

Summary
The molecular chaperone Hsp90 is a hub of protein homeostasis and regulatory circuitry. Hsp90 function is regulated by posttranslational modifications including acetylation in mammals; however, whether this regulation is conserved remains unknown. In fungi, Hsp90 governs the evolution of drug resistance by stabilizing signal transducers. Here, we establish that pharmacological inhibition of lysine deacetylases (KDACs) blocks the emergence and maintenance of Hsp90-dependent resistance to the most widely deployed antifungals, the azoles, in the human fungal pathogen Candida albicans and the model yeast Saccharomyces cerevisiae. S. cerevisiae Hsp90 is acetylated on lysine 27 and 270, and key KDACs for drug resistance are Hda1 and Rpd3. Compromising KDACs alters stability and function of Hsp90 client proteins, including the drug-resistance regulator calcineurin. Thus, we establish acetylation as a mechanism of posttranslational control of Hsp90 function in fungi, functional redundancy between KDACs Hda1 and Rpd3, as well as a mechanism governing fungal drug resistance with broad therapeutic potential.
Graphical Abstract
Highlights
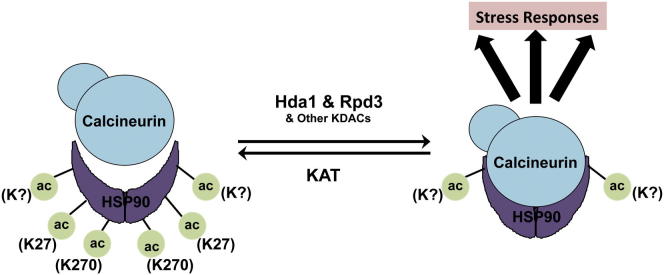
► KDACs govern Hsp90-dependent azole resistance in C. albicans and S. cerevisiae ► Hda1 and Rpd3 are the key KDACs for fungal drug resistance in S. cerevisiae ► Hsp90 is regulated by acetylation of lysine 27 and 270 in S. cerevisiae ► Hsp90 acetylation controls function of Hsp90 client proteins, including calcineurin
The molecular chaperone Hsp90 is a hub of protein homeostasis and orchestrates the evolution of antifungal drug resistance by stabilizing signal transducers. Hsp90 function is regulated by acetylation in mammals; however, whether this regulation is conserved remains elusive. Cowen and colleagues establish that Hsp90 is acetylated in the model yeast Saccharomyces cerevisiae, and key KDACs are Hda1 and Rpd3. In addition they show that Hsp90 acetylation governs drug resistance in the human fungal pathogen Candida albicans, with broad therapeutic potential.
Introduction
Hsp90 is an essential molecular chaperone that is conserved among all eukaryotes. Hsp90 has a specialized role in regulating the form and function of diverse client proteins by binding to metastable regulators of cellular signaling and key components of multiprotein complexes (Taipale et al., 2010; Trepel et al., 2010). Hsp90 is constitutively expressed at much higher levels than required to fulfill its normal functions, constituting a large but highly specific protein-folding reservoir. As a consequence, Hsp90 modulates the relationship between genotype and phenotype by acting as a capacitor and potentiator for the storage and release of genetic variation in response to environmental stress (Jarosz and Lindquist, 2010). By governing the expression of phenotypic diversity in an environmentally contingent manner, Hsp90 is poised to enable evolution (Jarosz et al., 2010).
In diverse fungal species, Hsp90 potentiates the evolution of drug resistance by enabling the phenotypic effects of resistance mutations (Cowen and Lindquist, 2005). Fungal pathogens are a leading cause of human mortality worldwide, especially in immunocompromised individuals. Chief among the opportunistic invaders is the fungal pathogen Candida albicans, which is a leading causal agent of mycotic death (Pfaller and Diekema, 2010). Hsp90 enables the emergence of resistance to the most widely deployed antifungal drugs in the clinic, the azoles. The azoles inhibit lanosterol demethylase, encoded by ERG11, causing a block in the production of ergosterol and the accumulation of a toxic sterol produced by Erg3 (Ostrosky-Zeichner et al., 2010; Shapiro et al., 2011). Genetic or pharmacological inhibition of Hsp90 blocks the emergence of azole resistance and abrogates resistance that had already been acquired by diverse mutations in both C. albicans and the model yeast Saccharomyces cerevisiae (Cowen et al., 2006; Cowen and Lindquist, 2005). For example, loss of function of Erg3 in the ergosterol biosynthetic pathway blocks the accumulation of toxic sterols and confers resistance that is contingent upon Hsp90-mediated stress responses (Cowen, 2008). Hsp90’s role in azole resistance depends on the mode of selection. A more gradual selection regimen favors the evolution of Hsp90-independent resistance where drug-induced stress is bypassed by upregulation of multidrug transporters that efflux drugs from the cell (Cowen and Lindquist, 2005).
Hsp90 orchestrates cellular circuitry required for responses to drug-induced stress and can be targeted for the treatment of life-threatening fungal infections. The Hsp90 client protein calcineurin, a signal transducer and sensor of cellular stress, is a key mediator of Hsp90-dependent azole resistance in C. albicans and S. cerevisiae (Cowen and Lindquist, 2005; Singh et al., 2009). By chaperoning calcineurin, Hsp90 regulates membrane stress responses that are crucial for cells to survive azole exposure, thereby enabling the phenotypic consequences of new resistance mutations. Targeting Hsp90 has broad therapeutic potential because Hsp90 inhibitors that are well tolerated in humans are synergistic with azoles against C. albicans in a metazoan model of fungal disease (Cowen et al., 2009), and in a rat catheter model of biofilm infection (Robbins et al., 2011). Hsp90 governs not only the cellular circuitry required for drug resistance but also that required for pathogenesis. Hsp90 orchestrates temperature-dependent C. albicans morphogenesis, a key virulence trait (Gow et al., 2012; Shapiro et al., 2011), and depletion of C. albicans Hsp90 results in clearance of a disseminated infection in a mouse model (Shapiro et al., 2009). Hence, changes in a global regulator of signaling can potentiate evolution with profound implications for abrogating drug resistance and treating infectious disease.
Despite the fundamental importance of Hsp90 as a hub of protein homeostasis and regulatory circuitry, and its profound impact on diverse facets of biology, disease, and evolution, an understanding of how Hsp90 function is regulated remains in its infancy. Recently, it has become appreciated that Hsp90 is subject to complex regulation by posttranslational modification. In both mammals and yeast, Hsp90 function is regulated by phosphorylation of several serine, threonine, and tyrosine residues (Taipale et al., 2010; Trepel et al., 2010). Phosphorylation of S. cerevisiae Hsp90 on tyrosine 24 by the Swe1 kinase and on threonine 22 by protein kinase CK2 is important for Hsp90’s ability to interact with and stabilize several cochaperones and client proteins (Mollapour et al., 2010, 2011). Moreover, in mammalian cells the class IIb histone deacetylase 6 (HDAC6) complex regulates Hsp90 deacetylation, which is required for Hsp90 interaction with select cochaperones and for stabilization of several Hsp90 client proteins (Bali et al., 2005; Kovacs et al., 2005; Scroggins et al., 2007). Given that many key targets of histone deacetylases are now appreciated to be nonhistone proteins (Choudhary et al., 2009), we conform with the more appropriate lysine deacetylase (KDAC) terminology. Although Hsp90 lysine acetylation is well established in mammalian systems, whether this mechanism of posttranslational control is conserved in other species remains unknown.
Targeting Hsp90 provides a powerful strategy for the treatment of life-threatening fungal infections; however, there are therapeutic challenges given that compromise of host Hsp90 function has deleterious effects in the context of acute systemic fungal infections (Cowen et al., 2009), motivating the present study to identify novel regulators of Hsp90 function and fungal drug resistance. Given that Hsp90 function is regulated by acetylation in mammals, we investigated whether this regulatory mechanism is conserved in fungi and if it influences fungal drug resistance. Our results implicate acetylation as a mechanism of posttranslational control of Hsp90 in fungi, establish functional redundancy between Hda1 and Rpd3, identify lysine 27 and lysine 270 as functionally important acetylated residues on Hsp90, and establish a mechanism of fungal drug resistance with broad implications for treatment of life-threatening fungal disease.
Results
KDAC Inhibition Blocks the Emergence and Maintenance of Hsp90-Dependent Azole Resistance
Given that KDACs regulate Hsp90 function in mammalian cells (Aoyagi and Archer, 2005) and that Hsp90 potentiates the emergence and maintenance of fungal drug resistance (Cowen and Lindquist, 2005), we hypothesized that KDACs govern fungal drug resistance by regulating Hsp90 function in fungi. First, we tested if pharmacological inhibition of KDAC function with trichostatin A blocked the emergence of azole resistance in C. albicans. To select for resistant mutants, we used a rapid selection regimen and plated 1 × 105 cells onto rich medium containing a high concentration of the azole miconazole. Cells underwent several doublings before growth was arrested, producing small abortive colonies (Figure 1A). Large colonies were also recovered that upon further retesting displayed elevated resistance to azoles (data not shown). Including a concentration of trichostatin A in the medium that had no impact on growth on its own completely blocked the emergence of colonies in the presence of miconazole (Figure 1A), as was the case with the Hsp90 inhibitor radicicol (Figure 1A; Cowen and Lindquist, 2005). Thus, the rapid emergence of azole resistance in C. albicans is critically dependent upon Hsp90 and KDAC function.
Figure 1.

Trichostatin A Blocks the Emergence and Maintenance of Drug Resistance in C. albicans and S. cerevisiae
(A) A total of 1 × 105 cells of a wild-type C. albicans strain was plated onto YPD with no drug (−), 800 nM of the azole miconazole (ML), 1 μM of the Hsp90 inhibitor radicicol (RAD), 6 μg/ml of the KDAC inhibitor trichostatin A (TSA), or a combination of azole and inhibitor, as indicated.
(B) Fluconazole (FL) resistance of C. albicans clinical isolates is abrogated when KDACs are inhibited. MIC assays were conducted in YPD with no inhibitor (−), with the Hsp90 inhibitor geldanamycin (GdA, 5 μM), or with TSA (6 μg/ml). C. albicans clinical isolates (CaCis) are ordered sequentially with those recovered early in treatment at the top. Data are quantitatively displayed with color using TreeView (see bar).
