at 2,500 rpm for 5 min to remove unbroken cells. To isolate carbonate membranes (membrane sheets containing integral membrane proteins), equal protein amount from control and X-ALD cell homogenates were diluted with an ice-cold solution of 0.1 M sodium carbonate (pH 11.5) containing 30 mM iodoacetamide. After 30 minutes of incubation at 4°C, carbonate membranes were sedimented by centrifugation at 35,000 rpm for 1 hour in a 70Ti rotor (Beckman-Coulter, Fullerton, CA). The sedimented membranes were washed twice with cold water, lyophilized and stored at −70°C until use.
2.8 Western Blot Analysis
The cells were washed with cold Tris-buffered saline (20 mM Trizma base and 137 mM NaCl, pH 7.5) and lysed in 1X SDS sample-loading buffer (62.5 mM Trizma base, 2% [w/v] SDS,10% glycerol), and after sonication and centrifugation at 15,000 g for 5 min, the supernatant was used for the immunoblot assay. The protein concentration of samples was determined with the detergent compatible protein assay reagent (Bio-Rad) using BSA as the standard. The sample was boiled for 3 min with 0.1 volumes of 10% β-mercaptoethanol and 0.5% bromphenol blue mix. Then, 40 μg of total cellular protein was resolved by electrophoresis on 8 or 12% polyacrylamide gels, electrotransferred to polyvinylidene difluoride filter, and blocked with Tween 20-containing Tris-buffered saline (TBST; 10 mM Trizma base, pH 7.4, 1% Tween 20, and 150 mM NaCl) with 5% skim milk. After incubation with antiserum raised against mice ALDP, ALDRP, 5-LOX, p65, Na+K+ATPase and iNOS, the membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit or mouse IgG for 1 h. The membranes were detected by autoradiography using ECL-plus (Amersham Biosciences) after washing with TBST buffer.
2.9 RNA Extraction and cDNA Synthesis
Following total RNA extraction using TRIzol (Invitrogen) per the manufacturer’s protocol, single-stranded cDNA was synthesized from total RNA. Five microgram of total RNA was treated with 2 units of DNase I (bovine pancreas; Sigma) for 15 min at room temperature in an 18 μl-volume containing 1X PCR buffer and 2 mM MgCl2. This was then inactivated by incubation with 25 mM EDTA (2 μl) at 65 °C for 15 min. Next, 2 μl of random primers were added and annealed to the RNA according to the manufacturer’s protocol. cDNA was synthesized in a 50-μl reaction containing 5 μg of total RNA and 50–100 units of reverse transcriptase by incubating the tubes at 42 °C for 60 min.
2.10 Real-time PCR
Total RNA isolation from fibroblast (control, X-ALD and CAPE-treated X-ALD fibroblast) and astrocytes (control, scramble and Abcd-1/2-silenced) cultures was performed using TRIzol (Invitrogen) according to the manufacturer’s protocol. Real time PCR was conducted using Bio-Rad iCycler (iCycler iQ Multi-Color Real Time PCR Detection System; Bio-Rad). Single-stranded cDNA was synthesized from total RNA as described. The primer sets for use were designed (Oligoperfect™ designer, Invitrogen) and synthesized from Integrated DNA Technologies (Coralville, IA). IQ™ SYBR Green Supermix was purchased from Bio-Rad. Primers for human and mouse ABCD1, ABCD2, ABCD3, and ELOVL1, were purchased from Qiagen. Thermal cycling conditions were as follows: activation of DNA polymerase at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 30 s and 60 °C for 30 s. The normalized expression of target gene with respect to glyceraldehyde-3-phosphate dehydrogenase or 18S RNA was computed for all samples using Microsoft Excel data spreadsheet.
2.11 Determination of TNF-α in culture supernatants
Cells were silenced with Abcd1/2 siRNA or ScrRNA and concentrations of TNF-α was measured in culture supernatants using high-sensitivity enzyme-linked immunosorbent assay (R&D Systems).
2.12 Measurement of reactive oxygen species (ROS)
ROS were determined using the membrane permeable fluorescent dye 6-carboxy 20, 70-dichlorodihydrofluorescein diacetate (DCFH2-DA) in serum-free medium as described earlier [10]. The cultured cells with or without treatment were incubated with 5 mM DCF dye in PBS for 2 hr at 37 °C. The change in fluorescence was determined at excitation 485 nm and emission 530 nm using a Soft Max Pro spectrofluorometer (Molecular Devices, Sunnyvale, CA).
2.13 Measurement of Nitrite levels
Production of nitric oxide (NO) was determined by assaying culture supernatants for nitrite, a stable reaction product of NO and molecular oxygen. Briefly, 100 μl of culture supernatant was allowed to react with 100 μl of Griess reagent [10] and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank in all experiments. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in the assay.
2.14 Statistical analysis
Using the Student Newman-Keuls test and ANOVA, p values were determined for the respective experiments from three identical experiments using GraphPad software (GraphPad Software Inc. San Diego, CA). The criterion for statistical significance was p<0.05.
3. Results
3.1 CAPE induces the ABCD2 and ABCD3 mRNA expression in control human skin fibroblasts
ABCD2 as well as ABCD3 have been shown to be functionally redundant because their overexpression in X-ALD fibroblasts allows VLCFA β-oxidation to be restored [15, 22, 38–40] suggesting partially overlapping functions for these proteins [41, 42]. Furthermore overexpression of Abcd2 results in prevention of onset of the clinical symptoms in Abcd1 knockout mice [16]. Therefore, ABCD2/ALDRP is an attractive candidate gene for pharmacological gene therapy. We therefore first studied the effect of CAPE on ABCD1/ALDP, ABCD2/ALDRP and ABCD3/PMP70 expression in normal control human skin fibroblasts. The fibroblasts were treated with CAPE in normal medium (media with FBS and antibiotic cocktail) with varying doses and times and the gene expressions for ABCD1, ABCD2 and ABCD3 were analyzed by realtime RT-PCR. Treatment with CAPE significantly induced the expression of both ABCD2 (P<0.01) and ABCD3 (P<0.001) (Fig 1B–C) but not the expression of ABCD1 (Fig 1A). While ABCD2 expression was induced 3.4-fold (Fig. 1B), there was a 3.6-fold induction of ABCD3 expression (Fig. 1C) at maximum dose of 5μM CAPE treated for 3 days. There was no significant change in cell viability at the dose and duration of treatment used.
Figure 1.
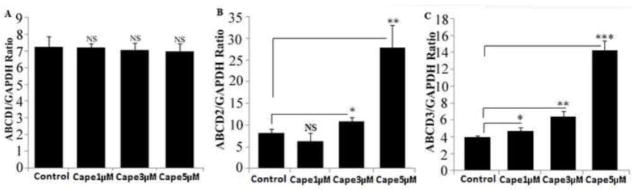
ABCD2 and ABCD3 mRNA is induced by CAPE in control normal human skin fibroblasts. ABCD1 (A), ABCD2 (B) and ABCD3 (C) expression levels in control normal human (n =3) treated with CAPE in a dose-dependent manner. ABCD2 and ABCD3 mRNA levels were determined by quantitative realtime RT–PCR and normalized to GAPDH. Data are represented as mean±SD. *P<0.05; **P<0.01; ***P<0.001; NS, non-significant.
3.2 CAPE increases the peroxisomal and mitochondrial β-oxidation activities in control human skin fibroblasts
Fibroblasts from X-ALD patients with loss/mutation of ABCD1 have decreased VLCFA β-oxidation activity and reduced accumulation of VLCFAs (C26:0 and C24:0). Overexpression of ABCD2 in fibroblasts from Abcd1-deficient mice or from X-ALD patients is shown to restore peroxisomal β-oxidation and to reduce the levels of VLCFA [15, 22, 38, 40]. Since CAPE upregulated the mRNA expression of ABCD2 and ABCD3 in control human fibroblasts, we thus examined the effect of this induction on β-oxidation of lignoceric acid (C24:0) as well as palmitic acid (C16:0) in control human fibroblasts. While ligoceric acid oxidation is a peroxisomal function, oxidation of palmitic acid occurs largely in mitochondria. Treatment with CAPE increased the lignoceric acid oxidation in a dose-dependent manner (Fig. 2). The highest dose of CAPE (5μM) was found to stimulate palmitic acid (Fig. 2A) and lignoceric acid (Fig. 2B) β-oxidation by approximately 53% and 50% respectively in 3 days. Increase of both lignoceric and palmitic acids β-oxidation activity in CAPE treated fibroblasts suggests that CAPE has an overall stimulating effect on fatty acid oxidation.
Figure 2.
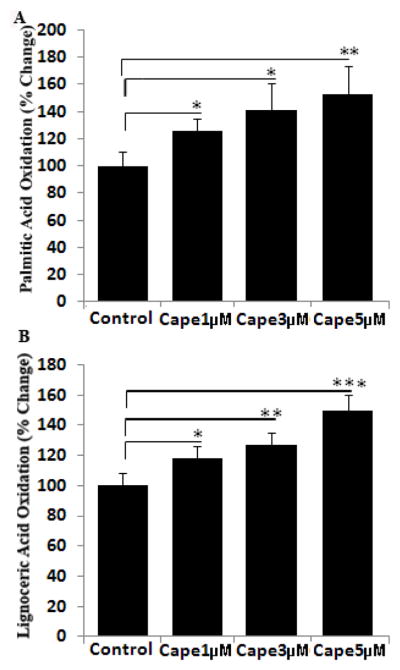
CAPE stimulates the β-oxidation of lignoceric and palmitic acid in cultured control normal human skin fibroblasts in a dose-dependent manner. Cells were incubated in serum-containing DMEM with different concentrations of CAPE for 3 days. After every 24 h, medium was replaced with the addition of fresh reagents. Palmitic (A) and lignoceric acid (B) β-oxidation activity was measured as mentioned in Materials and Methods. Values are mean±S.D. of three different experiments. *P<0.05, **P<0.01, ***P<0.001.
3.3 CAPE upregulates ABCD2 and ABCD3 mRNA expression and protein levels in fibroblasts from X-ALD patients
Human X-ALD fibroblasts were cultured in the presence of CAPE for 3 days and ABCD2 mRNA expression was quantified by real time RT-PCR. Treatment with CAPE resulted in a dose-dependent increase in ABCD2 mRNA expression in three different X-ALD cell lines (Fig. 3). Highest dose of CAPE (5μM) increased the ABCD2 mRNA expression by 12.4 (GM04932), 16.0 (GM04934) and 2.8 (GM07530)-fold in 3 days in 3 different cell lines. Much like ABCD2, the overxpression of ABCD3 has also been shown to be able to correct the defect in peroxisomal β-oxidation activity in ABCD1 deficient cells from X-ALD patients [43]. In all 3 cell lines, we observed a maximum 5.35 (GM04932), 11.0 (GM04934), and 2.77 (GM07530)-fold increase in ABCD3 mRNA expression after treatment with 5μM CAPE for 3 days (Fig. 4).
Figure 3.
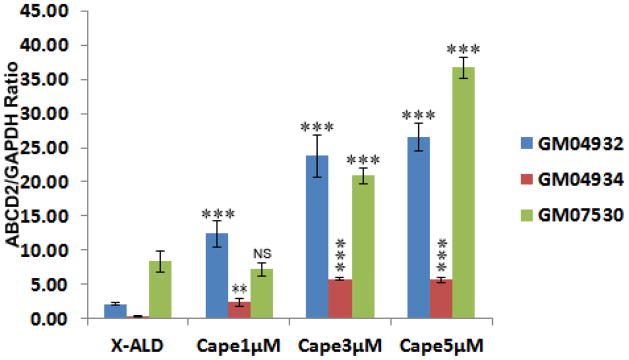
ABCD2 mRNA expression is induced by CAPE in human X-ALD skin fibroblasts. ABCD2 expression levels in human skin fibroblasts derived from X-ALD (GM04932, GM04934), and AMN (GM07530) treated with CAPE in a dose dependent manner. ABCD2 mRNA levels were determined by quantitative realtime RT–PCR and normalized to GAPDH. Data are represented as mean±SD of three different experiments. *P<0.05, **P<0.01; ***P<0.001 CAPE treated compared to respective untreated X-ALD fibroblasts; NS, non-significant
Figure 4.
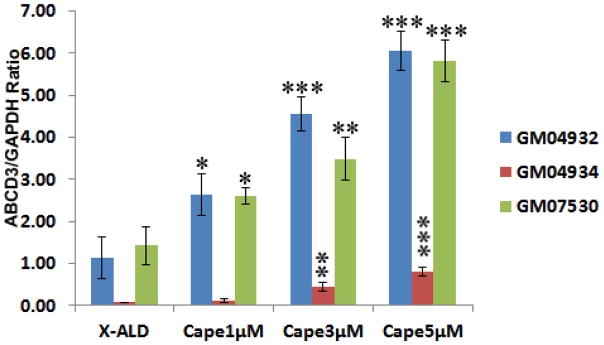
ABCD3 mRNA expression is induced by CAPE in human X-ALD skin fibroblasts. ABCD3 expression levels in human skin fibroblasts derived from X-ALD (GM04932, GM04934) and AMN (GM07530) treated with CAPE in a dose dependent manner. ABCD3 mRNA levels were determined by quantitative realtime RT–PCR and normalized to GAPDH. Data are represented as mean±SD of three different experiments. *P<0.05, **P<0.01, ***P<0.001 CAPE treated compared to respective untreated X-ALD fibroblasts; NS, non-significant.
Western blots for ABCD2 and ABCD3 were performed on carbonate membranes (integral membrane proteins) obtained from X-ALD fibroblast (GM04932) with or without CAPE treatment. ABCD2 was increased 2.4-fold while ABCD3 was induced 3-fold (Fig. 5) in CAPE-treated X-ALD fibroblasts. Screening for Na+/K+-ATPase (plasma membrane protein), used as indicator of protein loading for membrane fractions, did not show an appreciable difference between the same cell samples.
Figure 5.
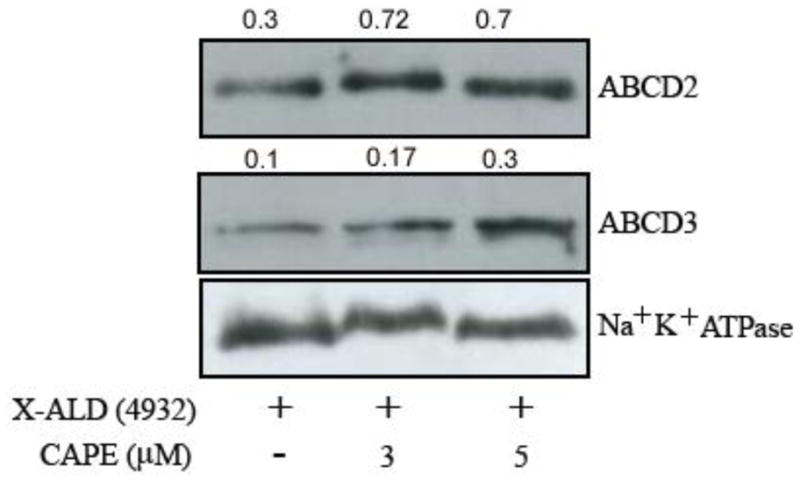
CAPE upregulates the levels of ABCD2 and ABCD3 proteins in X-ALD human skin fibroblasts. Human fibroblasts derived from an X-ALD patient (GM04932) were cultured and used to analyze the levels of peroxisomal integral membrane protein transporters ABCD2 and ABCD3 by Western blot in membranes fractions obtained by carbonate treatment (membrane preparation containing integral membrane proteins), as indicated in Methods section. Na+/K+-ATPase (plasma membrane protein) was used as indicator of protein loading for plasma membrane fractions.