(C) TSA specifically reduces FL resistance of C. albicans and S. cerevisiae Hsp90-dependent azole-resistant mutants. MIC assays were conducted in YPD with no inhibitor (−), with GdA (1 μM for C. albicans, 5 μM for S. cerevisiae), or with TSA (6 μg/ml). Growth and analysis are as in (B). PDR5⇑ represents a S. cerevisiae strain that acquired FL resistance by upregulation of the drug efflux pump Pdr5. erg3 represents a S. cerevisiae strain that acquired FL resistance due to a loss-of-function mutation in ERG3.
See also Table S1.
Next, we assessed whether KDACs enabled the maintenance of drug resistance that had already evolved in C. albicans clinical isolates (CaCis) collected from a single patient infected with HIV who was undergoing fluconazole therapy (White, 1997). We monitored resistance to the azole fluconazole in the presence or absence of pharmacological inhibitors of KDAC or Hsp90 function by minimum inhibitory concentration (MIC) assays. Inhibition of Hsp90 with geldanamycin or KDACs with trichostatin A markedly reduced resistance of all clinical isolates, especially for clinical isolates recovered earlier during treatment (Figure 1B). Azole resistance in this series evolved from a state of Hsp90 dependence toward independence accompanying the accumulation of additional mutations in the azole target ERG11 and overexpression of the drug pump Cdr1 (Cowen and Lindquist, 2005; White, 1997). The transition to KDAC independence perfectly recapitulates that observed with Hsp90 independence, suggesting functional relationships between these cellular regulators.
Given that clinical isolates often harbor multiple mechanisms of resistance, we next tested the impact of KDAC inhibition on azole resistance of laboratory-derived mutants to evaluate the dependence of specific resistance mechanisms on KDAC function. Furthermore, to determine whether KDACs have a conserved impact on drug resistance in other fungi, we turned to the genetically tractable model yeast S. cerevisiae. Pharmacological inhibition of KDACs with trichostatin A abrogated azole resistance of C. albicans and S. cerevisiae erg3 loss-of-function mutants, concordant with the impact of Hsp90 inhibition (Figure 1C; Cowen et al., 2006; Cowen and Lindquist, 2005; LaFayette et al., 2010). In contrast, KDAC inhibition did not affect S. cerevisiae azole resistance that had been acquired by an activating mutation in the transcription factor Pdr1 that results in overexpression of the multidrug transporter Pdr5 (Figure 1C), consistent with a robust azole-resistance phenotype despite Hsp90 inhibition (Cowen and Lindquist, 2005). Taken together, these results demonstrate that KDAC inhibition affects azole resistance in a manner that phenocopies Hsp90 inhibition, supporting the model that compromising KDAC function leads to impaired Hsp90 function, thereby blocking stress responses critical for azole resistance.
Compromising KDAC Function Does Not Decrease the Expression of Hsp90 or Key Drug-Resistance Determinants
Given that a predominant function of KDACs is to deacetylate lysines in core histones and thereby regulate gene expression (Shahbazian and Grunstein, 2007), we tested if compromising KDAC function led to reduction of HSP90 levels. Treatment of C. albicans with trichostatin A did not reduce HSP90 transcript levels as measured by quantitative RT-PCR but rather led to a small increase (p = 0.0011, t test; Figure 2A). Similarly, in S. cerevisiae, trichostatin A did not reduce transcript levels of either HSC82 or HSP82, the two genes encoding Hsp90: there was no effect on HSP82 and only a very small increase of HSC82 (p = 0.0101; Figure 2A). Western analysis for both species confirmed that trichostatin A did not affect Hsp90 protein levels relative to a tubulin-loading control (Figure 2B). Thus, pharmacological KDAC inhibition does not reduce Hsp90 transcript or protein levels.
Figure 2.

Trichostatin A Does Not Decrease the Expression of Hsp90 or Key Drug-Resistance Determinants
(A) Treatment of C. albicans or S. cerevisiae with trichostatin A (TSA) does not reduce Hsp90 gene expression. Strains were grown in rich medium to log phase without (−) or with TSA. Transcript levels of gene(s) encoding Hsp90 were measured by quantitative RT-PCR and normalized to GPD1 (C. albicans) or ACT1 (S. cerevisiae). Levels are expressed relative to the untreated sample, which is set to 1. Data are mean ± SD for triplicates.
(B) Treatment of S. cerevisiae or C. albicans with TSA does not alter Hsp90 protein levels. Cultures were grown as in (A). Total protein was resolved by SDS-PAGE, and blots were hybridized with α-Hsp90 or α-tubulin.
(C) TSA does not block the expression of azole-resistance determinants. Transcript levels of C. albicans or S. cerevisiae erg3 mutants were measured by quantitative RT-PCR after growth to log phase with no treatment, TSA, fluconazole (FL), or the combination of TSA and FL. Transcript levels are normalized as in (A).
See also Table S2.
Our findings suggest that the relevant effects of trichostatin A on azole resistance are not due to compromised gene expression because it had no impact on resistance that is dependent on the overexpression of drug transporters (Figure 1C), nor did it compromise expression of HSP90. Given that one study demonstrated that trichostatin A blocks azole-induced upregulation of resistance determinants in C. albicans (Smith and Edlind, 2002), we measured transcript levels of the same resistance determinants, including key multidrug transporters and the azole target ERG11. In C. albicans, trichostatin A did not impair basal expression or the fluconazole induction of the multidrug transporters CDR1, CDR2, or MDR1, or of the azole target ERG11; for CDR2 and ERG11 there was no effect of trichostatin A in the presence of fluconazole, whereas for CDR1 and MDR1, trichostatin A led to an increase in fluconazole-dependent induction of transcript levels (p < 0.01, ANOVA, Bonferroni’s multiple comparison test; Figure 2C). Similarly, in S. cerevisiae, trichostatin A did not reduce the basal expression, or the fluconazole induction, of the multidrug transporter PDR5 or the azole target ERG11 (p > 0.05; Figure 2C). Thus, KDAC inhibition does not block the expression of known resistance determinants but rather phenocopies Hsp90 inhibition and reduces drug resistance, consistent with compromised chaperone function.
The KDACs HDA1 and RPD3 Regulate Hsp90-Dependent Azole Resistance in S. cerevisiae
To genetically confirm the pharmacological effects of trichostatin A on erg3-mediated resistance and to determine which KDACs were the key targets of trichostatin A for azole resistance, we exploited the genetic tractability of S. cerevisiae. Based on homology with the class IIb HDAC6 complex in mammalian cells, we predicted that Hda1 was a key KDAC for azole resistance and that deletion of HDA1 from an erg3 mutant would abrogate azole resistance, as with trichostatin A. However, deletion of HDA1 had no impact on azole resistance of an erg3 mutant (Figure 3A). Deletion of other catalytic subunits of KDAC complexes including RPD3, HOS1, HOS2, or HOS3 in an erg3 mutant also did not abrogate azole resistance (Figures 3A and S1). However, deletion of both HDA1 and RPD3 in an erg3 mutant reduced fluconazole resistance back to wild-type levels, similar to the impact of trichostatin A (Figures 1C and 3A). Notably, this effect was specific for the combined deletion of RPD3 with HDA1 because deletion of other KDAC complexes in an erg3Δhda1Δ mutant had no impact on azole susceptibility (Figure S1). These findings suggest functional redundancy between Hda1 and Rpd3 in the regulation of Hsp90-dependent azole resistance.
Figure 3.

Deletion of RPD3 and HDA1, Encoding Catalytic Subunits of Distinct KDAC Complexes, Decreases Azole Resistance of Hsp90-Dependent Resistant Mutants in S. cerevisiae
(A) To identify the key targets of trichostatin A (TSA) that influence fluconazole (FL) resistance, we tested the impact of deletion of KDACs individually and in combination in an erg3 mutant in an FL MIC assay. The multiple erg3Δhda1Δrpd3Δ strains shown are independent meiotic progeny with the confirmed genotype. Assays are performed as in Figure 1. See also Figure S1.
(B) Deletion of both HDA1 and RPD3 has no effect on azole resistance caused by drug pump overexpression. Growth is as in (A) and analysis as in Figure 1.
Figure S1.

Genetic Deletion of HOS1, HOS2, or HOS3 Has No Effect on erg3-Mediated Azole Resistance, Related to Figure 3
To determine if other KDACs contribute to fluconazole (FL) resistance, we tested the impact of the deletion of HOS1, HOS2 or HOS3 individually and in combination with HDA1 in an erg3 mutant background in a FL MIC assay. Cultures were grown in YPD at 30° for 48 hr and growth was measured as in Figure 1.
To determine if these KDACs specifically regulate azole resistance in general or if their effects are specific to Hsp90-dependent resistance mechanisms, as with trichostatin A, we deleted HDA1 and RPD3 in a strain that overexpresses the drug pump Pdr5 due to an activating mutation in the transcription factor Pdr1. Deletion of HDA1 or RPD3 individually or in combination did not alter azole resistance caused by drug pump overexpression (Figure 3B). Therefore, Hda1 and Rpd3 are not global regulators of azole resistance but rather have a specialized role in enabling Hsp90-dependent drug resistance, consistent with the model that they influence resistance by modulating Hsp90 function.
Hsp90 Is Acetylated in S. cerevisiae, and the KDACs Hda1 and Rpd3 Regulate Hsp90 Function
If deletion of HDA1 and RPD3 compromises Hsp90 function, then these mutants should be hypersensitive to Hsp90 inhibition. To test this, we measured susceptibility of the wild-type strain and KDAC mutants to the pharmacological Hsp90 inhibitor geldanamycin. As predicted, deletion of both HDA1 and RPD3 increased sensitivity to geldanamycin relative to individual gene deletions (Figure 4A). Western analysis confirmed that the hypersensitivity was not due to reduced Hsp90 protein levels relative to a tubulin-loading control (Figure 4A). Thus, Hda1 and Rpd3 likely regulate Hsp90 function at a posttranslational level.
Figure 4.