3.4 Induction of ABCD2 and ABCD3 by CAPE correlated with normalization of X-ALD biochemical phenotype
To assess the therapeutic potential of CAPE-mediated ALDRP stimulation for X-ALD, we examined the effect of CAPE treatment on peroxisomal and mitochondrial β-oxidation activity in cultured XALD fibroblasts using radiolabeled fatty acids. Lignoceric acid β-oxidation activity was significantly low in all the X-ALD cell lines (approximately 47% compared to control human fibroblasts) due to the deletion/mutation of ABCD1 [44]. Treatment of X-ALD fibroblasts with 1–5μM CAPE for 3 days showed a dose-dependent increase in lignoceric acid oxidation with maximum of 71%, 78% and 58% respectively in GM04932, GM04934 and GM07530 (Fig. 6A). Palmitic acid β-oxidation activity was also increased in a dose-dependent manner (114%, 108% and 117% respectively in GM04932, GM04934 and GM07530) following CAPE treatment for 3 days (Fig. 6B). The induction of lignoceric acid and palmitic acid β-oxidation by CAPE suggests that it affects both mitochondrial and peroxisomal β-oxidation. Although the precise function of ALDP in the metabolism of VLCFA is not known at the present time; however, accumulation of VLCFA in X-ALD cells with loss or mutations in ALDP and their normalization following transfection of cDNA for ALDP indicate a role of ALDP in the metabolism of VLCFA [16]. Levels of C26:0 and the ratios of C26:0/C22:0 or C24:0/C22:0 are widely used for the diagnosis of X-ALD and other peroxisomal disorders. C26:0/C22:0 ratio was increased 3.96, 5.4 and 4.6-fold respectively in three X-ALD patient fibroblast cell lines (GM04932, GM04934 and GM07530 respectively) compared to normal healthy control fibroblasts (Fig. 6C). Normalization of C26:0/C22:0 following transfection of cDNA for ALDP indicates a role of ALDP in the metabolism of VLCFA. Also, overexpression of ABCD2 leads to reduced VLCFA accumulation in cultured fibroblasts from X-ALD patients [22]. Therefore, we next attempted to examine whether CAPE was able to lower the levels of VLCFA in X-ALD fibroblast cell lines. Treatment of X-ALD fibroblasts with CAPE (5μM) for 3 days significantly reduced (20.2%, 65.21% and 45.2% respectively) the levels of VLCFA (C26:0) in the three X-ALD cell lines (Fig. 6C). In addition to saturated VLCFA, X-ALD fibroblasts have significantly higher levels (37.5%, P<0.01) of monounsaturated VLCFA (C26:1) (Fig. 6D), indicating that β-oxidation of C26:1 is also reduced in X-ALD fibroblasts, and the plasma and brain of X-ALD patients [4, 17, 45, 46]. Treatment of human X-ALD fibroblasts with CAPE also resulted in a significant decrease (54.54% (P<0.01)) in the levels of C26:1 in a dose-dependent manner (Fig. 6D).
Figure 6.

CAPE stimulates the β-oxidation activities and lowers the levels of saturated and monounsaturated VLCFA in human X-ALD skin fibroblasts. Human skin fibroblasts from ALD (GM04932, GMo04934) and AMN (GM07530) patients were incubated in serum-containing DMEM with different concentrations of CAPE and lignoceric (A) and palmitic (B) acid β-oxidation activities were measured after 3 days as described in Materials and Methods. After every 24 h, medium was replaced with the addition of fresh reagents. FAME was prepared directly from cells as described in Material and Methods. FAs were analyzed by GC after adding C27:0 as an internal standard. C26.0, C26:1, and C22:0 were measured as a percent of total FAs and expressed as ratio of C26:0/C22:0 (C) and C26:1/C22:0 (D). Results represent the means ± SD of duplicates from three different experiments.*P<0.05; **P<0.01; ***P<0.001; NS, non- significant.
3.5 CAPE reduces the expression of ELOVL1 in X-ALD fibroblasts
The VLCFA which accumulate in X-ALD are partly absorbed from the diet [47], but mostly result from endogenous synthesis through elongation of long chain fatty acids [4]. Seven elongases have been identified in mammals and are designated as ELOVL1–7 [48]. Knockout studies identified ELOVL1 as the single elongase catalyzing the synthesis of both saturated (C26:0) and monounsaturated (C26:1) VLCFA in humans [4]. There was no significant difference in the levels of ELOVL1 in control and X-ALD fibroblasts (Fig. 7) as reported earlier [4], indicating that availability of substrate is the limiting factor for their elongation. CAPE treatment of control and X-ALD human fibroblasts significantly decreased the ELOVL1 expression (32.25% (P<0.005)) for X-ALD fibroblasts at 5μM dose. The results highlight the dual effect of CAPE on induction of β-oxidation activity and inhibition of VLCFA elongation, ultimately resulting in a net effect of lowering of the levels of both saturated and unsaturated VLCFAs.
Figure 7.
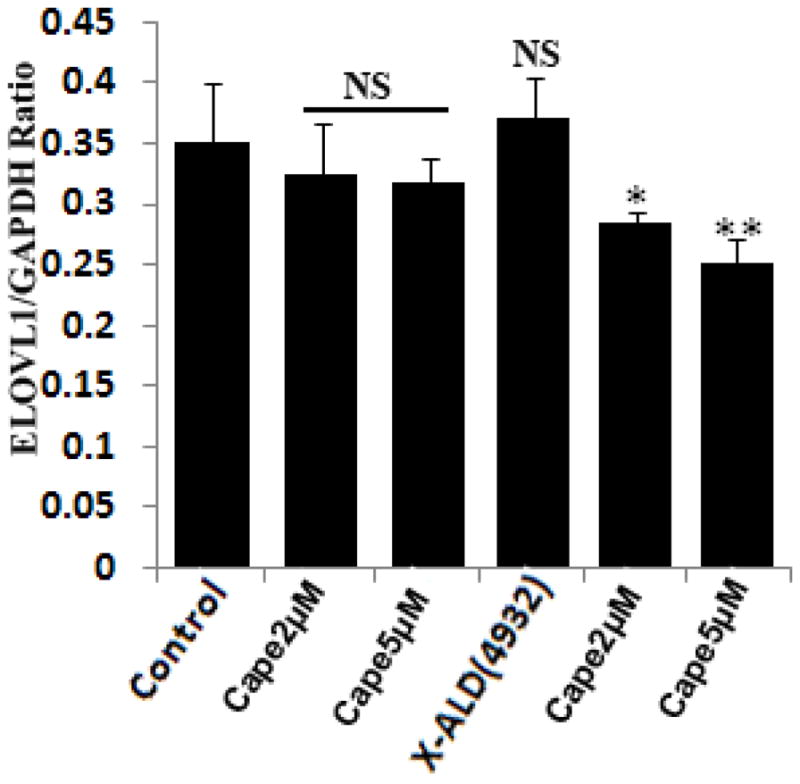
CAPE treatment lowers the mRNA expression of ELOVL1 in X-ALD human skin fibroblasts. Control and X-ALD human skin fibroblasts were treated dose-dependently with CAPE for 3 days and mRNA expression of ELOVL1 was quantified by real time RT-PCR normalized to GAPDH. Data are represented as mean±SD. *P<0.05, **P<0.01 CAPE treatment compared with untreated human X-ALD fibroblasts; NS, non-significant.
3.6 Abcd2 expression is upregulated by CAPE in mouse primary mixed glial cells and Abcd1-deficient U87 astrocytes and B12 oligodendrocytes
A possible role for ABCD2 in clinical course of the X-ALD disease has already been suggested because its expression is maximal in the brain and adrenals [49]. We, therefore, investigated the effect of CAPE treatment on Abcd2 expression in primary mouse mixed glial cultures, and Abcd1-deficient human U87 astrocytes and rat B12 oligodendrocytes stably silenced using lentiviral vectors. Abcd2 expression was significantly (1.5-fold, P<0.01) increased by CAPE in mouse primary mixed glial cultures (Fig. 8A). Also, CAPE significantly (4.28-fold, P<0.001) upregulated the MBP expression in mouse primary mixed glial cultures (Fig. 8B).