Hsp90 Is Acetylated in S. cerevisiae, and Compromising KDAC Function Results in Hypersensitivity to Further Hsp90 Inhibition
(A) Deletion of HDA1 and RPD3 in S. cerevisiae causes hypersusceptibility to geldanamycin (GdA) without altering Hsp90 protein levels. A titration of GdA was generated, and growth was measured as in Figure 1 (left panel). Total protein was resolved by SDS-PAGE, and blots were hybridized with α-Hsp90 or α-tubulin (right panels).
(B) Hsp90 is acetylated in S. cerevisiae. Immunoprecipitation of 6×His-Hsp82 with NiNTA agarose pulled down acetylated Hsp90 when immunoprecipitated samples were hybridized with α-Hsp90 or α-acetylated lysine (AcK). Reciprocally, Hsp90 was immunoprecipitated when protein extracts were incubated with protein A agarose coupled to an AcK antibody. C, a control lane where protein extracts were incubated with protein A agarose in the absence of AcK antibody; IB, immunoblot; IP, immunoprecipitate.
(C) Schematic of Hsp90 domain structure indicating candidate acetylated lysine residues. Mass spectrometry analysis identified a putative acetylation site at K27 of S. cerevisiae Hsp82 in an hda1Δrpd3Δ mutant, indicated by an asterisk. K270 has been identified in mammalian cells as acetylated. Sequences containing the putative acetylated lysines (bolded and underlined) are shown below. Hsp90 was immunoprecipitated with an AcK antibody as described in (B).
(D) K27 and K270 are functionally important Hsp90 residues that are modified by acetylation. Growth curves were conducted in YPD with or without GdA over 48 hr, and the area under the curve (AUC) was calculated. Data are mean ± SD of quadruplicates.
To determine if Hsp90 is acetylated in yeast, we performed immunoprecipitation assays coupled to western analysis. Immunoprecipitation of 6×His-Hsp82 from all strains harboring the tagged protein successfully pulled down Hsp90 (Figure 4B, top-left panel). When the same immunoprecipitated samples were probed with an antibody specific for acetylated-lysine residues, bands were detected that corresponded to ∼90 kDa (Figure 4B, middle-left panel). Notably, deletion of HDA1 and RPD3 did not intensify the acetylation signal relative to a wild-type strain or the individual KDAC deletion mutants. Reciprocally, protein extracts were immunoprecipitated using an acetylated-lysine antibody coupled to protein A agarose. When immunoprecipitated samples were probed with an Hsp90 antibody via western analysis, Hsp90 was pulled down approximately equally among all strains (Figure 4B, right panel). This demonstrates Hsp90 acetylation in fungi and suggests that Hsp90 is acetylated on numerous lysine residues. Our results suggest that Hda1 and Rpd3 may only regulate deacetylation of one or a few of these residues, leading to compromised Hsp90 chaperone function.
To identify the acetylated Hsp90 residues in the hda1Δrpd3Δ mutant, we turned to mass spectrometry. Hsp90 was immunoprecipitated from a wild-type strain and an hda1Δrpd3Δ mutant. After SDS-PAGE separation, bands corresponding to ∼90 kDa were excised and subjected to liquid chromatography-mass spectrometry to identify posttranslational modifications. Lysine 27 (K27) on Hsp90 was identified as acetylated in two biological hda1Δrpd3Δ replicates and was not identified as acetylated in either of the two wild-type biological replicates (Figure 4C). This residue is located in the N-terminal nucleotide-binding domain, corresponding to a region regulated by phosphorylation in S. cerevisiae (Mollapour et al., 2010, 2011). This K27 was the only residue identified as acetylated in both hda1Δrpd3Δ replicates and in neither wild-type replicate, suggesting that modification of this residue might influence Hsp90 function.
To explore the impact of the K27 residue on Hsp90 function, we constructed alleles of HSC82 with mutations of K27 to mimic a constitutively acetylated state (glutamine) or a state that cannot be acetylated (arginine). Vectors harboring a wild-type allele of HSC82 or the mutant alleles were transformed into a S. cerevisiae strain with the only native allele encoding Hsp90 expressed on a counterselectable plasmid and that is resistant to azoles due to deletion of ERG3. Geldanamycin and fluconazole susceptibility of strains in which the only allele encoding Hsp90 was expressed from the vectors was assessed. We predicted that mutating Hsp90 lysine residues that are important for its function would confer increased geldanamycin and fluconazole sensitivity in an erg3 background, as is observed with the erg3Δhda1Δrpd3Δ mutant (Figures 3A and 4A). However, mutations in K27 alone did not alter susceptibility to geldanamycin (Figure 4D) or fluconazole (Figure S2). Hence, deacetylation of this residue cannot be solely responsible for the effects of Hda1 and Rpd3 on Hsp90 function. In mammalian systems lysine 294 is a key acetylated Hsp90 residue that regulates interaction with cochaperones and client proteins (Scroggins et al., 2007). To determine if this residue is also acetylated in fungi, we created point mutations of K270, the S. cerevisiae counterpart to K294, to mimic acetylated and deacetylated states individually as well as in combination with K27. Lysine to arginine mutation of both K27 and K270 significantly reduced the amount of Hsp90 immunoprecipitated using an acetylated-lysine antibody (Figure 4C), demonstrating two key residues on which Hsp90 is acetylated in yeast. Functional characterization revealed that mutating both of these residues to either glutamine or arginine conferred hypersensitivity to geldanamycin compared to a wild-type strain (p < 0.01, ANOVA, Bonferroni’s multiple comparison test; Figure 4D), indicative of compromised chaperone function. Notably, mutating K270 to arginine alone caused geldanamycin hypersensitivity, which was further enhanced upon mutation of K27. Mutation of K27 and K270 did not increase susceptibility to fluconazole (Figure S2), implicating additional targets of Hda1 and Rpd3 in azole resistance. Taken together, these results suggest that Hsp90 is acetylated in yeast on K27 and K270 and that these residues have a profound impact on Hsp90 function.
Figure S2.

K27 and K270 Point Mutations in Hsp90-Mimicking Acetylation or Deacetylation Do Not Alter Fluconazole Susceptibility, Related to Figure 4
To test the functional importance of candidate acetylated lysines, site-directed mutagenesis was performed to mutate K27 and K270 of Hsp90 to mimic permanent acetylation (lysine (K) to glutamine (Q)) or deacetylation (lysine (K) to arginine (R)). These mutant alleles were expressed as the only source of Hsp90 in the cell. We tested the impact of these mutations on fluconazole (FL) susceptibility in an MIC assay. Cultures were grown in YPD at 30°C for 48 hr and growth was measured as in Figure 1.
Compromised KDAC Function Blocks the Interaction of Hsp90 with Its Client Protein Calcineurin, Thereby Compromising Calcineurin Function
In order to assess whether trichostatin A abrogates Hsp90-dependent azole resistance via impairment of Hsp90 client protein function, we focused on the protein phosphatase calcineurin. Upon calcineurin activation in response to calcium, calcineurin dephosphorylates Crz1 to drive expression of genes containing calcineurin-dependent response elements (CDREs) in their promoters (Karababa et al., 2006; Stathopoulos and Cyert, 1997). If trichostatin A compromises Hsp90 function, then it should block calcium-induced calcineurin activation. To test this hypothesis, we used reporters that contained CDREs fused upstream of lacZ. As predicted, exposure of S. cerevisiae or C. albicans to calcium chloride activated calcineurin relative to the untreated control (p < 0.001, ANOVA, Bonferroni’s multiple comparison test; Figure 5A). Treatment with trichostatin A prior to and during calcium exposure blocked calcium-induced calcineurin activation to a similar extent as geldanamycin in both species (p < 0.001; Figure 5A). Thus, KDAC inhibition blocks calcineurin activation in both S. cerevisiae and C. albicans.
Figure 5.

Inhibition of KDACs Impairs Calcineurin Activation and Blocks the Interaction between Hsp90 and Calcineurin
(A) Geldanamycin (GdA) and trichostatin A (TSA) block calcium-induced calcineurin activation. Log phase cultures of a S. cerevisiae strain containing a 4×CDRE-lacZ reporter or a C. albicans strain harboring a UTR2p-lacZ construct were untreated (−), treated with 200 mM CaCl2 (Ca), treated with Ca and GdA, or treated with Ca and TSA. Data are mean ± SD from technical triplicates and are representative of biological duplicates. ∗p < 0.001.
(B) Deletion of HDA1 and RPD3 blocks the physical interaction between Hsp90 and calcineurin. Protein from S. cerevisiae strains harboring a HA-tagged catalytic subunit of calcineurin (Cna1) was immunoprecipitated with anti-HA agarose beads, resolved by SDS-PAGE, and blots were hybridized with α-HA or α-Hsp90 antibodies. Experiment was quantified using ImageJ, and data are mean ± SD of biological duplicates.
Next, we assessed the physical interaction between Hsp90 and calcineurin, which is important for calcineurin stability and activity (Imai and Yahara, 2000), in our S. cerevisiae KDAC mutants and wild-type controls. Cna1 was immunoprecipitated using anti-hemagglutinin (HA) agarose beads from all strains containing the catalytic subunit of calcineurin, CNA1, HA tagged (Figure 5B, top panel). Hsp90 copurified in all strains except when both HDA1 and RPD3 were deleted, despite equivalent levels of Hsp90 in all inputs (Figure 5B). Hence, deletion of HDA1 and RPD3 blocks Hsp90 interaction with calcineurin, thereby blocking calcineurin activation and providing a compelling mechanism by which KDACs regulate Hsp90-dependent azole resistance.
Compromised KDAC Function Leads to Altered Activity and Stability of Numerous Hsp90 Client Proteins
Next, we addressed whether compromised KDAC function had more global effects on Hsp90 client protein regulation. First, we exploited reporter constructs that monitor the heat shock response. In S. cerevisiae, Hsp90 exerts a repressive effect on the transcription factor Hsf1, such that compromising Hsp90 leads to upregulation of heat shock proteins and other targets with heat shock elements (HSEs) in their promoters (Guo et al., 2001). Comparable regulation is conserved in C. albicans (Shapiro et al., 2009). For S. cerevisiae and C. albicans we used reporters with HSEs driving lacZ expression and monitored β-galactosidase activity upon Hsp90 inhibition with geldanamycin or KDAC inhibition with trichostatin A. Treatment with the inhibitors led to a significant increase in lacZ expression relative to the untreated control in both species (p < 0.001, ANOVA, Bonferroni’s multiple comparison test; Figure 6A). We further confirmed the pharmacological effects we observed in S. cerevisiae genetically. When both HDA1 and RPD3 were deleted, activation of the heat shock response was higher than with a wild-type control or with the individual deletion mutants at 30°C (data not shown), and at the elevated temperature of 42°C (p < 0.001; Figure 6B). Thus, compromise of KDAC function leads to activation of the heat shock response in both S. cerevisiae and C. albicans, consistent with impaired Hsp90 function.