Figure 8.
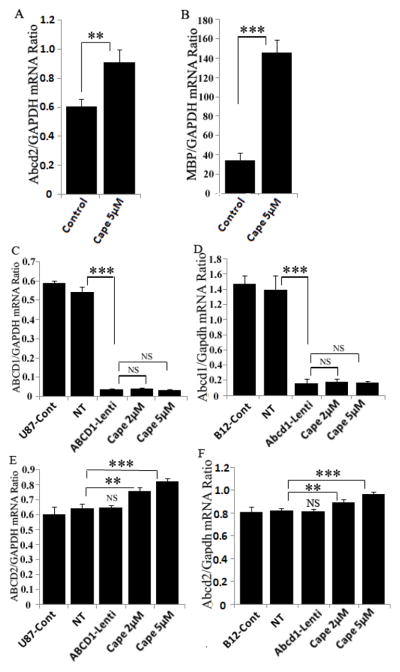
CAPE treatment upregulates Abcd2 expression in mouse primary mixed glial cells and Abcd1-deficient U87 and B12 oligodendrocytes. Mouse mixed glial cultures were treated with CAPE everyday for three days and mRNA expression of Abcd2 (A) and MBP (B) was quatified with real time RT-PCR. Pool of three GFP-tagged lentiviral-shRNAs for Abcd1 was used for transduction of human U87 astrocytoma (C) and rat B12 oligodendrocytes (D). Cells were tranduced with either non-targeting (NT) or Abcd1-Lentiviral (Abcd1-Lenti) particles. Lentiviral-shRNAs induced gene silencing, indicated by a significant decrease in Abcd1 expression, analyzed by qRT-PCR (C and D). There was no change in Abcd1 mRNA expression (C and D) in Abcd1-deficient B12 oligodendrocytes treated with CAPE. Abcd2 mRNA expression in control, NT and Abcd1-deficient U87 astrocytes (E) and B12 oligodendrocytes (F) (n =3) treated with CAPE dose-dependently. Data are represented as mean±SD. **P<0.01; ***P<0.001; NS, Non-significant.
Cells silenced for Abcd1 were selected with puromycin (3μg and 2μg/ml for U87 astrocytes and B12 oligodendrocytes, respectively) and the selected cells were maintained in 0.5μg/ml puromycin. Cells were harvested for mRNA quantification of Abcd1, Abcd2 and Abcd3. Abcd1 expression was significantly reduced (P<0.001) in both U87 astrocytes and B12 oligodendrocytes (Fig 8C and D). CAPE treatment had no effect on Abcd1 and Abcd3 (data not shown) expression however there was a small but significant increase in Abcd2 expression (1.26-fold for U87 and 1.18-fold for B12 oligodendrocyte and 5μM dose) in both the cell types (Fig. 8E and F). Thus Abcd2 is responsive to CAPE in glial cells.
3.7 CAPE treatment upregulates peroxisomal and mitochondrial β-oxidation activities and inhibits ELOVL1 expression resulting in downregulation of VLCFA levels
Loss or mutation of ABCD1 results in deficient β-oxidation of VLCFA [44]. Since CNS is the target organ for X-ALD therapy we investigated the peroxisomal (lignoceric acid, C24:0) and mitochondrial (palmitic acid, C16:0) β-oxidation activities in Abcd1-deficient U87 astrocytes and B12 oligodendrocytes. While Abcd1-deletion did not effect palmitic acid oxidation, the lignoceric β-oxidation was significantly (40%, P<0.01) reduced in Abcd1-deficient U87 astrocytes (Fig. 9A). In Abcd1-deficient B12 oligodendrocytes both palmitic acid (17%, P<0.05) and lignoceric acid (44.44%, P<0.001) β-oxidations were significantly reduced (Fig. 9B). Since CAPE treatment increased the plamitic and lignoceric acid β-oxidation in X-ALD fibroblasts (Fig. 6A) and increased the Abcd2 expression in Abcd1-deficient U87 astrocytes and B12-oligodendrocytes, we evaluated the effect of this induction on β-oxidation of lignoceric and palmitic acid. Treatment with CAPE significantly increased the lignoceric acid β-oxidation in both Acd1-deficient U87 astrocytes (55%, P<0.01) and B12 oligodendrocytes (60%, P<0.001) (Fig. 9A and B respectively). Furthermore, the levels of VLCFA are also regulated by elongases (ELOVL1 and ELOVL3) [4, 17]. Abcd1-deletion had no effect on ELOVL1 expression in U87 astrocytes but significantly induced ELOVL1 expression (10.5-fold) in B12 oligodendrocytes. Treatment with CAPE lowered the levels of ELOVL1 in both ABCD1-deficient U87 astrocytes (16.76%) and B12-oligodendrocytes (90%) (Fig. 9C and D) but had no effect on ELOVL3 expression that was induced in both Abcd1-deficient U87 astrocytes and B12 oligodendrocytes (data not shown). The results highlight the overall beneficial potential of CAPE to affect the fatty acid machinery in X-ALD by increasing the β-oxidation acitivites and lowering the expression of ELOVL1.
Figure 9.

CAPE stimulates the β-oxidation activities of lignoceric and palmitic acid, and inhibits ELOVL1 expression in Abcd1-deficient U87 astrocytes and B12 oligodendrocytes. Palmitic and lignoceric β-oxidation activities were measured in Abcd1-deficient U87 astrocytes (A) and B12 oligodendrocytes (B) incubated in serum-containing DMEM with different concentrations (μM) of CAPE for 3 days as described in Materials and Methods. After every 24 h, medium was replaced with the addition of fresh reagents. Abcd1-deficient U87 astrocytes (C) B12 oligodendrocytes (D) were treated dose-dependently with CAPE for 3 days and mRNA expression of ELOVL1 was quantified in control, NT, and untreated/treated Abcd1-deficient cells by qRT-PCR normalized to GAPDH. FAME was prepared directly from cells as described in Material and Methods. FAs were analyzed in U87 astrocytes (E) and B12 oligodendrocytes (F) by GC after adding C27:0 as an internal standard. C26.0, C26:1, and C22:0 were measured as a percent of total FAs and expressed as ratio of C26:0/C22:0. *P<0.05; **P<0.01; ***P<0.001; NS, non-significant.
Figure 9E shows a 2.1-fold increase in the C26:0/C22:0 ratio in Abcd1-deficient U87 astrocytes and a 4.1-fold increase in Abcd1-deficient B12 oligodendrocytes when compared with control and NT-treated astrocytes. Since CAPE increased the β-oxidation activity of lignoceric acid in Abcd1-deficient U87 astrocytes and B12 oligodendrocytes, we also examined the effect of CAPE on the levels of VLCFA in these cells. A significant decrease was observed in VLCFA levels in CAPE (5μM) treated Abcd1-deficient U87 astrocytes (1.5-fold, P<0.01) as well as B12 oligodendrocytes (2.63-fold, P<0.01) (Fig. 9E and F). These observations document that increased accumulation of VLCFA as a result of Abcd1-deficiency in astrocytes and oligodendrocytes can be attenuated by CAPE treatment.