Figure 6.

Inhibition of KDACs Impairs Hsp90 Client Protein Activation and Stability
(A) Geldanamycin (GdA) and trichostatin A (TSA) induce the heat shock response in S. cerevisiae and C. albicans. Log phase cultures of strains with an HSE-lacZ reporter (S. cerevisiae) or HSP70p-lacZ reporter (C. albicans) were untreated (−) or treated with GdA or TSA. β-Galactosidase data are mean ± SD for technical triplicates and are representative of biological duplicates.
(B) Deletion of HDA1 and RPD3 enhances the heat shock response. Log phase S. cerevisiae harboring an HSE-lacZ reporter was grown at 42°C for 3 hr.
(C) TSA blocks glucocorticoid receptor (GR) maturation in S. cerevisiae. Wild-type S. cerevisiae containing a GR expression plasmid and a GR reporter plasmid with a GR response element (GRE) driving lacZ expression was used. Maturation of GR upon addition of deoxycorticosterone (DOC, 10 μM) was assessed in the absence and presence of GdA or TSA.
(D) TSA leads to destabilization of Ste11. Wild-type S. cerevisiae was grown in SD with glucose (glu) as a negative control or galactose (gal) to induce 6×His-Ste11 expression. Cultures grown in galactose were grown without inhibitor (−) or with GdA or TSA. Total protein was resolved by SDS-PAGE, and blots were hybridized with α-His or with α-Hsp90 as a loading control.
(E) TSA does not rescue v-Src-induced lethality in S. cerevisiae. S. cerevisiae containing a vector with v-Src under the control of a galactose-inducible promoter was spotted in 5-fold dilutions onto SD with 2% glucose or 2% galactose with no drug (−), GdA, or TSA. Images are representative of biological duplicates.
∗p < 0.001.
We then examined the effect of KDAC inhibition on glucocorticoid receptor (GR) activation. GR is a well-characterized Hsp90 client protein in mammalian cells, and reporters of its activity have been used extensively in yeast (Pratt et al., 2004). We transformed S. cerevisiae with a vector containing GR under a constitutive promoter and with a plasmid containing a GR response element driving lacZ expression. When Hsp90 is functional and can assist in proper folding of GR, addition of the hormone deoxycorticosterone leads to activation of GR such that it can dimerize, translocate to the nucleus, and bind to response elements. We observed that treatment of S. cerevisiae with deoxycorticosterone led to activation of the lacZ reporter relative to the untreated control (p < 0.001, ANOVA, Bonferroni’s multiple comparison test; Figure 6C), However, the KDAC inhibitor trichostatin A blocked activation of GR to a similar extent as inhibition of Hsp90 (p < 0.001; Figure 6C). Therefore, pharmacological KDAC inhibition impairs GR activation.
Next, we assessed the stability of the endogenous yeast client protein kinase Ste11. We transformed S. cerevisiae with a construct containing a constitutively active variant of Ste11 that depends on Hsp90 for stability, Ste11ΔN, which is expressed under a galactose inducible promoter. Upon growth of S. cerevisiae in galactose, Ste11 was detectable by western analysis (Figure 6D). When cultures were grown in galactose with geldanamycin or trichostatin A, Ste11 levels were reduced, despite equivalent total protein as confirmed by an Hsp90-loading control (Figure 6D). Thus, KDAC inhibition leads to destabilization of the Hsp90 client kinase Ste11.
Finally, we examined the effect of KDAC inhibition on the v-Src oncogenic tyrosine kinase, a well-established Hsp90 client protein whose expression is toxic in yeast and whose stability is highly dependent on Hsp90 (Nathan and Lindquist, 1995). A strain of S. cerevisiae was transformed with a vector containing v-Src under the control of a galactose-inducible promoter. Cells were spotted onto glucose- or galactose-containing medium with no inhibitor, with geldanamycin, or with trichostatin A. v-Src-induced lethality was rescued upon Hsp90 inhibition, but not KDAC inhibition (Figure 6E), suggesting that KDAC function does not influence Hsp90’s ability to chaperone v-Src. Overall, these findings suggest that KDAC regulation of Hsp90 controls its ability to chaperone a distinct set of clients (Figure 6), similar to Hsp90 regulation by phosphorylation (Mollapour et al., 2010, 2011).
Discussion
Our study provides one of the premier examples of regulation of a nonhistone protein via acetylation in fungi, complementing the emerging paradigm from mammalian cells that acetylation may be as ubiquitous as phosphorylation (Choudhary et al., 2009). Specifically, our results implicate acetylation as a mechanism of posttranslational control of fungal Hsp90 function, establishing a mechanism of drug resistance with broad therapeutic potential. There is precedent for regulation of Hsp90 function by deacetylation in mammalian cells, where the class IIb HDAC6 complex deacetylates Hsp90, affecting its interaction with cochaperones and regulation of client protein function (Bali et al., 2005; Kovacs et al., 2005; Scroggins et al., 2007). Our work implicates the class I KDAC, Rpd3, and the class II KDAC, Hda1, as key regulators of Hsp90 deacetylation and Hsp90-dependent azole resistance in S. cerevisiae. Conventionally, these KDACs act in distinct complexes to deacetylate histones on different lysine residues (Rundlett et al., 1996), and large-scale genomic analysis in yeast suggests that these KDACs have largely nonoverlapping roles in protein deacetylation (Kaluarachchi Duffy et al., 2012). Recently, one study demonstrated that Rpd3 and Hda1 may partially compensate for each other in response to DNA damage by regulating stability of the endonuclease Sae2 (Robert et al., 2011). Thus, our findings provide a compelling example of cooperation between traditionally disparate KDACs to regulate function of Hsp90, a global regulator of protein homeostasis and cellular circuitry.
The emerging paradigm is that distinct aspects of Hsp90 function are modulated by a posttranslational code defined by a multitude of regulators and target residues. We establish that Hda1 and Rpd3 deacetylate K27 of Hsp90 despite not having an impact on global Hsp90 acetylation levels, suggesting that additional lysine residues of Hsp90 are acetylated. Consistent with this possibility, although human Hsp90 K294 regulates aspects of Hsp90 function, human HSP90-α is thought to be acetylated on at least 14 lysines and HSP90-β on a minimum of 4 (Choudhary et al., 2009). Importantly, we demonstrate that combined mutation of both the S. cerevisiae counterpart of K294 (K270) and K27 reduces Hsp90 acetylation and compromises chaperone function. Notably, these mutations did not reduce fluconazole susceptibility, suggesting that Hda1 and Rpd3 deacetylate additional targets important for azole resistance. The finding that mutation of K270 and K27 reduces global Hsp90 acetylation, whereas deletion of HDA1 and RPD3 does not, implicates additional KDACs in Hsp90 deacetylation. One candidate KDAC is Hos2, based on the recent functional connection identified between Hos2 and Hsp90 in C. albicans, where Hos2 interacts genetically with Hsp90 under diverse conditions (Diezmann et al., 2012). Combinatorial control of acetylation status of a multitude of Hsp90 residues by diverse KDACs may delineate a complex code to rapidly and reversibly modulate function of a global cellular regulator.
Posttranslational modifications of Hsp90 define an emerging code poised to have a myriad of effects on diverse facets of biology, disease, and evolution. Examining Hsp90 regulation by phosphorylation in S. cerevisiae has led to several key insights regarding biochemical regulation of this molecular chaperone. Swe1 phosphorylates Hsp90 on tyrosine 24, and compromising Swe1 function destabilizes client kinases and induces the heat shock response; however, it has no impact on the GR (Mollapour et al., 2010). Similarly, protein kinase CK2 phosphorylates Hsp90 on threonine 22, and phosphomimic mutants or mutants that cannot be phosphorylated destabilize client kinases and alter GR activity (Mollapour et al., 2011), suggesting that cycling between phosphorylation states is critical for Hsp90 function. S. cerevisiae Hsp90 is phosphorylated on at least 11 residues (Mollapour et al., 2011), and there may be combinatorial control based on their phosphorylation status. Our results suggest that similar to phosphorylation, acetylation of Hsp90 on multiple residues controls its ability to chaperone distinct client proteins. Compromising KDAC function blocks calcineurin activation, destabilizes Ste11, activates the heat shock response, and blocks GR activation, although it has no effect on v-Src-induced toxicity. The precise mechanism by which these posttranslational modifications alter Hsp90 function to influence select clients remains elusive. It is likely that there is a dynamic and complex code defined by phosphorylation, acetylation, and perhaps other posttranslational modifications to dictate function of Hsp90 and other cellular regulators. Because Hsp90 interacts with ∼10% of the S. cerevisiae proteome (Zhao et al., 2005) and the C. albicans Hsp90 genetic interaction network reaffirms Hsp90’s high degree of connectivity (Diezmann et al., 2012), posttranslational control of Hsp90 function is likely to be fundamental for its capacity to modulate client proteins in a dynamic and environmentally contingent manner.