3.8 CAPE inhibits proinflammatory cytokines and oxidative stress in Abcd1/2-silenced mouse primary astrocytes
Accumulation of VLCFA in X-ALD patients leads to secondary injury of inflammatory demyelination due to a marked increase in the activation of microglia and astrocytes, and thus the production of proinflammatory cytokines (TNF-α and IL-1β) [50, 51]. Expression of TNF-α and iNOS was higher in inflammatory areas compared with normal areas of cALD brain [52]. Therapeutic reduction of the levels of VLCFA correlates with decreased expression of proinflammatory cytokines [10]. Our laboratory has recently reported the accumulation of VLCFA and increased production of ROS and proinflammatory cytokines (iNOS, TNF-α and IL- 1β) in Abcd1/2 silenced mouse primary astrocytes [10]. Expression of iNOS and TNF-α was significantly increased in Abcd1/2-silenced mouse astrocytes. Therefore, we evaluated the effect of CAPE on ROS and proinflammatory cytokines in Abcd1/2-silenced mouse primary astrocytes (Fig. 10). CAPE at 5μM concentration significantly inhibited (26%, P<0.001) ROS generation in Abcd1/2-silenced mouse astrocytes (Fig. 10A). Treatment with CAPE also downregulated the NO (92%), iNOS (50%) and TNF-α (42%, P<0.001) protein levels at 5μM dose in Abcd-1/2 silenced mouse astrocytes (Fig. 10B and C).
Figure 10.

CAPE inhibits reactive oxygen species (ROS) and expression of proinflammatory cytokines (iNOS and TNF-α) in Abcd-1/2-silenced mouse primary astrocytes. ROS generation in primary astrocytes after silencing or CAPE treatment (A). Nitrite (B) and TNF-α (C) were measured by ELISA in the supernatant of primary astrocytes after silencing or CAPE treatment. For the detection of iNOS, 5LOX and p65 protein expression by immunoblot (D) in response to Abcd1/2 silencing or CAPE treatment, cell lysate from astrocytes was prepared as described in Material methods. *P<0.05; **P<0.01; ***P<0.001; NS, non-significant.
A growing body of evidence suggests the role of certain immunomodulatory leukotrienes [53] in the signaling cascade of inflammatory gene expression [9]. 5-LOX expression was increased in Abcd1/2-silenced mouse astrocytes, which was significantly decreased (80%) by treatment with CAPE (Fig. 10D). Mechanisms of Abcd1/2-silencing-induced proinflammatory response involve activation of NF-κB [10]. Western blots showed increased levels of p65 protein in Abcd1/2-silenced mouse primary astrocytes (Fig. 10D). The level of p65 in the nucleus of Abcd1/2 silenced astrocytes was significantly reduced (63%) by CAPE at 5μM dose. Since CAPE has the ability to cross the blood brain barrier [29], downregulates inflammatory mediators and corrects abnormality of VLCFA, it may have the potential for X-ALD therapy.
4. Discussion
The present study documents the possible therapeutic intervention against pathognomic accumulation of VLCFA in X-ALD. CAPE lowered the cellular content of VLCFA levels in human skin fibroblasts from X-ALD patients. The mechanism leading to decreased VLCFA levels in X-ALD fibroblasts is likely due to higher expression of ABCD2 and/or ABCD3 resulting in increased peroxisomal β-oxidation and decreased chain elongation upon treatment with CAPE. We further observed CAPE induction of Abcd2 gene and lowering of VLCFA levels in Abcd1-deficient U87 astrocytes and B12 oligodendrocytes together with inhibition of proinflammatory cytokines in Abcd1/2-silenced mouse astrocytes.
Mutations in the ABCD1 gene encoding the peroxisomal ABC transporter ABCD1 are the initial cause of X-ALD regardless of clinical variant. The role of VLCFA in the pathogenesis of X-ALD is largely unknown. VLCFA levels in the plasma do not correlate with the patient’s phenotype. However, as VLCFA accumulation is the only biochemical abnormality identified in X-ALD, it seems plausible that this accumulation is somehow related to the eventual development of neurological symptoms [10]. Pharmacological therapy for a genetic disease is aimed at upregulating redundant genes to compensate for biochemical defects. CAPE did not induce the expression of ABCD1 in control normal human fibroblasts. This is in line with the previous findings where ABCD1 was not induced pharmacologically [54, 55]. Overexpression of Abcd2 is able to correct the defective catabolism of VLCFA in Abcd1-deficient mice and even to prevent the development of a late onset neurodegenerative phenotype caused by loss of function of Abcd1 [16, 56]. Treatment with CAPE increased the expression of ABCD2 in control and X-ALD fibroblasts. Beyond the demonstration that ABCD2 responds to CAPE treatment, our study also found increased expression of ABCD3 in CAPE treated control and X-ALD fibroblasts although to a lesser extent than ABCD2. Differential upregulation of ABCD2 and/or ABCD3 in three different X-ALD fibroblast cell lines in response to CAPE treatment could be attributed to individual background, genetic differences and/or kind of mutation or deletion in ABCD1. Additionally, fibroblasts in culture undergo random modification of gene expression by somatic mutation or epigenetic regulation [57]. Also, a dominant negative effect of ALDP mutation and/or metabolites may have an impact on the expression of other ABC transporters. A recent study [31] reported the upregulation of SMN2 gene by caffeic acid as a target therapy for spinal muscular atrophy (SMA) caused by loss of SMN1 gene. The possible mechanism suggested was the HDAC inhibition activity of caffeic acid. Previously HDAC inhibitors 4-PBA and VPA have been shown to upregulate ABCD2 expression [21, 22]. However, both the drugs were required in millimolar concentrations to observe the increase in β-oxidation activities. Furthermore, while VPA failed to lower the levels of VLCFA, in longterm studies with PBA the VLCFA levels returned to pretreatment levels [23]. Thus there is a continuous need to test newer drugs especially if they demonstrate a greater efficacy and low dose requirement.
Although ALDRP is apparently unable to compensate for ALDP deficiency at the intrinsic levels of expression in humans with X-ALD and Abcd1-deficient mice, its overexpression partially restores VLCFA β-oxidation [15, 17, 22, 58]. Previously, studies from our laboratory [44, 59–61] and others [62] have documented that VLCFA metabolism is restricted to peroxisomes while LCFA β-oxidation is attributable largely to mitochondria. Treatment with CAPE, at doses that upregulated ALDRP expression, significantly increased both VLCFA and LCFA β-oxidation in control and X-ALD fibroblasts. Treatment with CAPE at micromolar dose lowered the levels of C26:0 in a dose-dependent manner in human skin fibroblasts from X-ALD patients. In addition to saturated (C26:0), monounsaturated VLCFA (C26:1) also accumulate in X-ALD tissues, implicating both C26:0 and C26:1 in the X-ALD pathology [21]. Similar to C26:0 [44], C26:1 is also β-oxidized in peroxisomes and ABCD2 plays a prominent role in their catabolism [17, 21, 63, 64]. Accordingly, treatment with CAPE significantly reduced the C26:1 levels in X-ALD fibroblasts, suggesting a causal relationship between ALDRP expression and levels of monounsaturated VLCFA. A deficiency in ALDP impairs VLCFA β-oxidation activity and the resultant accumulation of VLCFA acyl-CoA esters is further elongated by ELOVL1, the human C26:0 specific elongase [4]. Furthermore, ELOVL1 knockdown [4] or pharmacological inhibition [17] lowers the level of C26:0. Treatment with CAPE significantly lowered the expression of ELOVL1 in X-ALD fibroblasts.
Glial cells (astrocytes and oligodendrocytes) are among the cell types mainly affected in X-ALD [52, 66]. We, therefore investigated the effect of CAPE on VLCFA metabolism in Abcd1-deficient U87 astrocytes and B12-oligodendrocytes. Treatment with CAPE treatment induced Abcd2 expression in both the cell types though the degree of increase was much less compared to X-ALD fibroblasts. More impressively CAPE significantly upregulated the C16:0 as well as C24:0 oxidation and lowered the VLCFA levels in both Abcd1-deficient U87 astrocytes and B12 oligodendrocytes. Interestingly, Abcd1 deletion significantly induced the expression of ELOVL1 in B12-oligodendrocytes (Fig. 9D). While the reason(s) for this differential increase in expression of ELOVL1 in Abcd1-deficient B12 oligodendrocyte remains to be investigated, it is in line with the previous finding that ELOVL1 expression was significantly increased in oligodendrocytes differentiated from cALD patient-derived induced pluripotent stem cells (iPSCs) [67]. Furthermore, Asheuer et al., [43] reported higher C26:0 levels in normal appearing white matter of patients with the childhood cerebral phenotype than in those with AMN. The individual contributions of Abcd2 induction and the inhibition of chain elongation activities toward the normalization of VLCFA load following pharmacological treatment are not known. Our data indicate that possibly both reduced ELOVL1 expression thus decreased synthesis of VLCFA and increased expression of Abcd2 thus increased peroxisomal β-oxidation of VLCFA following CAPE treatment contribute to the reduced levels of VLCFA in glial cells. For a therapy to progress beyond the experimental systems and move into clinic, especially for diseases involving CNS, the ability to access CNS through BBB is critical. In silico screening strategies have shown that CAPE possesses the biochemical properties necessary to cross the BBB [29].
The consistent hallmark of X-ALD is an excessive accumulation of VLCFA with subsequent involvement of CNS accompanied by induction of proinflammatory cytokines (TNF-α and IL-β). At present, it is not known how the inherited metabolic abnormality of VLCFA accumulation subsequently triggers a neuroinflammatory response in X-ALD brain. As the metabolic defect appears much prior to neuroinflammatory disease, VLCFA by themselves or as a constituent of complex lipids act as a trigger for the inflammatory response as was documented recently by our laboratory [10]. Therefore, drugs that inhibit the inflammatory response in brain would also help in ameliorating the disease. There is also growing evidence that oxidative stress contributes to the pathogenesis of X-ALD and that excess of VLCFAs plays a role in this process [66]. CAPE, unlike other antioxidants, specifically suppresses NF-κB binding to DNA without affecting IκBα degradation [26] and is also a specific lipoxygenase inhibitor [30]. Abcd1/2-silencing of wild type mouse primary astrocytes induced a proinflammatory response (iNOS and proinflammatory cytokines) [10], which was mediated by the activation of NF-κB and increased levels of ROS [10]. CAPE treatment of Abcd1/2 silenced mouse astrocytes reduced the expressions of iNOS and TNF-α (mRNA and protein), suppressed NF-κB activation and reduced the ROS production.
The observed increased activation of lipoxidative enzymes in X-ALD brain [9] and in Abcd1/2 silenced astrocytes [10] suggests the role of these proinflammatory mediators in the pathobiology of X-ALD. Treatment with CAPE also reduced the activation of 5-LOX in Abcd1/2-silenced astrocytes. Our observation of inhibition of NF-κB activation by CAPE in Abcd1/2-silenced astrocytes is consistent with previous studies from our [30] and other laboratories reporting inhibition of proinflammatory mediators and NF-κB activity by CAPE in glial cell cultures [68] and in mouse brain [69]. In summary, our observations describe upregulation of peroxisomal β-oxidation activity and inhibition of chain elongation activity, leading to the correction of VLCFA levels and inhibition of inflammatory response in Abcd1/2-silenced astrocytes by CAPE. The ability of CAPE to cross BBB [30] and the observed inhibition of iNOS in the brain [69], and the documented neuroprotection against ischemic brain injury [30] by CAPE indicate that it is a good candidate drug for neurological disorders in general and for X-ALD in particular.
Highlights.
CAPE upregulated ABCD2 expression in human skin fibroblasts from X-ALD patients.
ABCD2 expression was increased in Abcd1-deficient astrocytes and oligodendrocytes.
Both C26:0 and C26:1 VLCFA levels were reduced in X-ALD patient fibroblasts.
CAPE inhibited inflammatory response in Abcd1/2-silenced mouse primary astrocytes.
CAPE may have therapeutic potential for X-ALD.
Acknowledgments
We thank the members of our laboratory for their valuable comments and help during this study. We greatly appreciate the help of Ms. Joyce Bryan and Ms. Chara Williams for technical assistance. This study was supported in part by grants from the National Institutes of Health: NS-22576, NS-37766, C06 RR018823, C06 RR015455, and VA merit award BX1072-01.
Abbreviations used
- X-ALD
X-adrenoleukodystrophy
- ALDP
adrenoleukodystrophy protein
- ALDRP
adrenoleukodystrophy related protein
- AMN
adrenomyeloneuropathy
- cALD
cerebral adrenoleukodystrophy
- DMEM
dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HBSS
Hanks’ balanced salt solution
- FAME
fatty acid methyl ester
- ELOVL
elongase of very long chain fatty acid
- GC
gas chromatography
- ROS
reactive oxygen species
- Scr
scrambled
- CAPE
caffeic acid phenethyl Ester
- MBP
myelin basic protein
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- TNF-α
tumor necrosis factor-α
- VLCFA
very long chain fatty acid
- 5-LOX
5-lipoxygenase
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
- VPA
valproic acid
- SMN
survival of motor neuron
- NT
non-targeting
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moser HW. Lorenzo oil therapy for adrenoleukodystrophy: a prematurely amplified hope. Ann Neurol. 1993;34:121–122. doi: 10.1002/ana.410340202. [DOI] [PubMed] [Google Scholar]
- 2.Contreras M, Mosser J, Mandel JL, Aubourg P, Singh I. The protein coded by the X-adrenoleukodystrophy gene is a peroxisomal integral membrane protein. FEBS Lett. 1994;344:211–215. doi: 10.1016/0014-5793(94)00400-5. [DOI] [PubMed] [Google Scholar]
- 3.Contreras M, Sengupta TK, Sheikh F, Aubourg P, Singh I. Topology of ATP-binding domain of adrenoleukodystrophy gene product in peroxisomes. Arch Biochem Biophys. 1996;334:369–379. doi: 10.1006/abbi.1996.0467. [DOI] [PubMed] [Google Scholar]
- 4.Ofman R, Dijkstra IM, van Roermund CW, Burger N, Turkenburg M, van Cruchten A, van Engen CE, Wanders RJ, Kemp S. The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO Mol Med. 2010;2:90–97. doi: 10.1002/emmm.201000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Biase A, Di Benedetto R, Fiorentini C, Travaglione S, Salvati S, Attorri L, Pietraforte D. Free radical release in C6 glial cells enriched in hexacosanoic acid: implication for X-linked adrenoleukodystrophy pathogenesis. Neurochem Int. 2004;44:215–221. doi: 10.1016/s0197-0186(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 6.Fourcade S, Lopez-Erauskin J, Galino J, Duval C, Naudi A, Jove M, Kemp S, Villarroya F, Ferrer I, Pamplona R, Portero-Otin M, Pujol A. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet. 2008;17:1762–1773. doi: 10.1093/hmg/ddn085. [DOI] [PubMed] [Google Scholar]
- 7.Hein S, Schonfeld P, Kahlert S, Reiser G. Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet. 2008;17:1750–1761. doi: 10.1093/hmg/ddn066. [DOI] [PubMed] [Google Scholar]
- 8.Ho JK, Moser H, Kishimoto Y, Hamilton JA. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest. 1995;96:1455–1463. doi: 10.1172/JCI118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan M, Singh J, Gilg AG, Uto T, Singh I. Very long-chain fatty acid accumulation causes lipotoxic response via 5-lipoxygenase in cerebral adrenoleukodystrophy. J Lipid Res. 2010;51:1685–1695. doi: 10.1194/jlr.M002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh J, Khan M, Singh I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X-adrenoleukodystrophy. J Lipid Res. 2009;50:135–147. doi: 10.1194/jlr.M800321-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schluter A, Espinosa L, Fourcade S, Galino J, Lopez E, Ilieva E, Morato L, Asheuer M, Cook T, McLaren A, Reid J, Kelly F, Bates S, Aubourg P, Galea E, Pujol A. Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum Mol Genet. 2012;21:1062–1077. doi: 10.1093/hmg/ddr536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan M, Pahan K, Singh AK, Singh I. Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem. 1998;71:78–87. doi: 10.1046/j.1471-4159.1998.71010078.x. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro E, Krivit W, Lockman L, Jambaque I, Peters C, Cowan M, Harris R, Blanche S, Bordigoni P, Loes D, Ziegler R, Crittenden M, Ris D, Berg B, Cox C, Moser H, Fischer A, Aubourg P. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–718. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- 14.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 15.Netik A, Forss-Petter S, Holzinger A, Molzer B, Unterrainer G, Berger J. Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): implications for therapy. Hum Mol Genet. 1999;8:907–913. doi: 10.1093/hmg/8.5.907. [DOI] [PubMed] [Google Scholar]