Targeting Hsp90 has emerged as a powerful strategy for the treatment of diverse diseases such as cancer, neurodegenerative disease, and infectious disease caused by protozoan parasites and fungal pathogens, though there are therapeutic challenges that might be overcome by targeting Hsp90 indirectly. With fungal infections the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) transforms fluconazole from ineffective to highly efficacious in a rat catheter model of biofilm infection, where the infection and drug remain localized (Robbins et al., 2011). However, in a murine model of disseminated C. albicans infection, toxicity of systemic 17-AAG compromises its therapeutic utility (Cowen et al., 2009), despite the fact that genetic depletion of C. albicans Hsp90 clears the disseminated infection (Shapiro et al., 2009). Our findings suggest that targeting KDACs may provide a powerful alternative. KDACs are structurally and functionally distinct from Hsp90 and are inhibited by entirely different molecules, providing new opportunities for drug development. Furthermore, the divergence of KDACs between fungi and humans is greater than Hsp90. For example the noncatalytic subunits of the Hda1 complex are not conserved in metazoans (Yang and Seto, 2008). Fungal-specific targets provide critical avenues for exploitation to minimize host toxicity. In support of this a fungal-specific Hos2 inhibitor has recently been reported by Pfaller et al. (2009), though the biology underpinning its efficacy remains enigmatic. KDAC inhibitors have emerged as important anticancer agents with benefits in clinical trials as single or combination therapy agents (Bolden et al., 2006; Marks et al., 2001; Xu et al., 2007). Furthermore, treatment of patients with breast cancer with the KDAC inhibitor vorinostat leads to enhanced Hsp90 acetylation and induction of Hsp70 in tumor biopsies, suggesting functional Hsp90 inhibition in vivo and validating this strategy to modulate Hsp90 via KDACs (Ramaswamy et al., 2012). The challenge ahead for successfully targeting KDACs in the treatment of infectious disease lies in developing potent and specific inhibitors capable of discriminating between machineries of pathogen and host.
Experimental Procedures
Strains and Culture Conditions
Strains are listed in Table S1, and their construction is described in the Extended Experimental Procedures, as are culture conditions.
Extended Experimental Procedures.
Strain Construction
CaLC206. The plasmid pLC53 was digested with BssHII to liberate the C. albicans transactivator cassette and was transformed into SN95 (CaLC239). For NAT-resistant transformants, proper integration was verified by PCR using oLC239/218. The SAP2 promoter was induced to drive expression of FLP recombinase to excise the NAT marker cassette. Presence of the construct was verified after using excision using the same primers as above.
ScLC766. Made by mating ScLC9 (MATα erg3::KAN) with hda1::KAN from the Invitrogen MATa deletion library. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for hda1Δ by PCR using oLC643/101 and erg3Δ by PCR using oLC64/101. Absence of wild-type HDA1 in the genome was verified by PCR using oLC643/644 and absence of wild-type ERG3 in the genome was verified by PCR using oLC76/77.
ScLC965. Made by mating ScLC9 (MATα erg3::KAN) with hos1::KAN from the Invitrogen MATa deletion library. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101 and hos1Δ by PCR using oLC101/960. Absence of wild-type HOS1 in the genome was verified by PCR using oLC960/961 and absence of wild-type ERG3 in the genome was verified by PCR using oLC76/77.
ScLC967. Made by mating ScLC9 (MATα erg3::KAN) with hos2::KAN from the Invitrogen MATa deletion library. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101 and hos2Δ by PCR using oLC101/963. Absence of wild-type HOS2 in the genome was verified by PCR using oLC963/964 and absence of wild-type ERG3 in the genome was verified by PCR using oLC76/77.
ScLC979. Made by transforming ScLC387 (HSC82-TAP) with pLC436 (GALp-CNA1-HA) using standard lithium acetate protocol. Transformants were plated onto SD plates containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same media. Expression of the construct was verified by Western blotting.
ScLC1342. Made by mating ScLC9 (MATα erg3::KAN) with hos3::KAN from the Invitrogen MATa deletion library. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101 and hos3Δ by PCR using oLC101/1160. Absence of wild-type HOS3 in the genome was verified by PCR using oLC1160/1161 and absence of wild-type ERG3 in the genome was verified by PCR using oLC76/77.
ScLC1343. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC965. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos1Δ by PCR using oLC101/960. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS1 were present using oligos listed above.
ScLC1344. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC965. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos1Δ by PCR using oLC101/960. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS1 were present using oligos listed above.
ScLC1345. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC967. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos2Δ by PCR using oLC101/963. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS2 were present using oligos listed above.
ScLC1346. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC967. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos2Δ by PCR using oLC101/963. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS2 were present using oligos listed above.
ScLC1347. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC1342. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos3Δ by PCR using oLC101/1160. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS3 were present using oligos listed above.
ScLC1348. Made by mating ScLC767 (MATα erg3::KAN hda1::KAN) with ScLC1342. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and hos3Δ by PCR using oLC101/1160. PCR tests confirmed no extra alleles of ERG3, HDA1, or HOS3 were present using oligos listed above.
ScLC1364. The plasmid pLC536 was digested with NotI to liberate the NAT marker along with ∼500bp of upstream and downstream sequence homologous to the RPD3 locus, and was transformed into BY4742 (ScLC152) using standard protocol. For NAT-resistant transformants, proper integration was verified by PCR using oLC274/1176 and oLC659/1179. PCR tests confirmed no extra alleles of RPD3 using oLC798/799.
ScLC1365. The plasmid pLC536 was digested with NotI to liberate the NAT marker along with ∼500bp of upstream and downstream sequence homologous to the RPD3 locus, and was transformed into ScLC9 (MATα erg3::KAN) using standard protocol. For NAT-resistant transformants, proper integration was verified by PCR using oLC274/1176 and oLC659/1179. PCR tests confirmed no extra alleles of RPD3 using oLC798/799.
ScLC1366. ScLC9 (BY4742 erg3::KAN) was independently mated with hsc82::KAN or hsp82::KAN deletion strains from the MATa Invitrogen deletion library. Strains were grown in sporulation medium and tetrads were dissected to obtain erg3::KAN hsc82::KAN and erg3::KAN hsp82::KAN double mutants. These two strains were mated together and plated on SD + histidine, leucine, and uracil in order to obtain an erg3/erg3 HSC82/hsc82 HSP82/hsp82 diploid. The diploid was transformed with pLC495, which contains HSC82 under the control of a GPD promoter. The diploid was sporulated and tetrads were selected that contained deletion of erg3, hsc82, and hsp82. PCR tests confirmed the appropriate deletions using oLC64/101 (deletion of ERG3), oLC1/101 (deletion of HSC82) and (oLC29/101) deletion of (HSP82).
ScLC1377. Made by mating hda1::KAN from the Invitrogen MATa deletion library with ScLC1365. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and rpd3Δ by PCR using oLC274/1176. PCR tests confirmed no extra alleles of ERG3, HDA1, or RPD3 were present using oligos listed above.
ScLC1378. Made by mating hda1::KAN from the Invitrogen MATa deletion library with ScLC1365. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for erg3Δ by PCR using oLC64/101, hda1Δ by PCR using oLC101/643, and rpd3Δ by PCR using oLC274/1176. PCR tests confirmed no extra alleles of ERG3, HDA1, or RPD3 were present using oligos listed above.
ScLC1394. Made by transforming ScLC1366 with pLC407 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K270Q mutation.
ScLC1396. Made by transforming ScLC1366 with pLC408 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K270R mutation.
ScLC1644. The HYGMX resistance cassette was amplified from pLC3 using oLC1138/1139. The PCR fragment was transformed into ScLC4. HYGB resistant transformants were replica plated onto G418 in order to select transformants where the HYGMX cassette had in fact replaced the KANMX cassette.
ScLC1695. Made by PCR amplifying the hda1::KAN cassette from genomic DNA from ScLC766 using oLC643/758. The PCR product was electroporated into the ScLC1698. Transformants were tested for hda1Δ using oLC101/643. PCR tests confirmed no extra alleles of HDA1, or RPD3 were present using oligos listed above.
ScLC1697. Made by PCR amplifying the hda1::KAN cassette from genomic DNA from ScLC766 using oLC643/758. The PCR product was electroporated into the ScLC1644. Transformants were tested for hda1Δ using oLC101/643. PCR tests confirmed no extra alleles of HDA1 were present using oligos listed above.
ScLC1698. Made by mating ScLC1644 with ScLC1364. Diploids were sporulated in liquid medium and tetrads were dissected. Meiotic progeny were tested for rpd3Δ by PCR using oLC274/1176. PCR tests confirmed no extra alleles RPD3 were present using oligos listed above. Fluconazole-resistant spores were identified and sent for sequencing to confirm the C862W Pdr1 mutation.
ScLC1847. Made by transforming ScLC149 with pLC549 using standard lithium acetate protocol. Transformants were plated onto SD agar containing histidine, uracil, adenine, and leucine to select for tryptophan prototrophy. Tryprophan prototrophs were transformed with pLC550 using standard lithium acetate protocol. Transformants were plated onto SD agar containing histidine, adenine, and leucine to select for uracil prototrophy.
ScLC1876. Made by transforming ScLC387 rpd3::NAT with pLC436 (GALp-CNA1-HA) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same media. Expression of the construct was verified by Western blotting. Transformants were confirmed to have rpd3Δ using PCR with oLC274/1176.
ScLC1877. Made by transforming ScLC387 rpd3::NAT hda1::KAN with pLC436 (GALp-CNA1-HA) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium. Expression of the construct was verified by Western blotting. Transformants were confirmed to have rpd3Δ using PCR with oLC274/1176 and hda1Δ using PCR with oLC101/643.
ScLC1878. Made by transforming ScLC387 hda1::KAN with pLC436 (GALp-CNA1-HA) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium. Expression of the construct was verified by Western blotting. Transformants were confirmed to have hda1Δ using PCR with oLC101/643.
ScLC2062. Made by transforming pLC630 into BY4741 (ScLC151) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium.
ScLC2064. Made by transforming pLC630 into ScLC766 using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium.
ScLC2065. Made by transforming pLC630 into ScLC1365 using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium.
ScLC2066. Made by transforming pLC630 into ScLC1377 using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except uracil. Uracil prototrophs were confirmed by patching onto the same medium.
ScLC2068. Made by transforming pLC631 into W303 (ScLC149) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing histidine, leucine, uracil and adenine to select for tryptophan prototrophy. Tryptophan prototrophs were confirmed by patching onto the same medium.
ScLC2069. Made by transforming pLC637 into BY4741 (ScLC151) using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except leucine. Leucine prototrophs were confirmed by patching onto the same medium.
ScLC2071. Made by transforming pLC637 into ScLC766 using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except leucine. Leucine prototrophs were confirmed by patching onto the same medium.
ScLC2072. Made by transforming pLC637 into ScLC1365 using a standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except leucine. Leucine prototrophs were confirmed by patching onto the same medium.