- 16.Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N, Ruiz M, Pampols T, Giros M, Mandel JL. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet. 2004;13:2997–3006. doi: 10.1093/hmg/ddh323. [DOI] [PubMed] [Google Scholar]
- 17.Singh J, Khan M, Singh I. HDAC inhibitor SAHA normalizes the levels of VLCFAs in human skin fibroblasts from X-ALD patients and downregulates the expression of proinflammatory cytokines in Abcd1/2-silenced mouse astrocytes. J Lipid Res. 2011;52:2056–2069. doi: 10.1194/jlr.M017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh I, Pahan K, Khan M. Lovastatin and sodium phenylacetate normalize the levels of very long chain fatty acids in skin fibroblasts of X- adrenoleukodystrophy. FEBS Lett. 1998;426:342–346. doi: 10.1016/s0014-5793(98)00370-6. [DOI] [PubMed] [Google Scholar]
- 19.Uto T, Contreras MA, Gilg AG, Singh I. Oxidative imbalance in nonstimulated X-adrenoleukodystrophy-derived lymphoblasts. Dev Neurosci. 2008;30:410–418. doi: 10.1159/000191212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pai GS, Khan M, Barbosa E, Key LL, Craver JR, Cure JK, Betros R, Singh I. Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients. Mol Genet Metab. 2000;69:312–322. doi: 10.1006/mgme.2000.2977. [DOI] [PubMed] [Google Scholar]
- 21.Fourcade S, Ruiz M, Guilera C, Hahnen E, Brichta L, Naudi A, Portero-Otin M, Dacremont G, Cartier N, Wanders R, Kemp S, Mandel JL, Wirth B, Pamplona R, Aubourg P, Pujol A. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum Mol Genet. 2010;19:2005–2014. doi: 10.1093/hmg/ddq082. [DOI] [PubMed] [Google Scholar]
- 22.Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261–1268. doi: 10.1038/3242. [DOI] [PubMed] [Google Scholar]
- 23.McGuinness MC, Zhang HP, Smith KD. Evaluation of pharmacological induction of fatty acid beta-oxidation in X-linked adrenoleukodystrophy. Mol Genet Metab. 2001;74:256–263. doi: 10.1006/mgme.2001.3239. [DOI] [PubMed] [Google Scholar]
- 24.Nagaoka T, Banskota AH, Tezuka Y, Saiki I, Kadota S. Selective antiproliferative activity of caffeic acid phenethyl ester analogues on highly liver-metastatic murine colon 26-L5 carcinoma cell line. Bioorg Med Chem. 2002;10:3351–3359. doi: 10.1016/s0968-0896(02)00138-4. [DOI] [PubMed] [Google Scholar]
- 25.Sud’ina GF, Mirzoeva OK, Pushkareva MA, Korshunova GA, Sumbatyan NV, Varfolomeev SD. Caffeic acid phenethyl ester as a lipoxygenase inhibitor with antioxidant properties. FEBS Lett. 1993;329:21–24. doi: 10.1016/0014-5793(93)80184-v. [DOI] [PubMed] [Google Scholar]
- 26.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laranjinha J, Vierira O, Almeida L, Madeira V. Inhibition of metmyoglobin/H2O2-dependent low density lipoprotein lipid peroxidation by naturally occurring phenolic acids. Biochem Pharmacol. 1996;51:395–402. doi: 10.1016/0006-2952(95)02171-x. [DOI] [PubMed] [Google Scholar]
- 28.Boudreau LH, Maillet J, LeBlanc LM, Jean-Francois J, Touaibia M, Flamand N, Surette ME. Caffeic acid phenethyl ester and its amide analogue are potent inhibitors of leukotriene biosynthesis in human polymorphonuclear leukocytes. PLoS One. 2012;7:e31833. doi: 10.1371/journal.pone.0031833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barber SC, Higginbottom A, Mead RJ, Barber S, Shaw PJ. An in vitro screening cascade to identify neuroprotective antioxidants in ALS. Free Radic Biol Med. 2009;46:1127–1138. doi: 10.1016/j.freeradbiomed.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan M, Elango C, Ansari MA, Singh I, Singh AK. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J Neurochem. 2007;102:365–377. doi: 10.1111/j.1471-4159.2007.04526.x. [DOI] [PubMed] [Google Scholar]
- 31.Dayangac-Erden D, Bora-Tatar G, Dalkara S, Demir AS, Erdem-Yurter H. Carboxylic acid derivatives of histone deacetylase inhibitors induce full length SMN2 transcripts: a promising target for spinal muscular atrophy therapeutics. Arch Med Sci. 2011;7:230–234. doi: 10.5114/aoms.2011.22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grunberger D, Banerjee R, Eisinger K, Oltz EM, Efros L, Caldwell M, Estevez V, Nakanishi K. Preferential cytotoxicity on tumor cells by caffeic acid phenethyl ester isolated from propolis. Experientia. 1988;44:230–232. doi: 10.1007/BF01941717. [DOI] [PubMed] [Google Scholar]
- 33.Hoshi M, Kishimoto Y. Synthesis of cerebronic acid from lignoceric acid by rat brain preparation. Some properties and distribution of the -hydroxylation system. J Biol Chem. 1973;248:4123–4130. [PubMed] [Google Scholar]
- 34.Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I. Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 35.Won JS, Choi MR, Suh HW. Stimulation of astrocyte-enriched culture with C2 ceramide increases proenkephalin mRNA: involvement of cAMP-response element binding protein and mitogen activated protein kinases. Brain Res. 2001;903:207–215. doi: 10.1016/s0006-8993(01)02452-0. [DOI] [PubMed] [Google Scholar]
- 36.Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E, Singh AK, Contreras MA. Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res. 2004;1022:1–11. doi: 10.1016/j.brainres.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 37.Wilson R, Sargent JR. Lipid and fatty acid composition of brain tissue from adrenoleukodystrophy patients. J Neurochem. 1993;61:290–297. doi: 10.1111/j.1471-4159.1993.tb03567.x. [DOI] [PubMed] [Google Scholar]
- 38.Albet S, Bentejac M, Savary S, Gondcaille C, Netik A, Berger J, Szpirer C, Troffer-Charlier N, Bugaut M. Rat adrenoleukodystrophy-related (ALDR) gene: full-length cDNA sequence and new insight in expression. Biochim Biophys Acta. 2001;1517:257–269. doi: 10.1016/s0167-4781(00)00291-8. [DOI] [PubMed] [Google Scholar]
- 39.Braiterman LT, Watkins PA, Moser AB, Smith KD. Peroxisomal very long chain fatty acid beta-oxidation activity is determined by the level of adrenodeukodystrophy protein (ALDP) expression. Mol Genet Metab. 1999;66:91–99. doi: 10.1006/mgme.1998.2789. [DOI] [PubMed] [Google Scholar]
- 40.Flavigny E, Sanhaj A, Aubourg P, Cartier N. Retroviral-mediated adrenoleukodystrophy-related gene transfer corrects very long chain fatty acid metabolism in adrenoleukodystrophy fibroblasts: implications for therapy. FEBS Lett. 1999;448:261–264. doi: 10.1016/s0014-5793(99)00379-8. [DOI] [PubMed] [Google Scholar]
- 41.Holzinger A, Kammerer S, Roscher AA. Primary structure of human PMP69, a putative peroxisomal ABC-transporter. Biochem Biophys Res Commun. 1997;237:152–157. doi: 10.1006/bbrc.1997.7102. [DOI] [PubMed] [Google Scholar]
- 42.Kamijo K, Kamijo T, Ueno I, Osumi T, Hashimoto T. Nucleotide sequence of the human 70 kDa peroxisomal membrane protein: a member of ATP-binding cassette transporters. Biochim Biophys Acta. 1992;1129:323–327. doi: 10.1016/0167-4781(92)90510-7. [DOI] [PubMed] [Google Scholar]
- 43.Asheuer M, Bieche I, Laurendeau I, Moser A, Hainque B, Vidaud M, Aubourg P. Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X-linked adrenoleukodystrophy. Hum Mol Genet. 2005;14:1293–1303. doi: 10.1093/hmg/ddi140. [DOI] [PubMed] [Google Scholar]
- 44.Singh I, Moser AE, Goldfischer S, Moser HW. Lignoceric acid is oxidized in the peroxisome: implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc Natl Acad Sci U S A. 1984;81:4203–4207. doi: 10.1073/pnas.81.13.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharp P, Johnson D, Poulos A. Molecular species of phosphatidylcholine containing very long chain fatty acids in human brain: enrichment in X-linked adrenoleukodystrophy brain and diseases of peroxisome biogenesis brain. J Neurochem. 1991;56:30–37. doi: 10.1111/j.1471-4159.1991.tb02558.x. [DOI] [PubMed] [Google Scholar]
- 46.Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab. 2003;79:189–196. doi: 10.1016/s1096-7192(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 47.Kishimoto Y, Moser HW, Kawamura N, Platt M, Pallante SL, Fenselau C. Adrenoleukodystrophy: evidence that abnormal very long chain fatty acids of brain cholesterol esters are of exogenous origin. Biochem Biophys Res Commun. 1980;96:69–76. doi: 10.1016/0006-291x(80)91182-1. [DOI] [PubMed] [Google Scholar]
- 48.Tvrdik P, Westerberg R, Silve S, Asadi A, Jakobsson A, Cannon B, Loison G, Jacobsson A. Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. J Cell Biol. 2000;149:707–718. doi: 10.1083/jcb.149.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lombard-Platet G, Savary S, Sarde CO, Mandel JL, Chimini G. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc Natl Acad Sci U S A. 1996;93:1265–1269. doi: 10.1073/pnas.93.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGuinness MC, Griffin DE, Raymond GV, Washington CA, Moser HW, Smith KD. Tumor necrosis factor-alpha and X-linked adrenoleukodystrophy. J Neuroimmunol. 1995;61:161–169. doi: 10.1016/0165-5728(95)00084-f. [DOI] [PubMed] [Google Scholar]
- 51.Powers JM, Liu Y, Moser AB, Moser HW. The inflammatory myelinopathy of adreno-leukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol. 1992;51:630–643. doi: 10.1097/00005072-199211000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425–439. doi: 10.1016/j.nbd.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 53.Sala A, Zarini S, Bolla M. Leukotrienes: lipid bioeffectors of inflammatory reactions. Biochemistry (Mosc) 1998;63:84–92. [PubMed] [Google Scholar]
- 54.Baarine M, Ragot K, Genin EC, El Hajj H, Trompier D, Andreoletti P, Ghandour MS, Menetrier F, Cherkaoui-Malki M, Savary S, Lizard G. Peroxisomal and mitochondrial status of two murine oligodendrocytic cell lines (158N, 158JP): potential models for the study of peroxisomal disorders associated with dysmyelination processes. J Neurochem. 2009;111:119–131. doi: 10.1111/j.1471-4159.2009.06311.x. [DOI] [PubMed] [Google Scholar]
- 55.Berger J, Albet S, Bentejac M, Netik A, Holzinger A, Roscher AA, Bugaut M, Forss-Petter S. The four murine peroxisomal ABC-transporter genes differ in constitutive, inducible and developmental expression. Eur J Biochem. 1999;265:719–727. doi: 10.1046/j.1432-1327.1999.00772.x. [DOI] [PubMed] [Google Scholar]
- 56.Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer-Charlier N, Broccoli V, Callyzot N, Mooyer P, Selhorst J, Vreken P, Wanders RJ, Mandel JL, Pujol A. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet. 2005;14:3565–3577. doi: 10.1093/hmg/ddi384. [DOI] [PubMed] [Google Scholar]
- 57.Traynor J, Agarwal P, Lazzeroni L, Francke U. Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med Genet. 2002;3:12. doi: 10.1186/1471-2350-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh I, Khan M, Key L, Pai S. Lovastatin for X-linked adrenoleukodystrophy. N Engl J Med. 1998;339:702–703. doi: 10.1056/NEJM199809033391012. [DOI] [PubMed] [Google Scholar]
- 59.Hashmi M, Stanley W, Singh I. Lignoceroyl-CoASH ligase: enzyme defect in fatty acid beta-oxidation system in X-linked childhood adrenoleukodystrophy. FEBS Lett. 1986;196:247–250. doi: 10.1016/0014-5793(86)80256-3. [DOI] [PubMed] [Google Scholar]
- 60.Lazo O, Contreras M, Bhushan A, Stanley W, Singh I. Adrenoleukodystrophy: impaired oxidation of fatty acids due to peroxisomal lignoceroyl-CoA ligase deficiency. Arch Biochem Biophys. 1989;270:722–728. doi: 10.1016/0003-9861(89)90555-9. [DOI] [PubMed] [Google Scholar]
- 61.Lazo O, Contreras M, Hashmi M, Stanley W, Irazu C, Singh I. Peroxisomal lignoceroyl-CoA ligase deficiency in childhood adrenoleukodystrophy and adrenomyeloneuropathy. Proc Natl Acad Sci U S A. 1988;85:7647–7651. doi: 10.1073/pnas.85.20.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lageweg W, Sykes JE, Lopes-Cardozo M, Wanders RJ. Oxidation of very-long-chain fatty acids in rat brain: cerotic acid is beta-oxidized exclusively in rat brain peroxisomes. Biochim Biophys Acta. 1991;1085:381–384. doi: 10.1016/0005-2760(91)90144-7. [DOI] [PubMed] [Google Scholar]
- 63.Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PA, Pampols T, Dacremont G, Wanders RJ, Giros M, Pujol A. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab. 2009;296:E211–221. doi: 10.1152/ajpendo.90736.2008. [DOI] [PubMed] [Google Scholar]
- 64.Genin EC, Geillon F, Gondcaille C, Athias A, Gambert P, Trompier D, Savary S. Substrate specificity overlap and interaction between adrenoleukodystrophy protein (ALDP/ABCD1) and adrenoleukodystrophy-related protein (ALDRP/ABCD2) J Biol Chem. 2011;286:8075–8084. doi: 10.1074/jbc.M110.211912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moser SK, Watkins HW, Powers PA, Moser J, AB . X-linked adrenoleukodystrophy. In: Scriver ABCR, Sly WS, Valle D, editors. The Metabolic and MolecularBases of Inherited Disease. McGraw Hill; NewYork: 2001. pp. 3257–3301. [Google Scholar]
- 66.Singh I, Pujol A. Pathomechanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis. Brain Pathol. 2010;20:838–844. doi: 10.1111/j.1750-3639.2010.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, Yoon TM, Park IH, Hwang DY, Daley GQ, Kim DW. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann Neurol. 2011;70:402–409. doi: 10.1002/ana.22486. [DOI] [PubMed] [Google Scholar]
- 68.Wang G, Daniel BM, DeCoster MA. Role of nitric oxide in regulating secreted phospholipase A2 release from astrocytes. Neuroreport. 2005;16:1345–1350. doi: 10.1097/01.wnr.0000174403.79020.65. [DOI] [PubMed] [Google Scholar]
- 69.Fontanilla CV, Ma Z, Wei X, Klotsche J, Zhao L, Wisniowski P, Dodel RC, Farlow MR, Oertel WH, Du Y. Caffeic acid phenethyl ester prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration. Neuroscience. 2011;188:135–141. doi: 10.1016/j.neuroscience.2011.04.009. [DOI] [PubMed] [Google Scholar]