ScLC2073. Made by transforming pLC637 into ScLC1377 using standard lithium acetate protocol. Transformants were plated onto SD agar containing all amino acids except leucine. Leucine prototrophs were confirmed by patching onto the same medium.
ScLC2168. Made by transforming ScLC1366 with pLC638 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSP82 allele with the K27Q mutation.
ScLC2169. Made by transforming ScLC1366 with pLC639 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSP82 allele with the K27R mutation.
ScLC2311. Made by transforming ScLC1366 with pLC683 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K27Q and K270Q mutations. One of two tranformants used in study.
ScLC2312. Made by transforming ScLC1366 with pLC683 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K27Q and K270Q mutations. Second of two tranformants used in study.
ScLC2313. Made by transforming ScLC1366 with pLC684 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K27R and K270R mutations. One of two tranformants used in study.
ScLC2314. Made by transforming ScLC1366 with pLC684 using a standard lithium acetate protocol. Transformants were plated onto SD agar with histidine, lysine, and methionine to select for uracil and leucine prototrophs. Subsequently, transformants were struck out onto SD plates with 5-FOA with uracil, histidine, lysine, and methionine to cure the strain of the Ura-marked vector. Single colonies were isolated and uracil auxotrophy and leucine prototrophy were verified. Colonies were sequenced to ensure strain only contained an HSC82 allele with the K27R and K270R mutations. Second of two tranformants used in study.
Plasmid Construction
Recombinant DNA procedures were performed according to standard protocols and their construction is described below. Plasmids were sequenced to verify the absence of any nonsynonymous mutations. Primers used in this study are listed in Table S2.
pLC407. This plasmid is based on pLC28 (P415GPD-HSC82). It underwent site directed mutagenesis to mutate Lysine 270 to a glutamine residue (AAG to CAG) using primers oLC668 and oLC669 using standard Stratagene protocol. The clone was sequence verified with primers oLC10, oLC11, oLC43-oLC47.
pLC408. This plasmid is based on pLC28 (P415GPD-HSC82). It underwent site directed mutagenesis to mutate Lysine 270 to an arginine residue (AAG to AGG) using primers oLC670 and oLC671 using standard Stratagene protocol. The clone was sequence verified with primers oLC10, oLC11, oLC43-oLC47.
pLC495. pLC28 (P415GPD-HSC82) was digested with BamHI and SpeI. The HSP90 insert was isolated by gel purification and was inserted into pLC138 (p416GPD) that had also been digested with BamHI and SpeI. Transformants were selected on LB + Amp plates. The presence of the insert was confirmed by PCR with primers oLC805 and oLC11, and by sequencing over the entire insert to ensure no mutations.
pLC536. A DNA region, approximately 500 base pairs long, that was homologous to a region of DNA immediately upstream of the S. cerevisiae RPD3 start codon was amplified from BY4741 genomic DNA by PCR using oLC1174/1175 and cloned into pLC1 at the HindIII/SalI restriction sites. A DNA region, approximately 500 base pairs long, that was homologous to a region of DNA immediately downstream of the S. cerevisiae RPD3 stop codon was amplified from BY4741 genomic DNA with primers oLC1177/1178 and cloned into pLC1 containing the upstream homology at SacI. The presence of the inserts within the vector was tested by PCR with oLC1175/274 and oLC1178/659. The cassette to knock out RPD3 can be excised with NotI.
pLC638. HSP82 was amplified from BY4741 genomic DNA by PCR using oLC1807 and oLC1808 and cloned into pLC136 (p415GPD) at SpeI and BamHI. The presence of the insert within the vector was tested by PCR with oLC748/805. The vector was sequence verified to ensure HSP82 contained no mutations. Site directed mutagenesis was performed to mutate lysine 27 to a glutamine residue, using primers oLC1929 and oLC1930 using standard Stratagene protocol. The clone was sequence verified with primers oLC805, oLC43-oLC46 to ensure the only mutation in HSP82 was the K27Q mutation.
pLC639. HSP82 was amplified from BY4741 genomic DNA by PCR using oLC1807 and oLC1808 and cloned into pLC136 (p415GPD) at SpeI and BamHI. The presence of the insert within the vector was tested by PCR with oLC748/805. The vector was sequence verified to ensure HSP82 contained no mutations. We then performed site directed mutagenesis to mutate lysine 27 to an arginine residue using primers oLC1931 and oLC1932 using standard Stratagene protocol. The clone was sequence verified with primers oLC805, oLC43-oLC46 to ensure the only mutation in HSP82 was the K27Q mutation.
pLC683. This plasmid is based on pLC622 (P415GPD-HSC82K27Q). It underwent site directed mutagenesis to mutate Lysine 270 to an arginine residue (AAG to AGG) using primers oLC668 and oLC669 using standard Stratagene protocol. The clone was sequence verified with primers oLC10, oLC11, oLC43-oLC47.
pLC684. This plasmid is based on pLC623 (P415GPD-HSC82K27R). It underwent site directed mutagenesis to mutate Lysine 270 to an arginine residue (AAG to AGG) using primers oLC670 and oLC671 using standard Stratagene protocol. The clone was sequence verified with primers oLC10, oLC11, oLC43-oLC47.
Strains and Culture Conditions
Archives of C. albicans strains were maintained at −80°C in 25% glycerol. Strains were routinely maintained and grown in YPD liquid medium (1% yeast extract, 2% bactopeptone, 2% glucose) at 30°C unless otherwise indicated.
qRT-PCR
qRT-PCR was performed as described previously (Cowen et al., 2009).All reactions were performed in triplicate, using primers for the following genes: CaGPD1(oLC752/753), CaHSP90 (oLC754/755), ScACT1 (oLC1015/1016), ScHSC82 (oLC4/10), ScHSP82 (oLC45/2242), ScPDR5 (oLC1123/1124), ScERG11 (oLC1125/1126), CaCDR1 (oLC1127/1128), CaCDR2 (oLC1129/1130), CaMDR1 (oLC1131/1132), and CaERG11 (oLC1133/1134). Data were analyzed using iQ5 Optical System Software Version 2.0 (Bio-Rad Laboratories, Inc.). Statistical significance was evaluated using GraphPad Prism 4.0.
β-Galactosidase Assays
S. cerevisiae strains harboring a 4×CDRE-lacZ construct were grown overnight at 25°C in synthetic defined (SD) medium supplemented for auxotrophies. Cells were diluted to OD600 of 0.3 and were either left untreated or were treated with 200 mM CaCl2 for 3 hr at 25°C. When GdA or TSA were used as inhibitors in the assay, cultures were grown overnight in SD with the inhibitors at 25°C and diluted to OD600 of 0.3 in SD with or without GdA (5 μM) or TSA (6 μg/mL) in the presence of 200 mM CaCl2 for 3 hr at 25°C. C. albicans cultures harboring the UTR2p-lacZ construct were grown overnight in YPD at 30°C with or without GdA (10 μM) or TSA (12 μg/mL). Cells were diluted to OD600 of 0.5 with or without inhibitor and allowed to grow for 2 hr before being treated with 200 mM CaCl2 for 6 hr. S. cerevisiae cultures harboring the HSE-lacZ construct were grown overnight at 30°C in SD medium supplemented for auxotrophies with or without GdA (20 μM) or TSA (100 μg/mL) as indicated. Cells were diluted to OD600 of 0.3 in SD medium with or without inhibitor and left shaking at 30°C for 4 hr. C. albicans cultures harboring the HSP70p-lacZ construct were grown overnight at 30°C in YPD medium with or without GdA (10 μM) or TSA (12 μg/mL) as indicated. Cells were diluted to OD600 of 0.2 in YPD medium with or without inhibitor and left shaking at 30°C for 6 hr. S. cerevisiae cultures harboring the GRE-lacZ construct grown overnight at 30°C in SD medium supplemented for auxotrophies with or without GdA (20 μM) or TSA (20 μg/mL), as indicated. Cells were diluted to OD600 of 0.2 in SD medium with or without GdA, TSA, or the hormone deoxycorticosterone (DOC, 10 μM, Sigma Aldrich Co., D7000), as indicated, and left shaking at 30°C for 8 hr.
After treatment, cells were harvested and washed, protein was extracted, and protein concentrations were determined by Bradford analysis as described (LaFayette et al., 2010; Singh et al., 2009). Protein samples were diluted to the same concentration and β-galactosidase activity was measured using the substrate ONPG (O-nitrophenyl-β-D-galactopyranosidase, Sigma Aldrich Co.) as described (Singh et al., 2009). β-galactosidase activity is given in units of nanomoles ONPG converted per minute per milligram of protein. Statistical significance was evaluated using GraphPad Prism 4.0.
Immunoprecipitation
To monitor Hsp90 acetylation, cells were grown overnight at 30°C in SD supplemented for auxotrophies. Cells were diluted to OD600 of 0.1 in 50 ml and grown at 30°C to mid-log phase. To monitor the interaction between Hsp90 and calcineurin, cells were grown up overnight in SD supplemented with 2% glucose and supplemented for auxotrophies at 30°C. Cells were washed and diluted to OD600 of 0.1 in 50 ml of SD supplemented with 2% galactose and supplemented for auxotrophies. Cells were grown to mid-log phase.
Protein was harvested as described previously (Singh et al., 2009). In brief, cells were washed with sterile water and resuspended in 500 μl of lysis buffer J containing 20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl and 20% glycerol, with one protease inhibitor cocktail (complete, EDTA-free tablet, Roche Diagnostics) per 10 mL, as well as 1 mM PMSF (EMD Chemicals) and 20 mM sodium molybdate (Sigma Aldrich Co.) added fresh before use. Cells were mechanically disrupted by adding acid-washed glass beads and bead beating twice for 4 min with a 10 min break on ice between cycles. Total collected lysates were cleared by centrifugation at 20817xg for 10 min at 4°C and protein concentrations were determined by Bradford analysis.
Anti-His immunoprecipitations were conducted by diluting protein samples to 1 mg/mL in lysis buffer J and incubating with NiNTA agarose (QIAGEN, 30210) at 4°C for four hours. Unbound material was removed by three washes with lysis buffer J. Bound material was removed from the beads by incubating beads in 250 mM imidazole (Sigma Aldrich Co., I0125) for 5 min on ice. Supernatant was removed and was combined with one-sixth volume of 6X SDS-PAGE sample buffer containing 0.35 M Tris-HCl, 10% (w/v) SDS, 36% glycerol, 5% β- mercaptoethanol, and 0.012% bromophenol blue for SDS-PAGE. Anti-AcK immunoprecipitations were conducted by diluting protein samples to 1 mg/ml in lysis buffer J and incubating with protein A sepharose (GE Healthcare Bio-Sciences, CL-4B) for 30 min at 4°C to pre-clear the lysate. The supernatant (1mL) was combined with 10 μl of an anti-AcK antibody (Cell Signaling, 9814) and left rocking for 1 hr at 4°C. The entire mixture was then added to protein A agarose beads and left rocking overnight at 4°C. Unbound material was removed by washing five times with 1 ml lysis buffer J with 0.2% tween and protein was eluted by boiling the sample in one-sixth volume of 6X SDS-PAGE sample buffer. Anti-HA immunoprecipitations were done using Profound HA-Tag IP/Co-IP kit (PI23610) as per manufacturer’s instructions.
Immune Blot Analysis
To monitor Hsp90 protein levels, cells were grown overnight in YPD at 30°C with or without TSA (25 μg/mL for S. cerevisiae, 6 μg/mL for C. albicans) as indicated. In the morning, cells were diluted to OD600 of 0.2 in YPD with or without TSA and were grown to log-phase (∼5 hr) at 30°C. To monitor Ste11 stability, cells were grown overnight in SD supplemented with 2% glucose and supplemented for auxotrophies with or without GdA (20 μM) or TSA (20 μg/mL). Cells were washed in sterile water and resuspended to OD600 of 0.2 in SD media supplemented with 2% glucose or 2% galactose and supplemented for auxotrophies, with or without inhibitors. Cultures were grown to log-phase (∼6 hr) at 30°C.
Cells were harvested as described previously (LaFayette et al., 2010; Robbins et al., 2011). In brief, cells were harvested by centrifugation and were washed with sterile water. Cell pellets were resuspended in lysis buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM PMSF, and protease inhibitor cocktail (complete, EDTA-free tablet, Roche Diagnostics). Cells suspended in lysis buffer were mechanically disrupted by adding acid-washed glass beads and bead beating for 3 min. Protein concentrations were determined by Bradford analysis. Protein samples were mixed with one-sixth volume of 6X sample buffer containing 0.35 M Tris-HCl, 10% (w/v) SDS, 36% glycerol, 5% β- mercaptoethanol, and 0.012% bromophenol blue for SDS-PAGE. Samples were boiled for 5 min.
Protein was separated by SDS-PAGE using an 8% acrylamide gel. Proteins were electrotransferred to PVDF membranes (Bio-Rad Laboratories, Inc.) and blocked with 5% skimmed milk in phosphate buffered saline (PBS) with 0.1% tween. Blots were hybridized with antibodies against CaHsp90 (1:10000), generously provided by Brian Larsen (Burt et al., 2003), ScHsp90 (1:5000), generously provided by Susan Lindquist, His6 (1:10, P5A11), generously provided by Elizabeth Wayner, acetylated-lysine (1:1000, Cell Signaling 9814), HA (1:10, 12CA5), generously provided by Alan Cochrane, or against alpha-tubulin (1:1000; AbD Serotec, MCA78G).
Mass Spectrometry
All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version Mascot) and X! Tandem (http://www.thegpm.org; version 2007.01.01.1). X! Tandem was set up to search a subset of the Hsp82 database assuming digestion with the enzyme trypsin. Mascot was set up to search the Hsp82 database assuming the digestion enzyme trypsin. Mascot and X! Tandem were searched with a fragment ion mass tolerance of 0.50 Da and a parent ion tolerance of 3.0 Da. For protein identification, Scaffold (version Scaffold_2_06_02, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 90.0% probability as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Plasmid Construction
Recombinant DNA procedures were performed according to standard protocols. Primers used in this study are listed in Table S2. Plasmids used in this study are listed in Table S3. Plasmid construction is described in the Extended Experimental Procedures.
Selection of Azole-Resistant Mutants in C. albicans
For selection of azole resistance in C. albicans by a rapid, one-step regime, CaLC75 was grown overnight in YPD at 30°C. Cell counts were performed with a hemacytometer, and ∼105 cells were plated on YPD agar to estimate the number of viable colony-forming units, or YPD agar supplemented with 800 nM miconazole (Sigma-Aldrich), 1 μM of the Hsp90 inhibitor radicicol (AG Scientific; R-1130), 6 μg/ml of the KDAC inhibitor trichostatin A (AG Scientific; T-1052), or a combination of drugs as indicated. Each condition was performed in duplicate. Plates were photographed after 7 days at 30°C in the dark.
MIC Assays and Growth Curve Assays
Antifungal tolerance and resistance were determined in flat-bottom, 96-well microtiter plates (Sarstedt) using a modified broth microdilution protocol as described by LaFayette et al. (2010) and Singh et al. (2009). DMSO (Sigma-Aldrich) was the solvent for the Hsp90 inhibitor geldanamycin (ant-gl-5; Cedarlane) and trichostatin A; fluconazole (Sequoia Research Products) was dissolved in sterile ddH2O. MIC tests were set up in a total volume of 0.2 ml/well with 2-fold dilutions of fluconazole or geldanamycin. Plates were incubated in the dark for 48 hr. Optical density (OD) was determined at 600 nm using a spectrophotometer (Molecular Devices) and corrected for background from the corresponding medium. Each strain was tested in duplicate on at least three occasions. MIC data were quantitatively displayed with color using the program Java TreeView 1.1.1 (http://jtreeview.sourceforge.net). For growth curves, cells were grown overnight in YPD, diluted to an OD600 of 0.0625 with or without geldanamycin in 96-well plates, and grown at 30°C with continuous shaking (TECAN GENios). OD595 was measured every 15 min. Data were analyzed in Microsoft Excel.
β-Galactosidase Assays
All β-galactosidase assays were conducted as described previously by LaFayette et al. (2010) and Singh et al. (2009). Detailed protocols are provided in the Extended Experimental Procedures. Protein samples were diluted to the same concentration, and β-galactosidase activity was measured using the substrate ONPG (O-nitrophenyl-β-D-galactopyranosidase; Sigma-Aldrich) as described by Singh et al. (2009). β-Galactosidase activity is given in units of nanomoles ONPG converted per minute per milligram of protein. Statistical significance was evaluated using GraphPad Prism 4.0.
v-Src Lethality Assay
Cultures were grown to log phase in synthetic-defined (SD) medium supplemented for auxotrophies and with 2% glucose. Cultures were washed and resuspended in PBS. Five-fold dilutions (from 1 × 106 cells/ml) were spotted onto freshly made agar medium using a spotter (Frogger; V&P Scientific). SD agar plates were supplemented for auxotrophies and contained 2% glucose, 2% galactose, 10 μM geldanamycin, or 50 μg/ml trichostatin A, as indicated. Plates were photographed after 4 days in the dark at 30°C.
Immunoblot Analysis and Immunoprecipitation
Total protein extracts were prepared as described previously (LaFayette et al., 2010; Robbins et al., 2011). Immunoblotting was conducted as described previously by Singh et al. (2009). Detailed immunoprecipitation and western blotting protocols are provided in the Extended Experimental Procedures.
Quantitative RT-PCR
S. cerevisiae and C. albicans were grown overnight in YPD with or without trichostatin A (6 μg/ml), diluted to OD600 of 0.1, and grown for an additional 2 hr in duplicate. Fluconazole (16 μg/ml for S. cerevisiae or 8 μg/ml for C. albicans) was then added to one of the duplicates, and cultures were grown for 6 hr at 30°C. RNA was isolated and PCR performed as described (Cowen et al., 2009). See Extended Experimental Procedures for details on primers used.
Mass Spectrometry
Cells were grown overnight in SD with 2% glucose supplemented for auxotrophies at 30°C. Cells were diluted to an OD600 of 0.2 in SD and grown to log phase at 30°C. Protein was harvested and immunoprecipitated using NiNTA agarose, as above. Protein was separated by SDS-PAGE on an 8% acrylamide gel and stained with Coomassie blue. Bands were excised for liquid chromatography-mass spectrometry analysis at SickKids Mass Spectrometry facility. See Extended Experimental Procedures for detailed protocols.
Acknowledgments
We thank Luke Whitesell, Leonard Neckers, Jill Johnson, Brenda Andrews, and Walid Houry for reagents, Danielle Sexton for technical assistance, and Rick Collins, Andrew Emili, and L.E.C. lab members for helpful discussions. N.R. was supported by a Natural Sciences & Engineering Research Council Canada Graduate Scholarship, M.D.L. by a Sir Henry Wellcome Postdoctoral Fellowship (Wellcome Trust 096072), and L.E.C. by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund, by a Canada Research Chair in Microbial Genomics and Infectious Disease, by a Canadian Institutes of Health Research (CIHR) Priority Announcement III-103027, and by CIHR Grant MOP-86452.
Published online: October 4, 2012
Footnotes
Supplemental Information includes Extended Experimental Procedures, two figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2012.08.035.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 Unported License (CC-BY-NC-ND; http://creativecommons.org/licenses/by-nc-nd/3.0/legalcode).
Supplemental Information
References
- Aoyagi S., Archer T.K. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005;15:565–567. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Bali P., Pranpat M., Bradner J., Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja P. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Cowen L.E. The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nat. Rev. Microbiol. 2008;6:187–198. doi: 10.1038/nrmicro1835. [DOI] [PubMed] [Google Scholar]
- Cowen L.E., Lindquist S. Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science. 2005;309:2185–2189. doi: 10.1126/science.1118370. [DOI] [PubMed] [Google Scholar]
- Cowen L.E., Carpenter A.E., Matangkasombut O., Fink G.R., Lindquist S. Genetic architecture of Hsp90-dependent drug resistance. Eukaryot. Cell. 2006;5:2184–2188. doi: 10.1128/EC.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L.E., Singh S.D., Köhler J.R., Collins C., Zaas A.K., Schell W.A., Aziz H., Mylonakis E., Perfect J.R., Whitesell L., Lindquist S. Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA. 2009;106:2818–2823. doi: 10.1073/pnas.0813394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S., Michaut M., Shapiro R.S., Bader G.D., Cowen L.E. Mapping the Hsp90 genetic interaction network in Candida albicans reveals environmental contingency and rewired circuitry. PLoS Genet. 2012;8:e1002562. doi: 10.1371/journal.pgen.1002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow N.A., van de Veerdonk F.L., Brown A.J., Netea M.G. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat. Rev. Microbiol. 2012;10:112–122. doi: 10.1038/nrmicro2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Guettouche T., Fenna M., Boellmann F., Pratt W.B., Toft D.O., Smith D.F., Voellmy R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001;276:45791–45799. doi: 10.1074/jbc.M105931200. [DOI] [PubMed] [Google Scholar]
- Imai J., Yahara I. Role of HSP90 in salt stress tolerance via stabilization and regulation of calcineurin. Mol. Cell. Biol. 2000;20:9262–9270. doi: 10.1128/mcb.20.24.9262-9270.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz D.F., Lindquist S. Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science. 2010;330:1820–1824. doi: 10.1126/science.1195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz D.F., Taipale M., Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu. Rev. Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Kaluarachchi Duffy S., Friesen H., Baryshnikova A., Lambert J.P., Chong Y.T., Figeys D., Andrews B. Exploring the yeast acetylome using functional genomics. Cell. 2012;149:936–948. doi: 10.1016/j.cell.2012.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karababa M., Valentino E., Pardini G., Coste A.T., Bille J., Sanglard D. CRZ1, a target of the calcineurin pathway in Candida albicans. Mol. Microbiol. 2006;59:1429–1451. doi: 10.1111/j.1365-2958.2005.05037.x. [DOI] [PubMed] [Google Scholar]
- Kovacs J.J., Murphy P.J., Gaillard S., Zhao X., Wu J.T., Nicchitta C.V., Yoshida M., Toft D.O., Pratt W.B., Yao T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- LaFayette S.L., Collins C., Zaas A.K., Schell W.A., Betancourt-Quiroz M., Gunatilaka A.A., Perfect J.R., Cowen L.E. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010;6:e1001069. doi: 10.1371/journal.ppat.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P., Rifkind R.A., Richon V.M., Breslow R., Miller T., Kelly W.K. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Mollapour M., Tsutsumi S., Donnelly A.C., Beebe K., Tokita M.J., Lee M.J., Lee S., Morra G., Bourboulia D., Scroggins B.T. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M., Tsutsumi S., Truman A.W., Xu W., Vaughan C.K., Beebe K., Konstantinova A., Vourganti S., Panaretou B., Piper P.W. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell. 2011;41:672–681. doi: 10.1016/j.molcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.F., Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrosky-Zeichner L., Casadevall A., Galgiani J.N., Odds F.C., Rex J.H. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 2010;9:719–727. doi: 10.1038/nrd3074. [DOI] [PubMed] [Google Scholar]
- Pfaller M.A., Diekema D.J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- Pfaller M.A., Messer S.A., Georgopapadakou N., Martell L.A., Besterman J.M., Diekema D.J. Activity of MGCD290, a Hos2 histone deacetylase inhibitor, in combination with azole antifungals against opportunistic fungal pathogens. J. Clin. Microbiol. 2009;47:3797–3804. doi: 10.1128/JCM.00618-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt W.B., Galigniana M.D., Morishima Y., Murphy P.J. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004;40:41–58. doi: 10.1042/bse0400041. [DOI] [PubMed] [Google Scholar]
- Ramaswamy B., Fiskus W., Cohen B., Pellegrino C., Hershman D.L., Chuang E., Luu T., Somlo G., Goetz M., Swaby R. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res. Treat. 2012;132:1063–1072. doi: 10.1007/s10549-011-1928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N., Uppuluri P., Nett J., Rajendran R., Ramage G., Lopez-Ribot J.L., Andes D., Cowen L.E. Hsp90 governs dispersion and drug resistance of fungal biofilms. PLoS Pathog. 2011;7:e1002257. doi: 10.1371/journal.ppat.1002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T., Vanoli F., Chiolo I., Shubassi G., Bernstein K.A., Rothstein R., Botrugno O.A., Parazzoli D., Oldani A., Minucci S., Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundlett S.E., Carmen A.A., Kobayashi R., Bavykin S., Turner B.M., Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. USA. 1996;93:14503–14508. doi: 10.1073/pnas.93.25.14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggins B.T., Robzyk K., Wang D., Marcu M.G., Tsutsumi S., Beebe K., Cotter R.J., Felts S., Toft D., Karnitz L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell. 2007;25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M.D., Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shapiro R.S., Uppuluri P., Zaas A.K., Collins C., Senn H., Perfect J.R., Heitman J., Cowen L.E. Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr. Biol. 2009;19:621–629. doi: 10.1016/j.cub.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro R.S., Robbins N., Cowen L.E. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 2011;75:213–267. doi: 10.1128/MMBR.00045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.D., Robbins N., Zaas A.K., Schell W.A., Perfect J.R., Cowen L.E. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 2009;5:e1000532. doi: 10.1371/journal.ppat.1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W.L., Edlind T.D. Histone deacetylase inhibitors enhance Candida albicans sensitivity to azoles and related antifungals: correlation with reduction in CDR and ERG upregulation. Antimicrob. Agents Chemother. 2002;46:3532–3539. doi: 10.1128/AAC.46.11.3532-3539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos A.M., Cyert M.S. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997;11:3432–3444. doi: 10.1101/gad.11.24.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M., Jarosz D.F., Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Trepel J., Mollapour M., Giaccone G., Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997;41:1482–1487. doi: 10.1128/aac.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.S., Parmigiani R.B., Marks P.A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Yang X.J., Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R., Davey M., Hsu Y.C., Kaplanek P., Tong A., Parsons A.B., Krogan N., Cagney G., Mai D., Greenblatt J. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
Supplemental References
- Anderson, J.B., Sirjusingh, C., Parsons, A.B., Boone, C., Wickens, C., Cowen, L.E., and Kohn, L.M. (2003). Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 163, 1287–1298. [DOI] [PMC free article] [PubMed]
- Burt, E.T., Daly, R., Hoganson, D., Tsirulnikov, Y., Essmann, M., and Larsen, B. (2003). Isolation and partial characterization of Hsp90 from Candida albicans. Ann. Clin. Lab. Sci. 33, 86–93. [PubMed]
- Cowen, L.E., Singh, S.D., Köhler, J.R., Collins, C., Zaas, A.K., Schell, W.A., Aziz, H., Mylonakis, E., Perfect, J.R., Whitesell, L., and Lindquist, S. (2009). Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 106, 2818–2823. [DOI] [PMC free article] [PubMed]
- Fonzi, W.A., and Irwin, M.Y. (1993). Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728. [DOI] [PMC free article] [PubMed]
- Gelperin, D.M., White, M.A., Wilkinson, M.L., Kon, Y., Kung, L.A., Wise, K.J., Lopez-Hoyo, N., Jiang, L., Piccirillo, S., Yu, H., et al. (2005). Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 19, 2816–2826. [DOI] [PMC free article] [PubMed]
- Ghaemmaghami, S., Huh, W.K., Bower, K., Howson, R.W., Belle, A., Dephoure, N., O’Shea, E.K., and Weissman, J.S. (2003). Global analysis of protein expression in yeast. Nature 425, 737–741. [DOI] [PubMed]
- Giaever, G., Chu, A.M., Ni, L., Connelly, C., Riles, L., Véronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., André, B., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed]
- Keller, A., Nesvizhskii, A.I., Kolker, E., and Aebersold, R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392. [DOI] [PubMed]
- LaFayette, S.L., Collins, C., Zaas, A.K., Schell, W.A., Betancourt-Quiroz, M., Gunatilaka, A.A., Perfect, J.R., and Cowen, L.E. (2010). PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 6, e1001069. [DOI] [PMC free article] [PubMed]
- Mollapour, M., Tsutsumi, S., Truman, A.W., Xu, W., Vaughan, C.K., Beebe, K., Konstantinova, A., Vourganti, S., Panaretou, B., Piper, P.W., et al. (2011). Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell 41, 672–681. [DOI] [PMC free article] [PubMed]
- Nesvizhskii, A.I., Keller, A., Kolker, E., and Aebersold, R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658. [DOI] [PubMed]
- Noble, S.M., and Johnson, A.D. (2005). Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell 4, 298–309. [DOI] [PMC free article] [PubMed]
- Robbins, N., Collins, C., Morhayim, J., and Cowen, L.E. (2010). Metabolic control of antifungal drug resistance. Fungal Genet. Biol. 47, 81–93. [DOI] [PubMed]
- Robbins, N., Uppuluri, P., Nett, J., Rajendran, R., Ramage, G., Lopez-Ribot, J.L., Andes, D., and Cowen, L.E. (2011). Hsp90 governs dispersion and drug resistance of fungal biofilms. PLoS Pathog. 7, e1002257. [DOI] [PMC free article] [PubMed]
- Schena, M., and Yamamoto, K.R. (1988). Mammalian glucocorticoid receptor derivatives enhance transcription in yeast. Science 241, 965–967. [DOI] [PubMed]
- Shapiro, R.S., Uppuluri, P., Zaas, A.K., Collins, C., Senn, H., Perfect, J.R., Heitman, J., and Cowen, L.E. (2009). Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr. Biol. 19, 621–629. [DOI] [PMC free article] [PubMed]
- Singh, S.D., Robbins, N., Zaas, A.K., Schell, W.A., Perfect, J.R., and Cowen, L.E. (2009). Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 5, e1000532. [DOI] [PMC free article] [PubMed]
- Wach, A., Brachat, A., Pöhlmann, R., and Philippsen, P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed]
- White, T.C. (1997a). Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 41, 1482–1487. [DOI] [PMC free article] [PubMed]
- White, T.C. (1997b). The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14alpha demethylase in Candida albicans. Antimicrob. Agents Chemother. 41, 1488–1494. [DOI] [PMC free article] [PubMed]
- White, T.C., Pfaller, M.A., Rinaldi, M.G., Smith, J., and Redding, S.W. (1997). Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis. 3 (Suppl 1), S102–S109. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.