

SUMMARY
Development of T helper (Th) 17 cells requires transforming growth factor (TGF)-β and interleukin (IL)-6 and is independent of the Th1 pathway. Although T cells that produce interferon (IFN)-γ are a recognized feature of Th17 cell responses, mice deficient for STAT4 and T-bet—two prototypical Th1 transcription factors—are protected from autoimmunity associated with Th17 pathogenesis. To examine the fate and pathogenic potential of Th17 cells and origin of IFN-γ-producing T cells that emerge during Th17 immunity, we developed IL-17F reporter mice that identify cells committed to expression of IL-17F and IL-17A. Th17 cells required TGF-β for sustained expression of IL-17F and IL-17A. In the absence of TGF-β, both IL-23 and IL-12 acted to suppress IL-17 and enhance IFN-γ production in a STAT4- and T-bet-dependent manner, albeit with distinct efficiencies. These results support a model of late Th17 developmental plasticity with implications for auto-immunity and host defense.
INTRODUCTION
T helper 17 is a third major CD4+ T cell effector subset important in host protection and autoinflammatory disorders (McGeachy and Cua, 2008; Stockinger and Veldhoen, 2007; Weaver et al., 2007). Th17 cells are characterized by production of the cytokines, interleukin (IL)-17A, and IL-17F. Although originally proposed as a branch of the Th1 lineage (Bettelli and Kuchroo, 2005; McKenzie et al., 2006), IL-17-producing effectors are now thought to develop independently of both Th1 and Th2 lineages (Harrington et al., 2005; Park et al., 2005). Th17 cells differentiate from naive CD4+ T cell precursors in response to transforming growth factor (TGF)-β and IL-6; the Th1 lineage-associated factors signal transducer and activator of transcription (STAT)1, T-bet, and STAT4, as well as the Th2 factors STAT6 and GATA3, are dispensable for Th17 differentiation (Bettelli et al., 2006; Harrington et al., 2005; Mangan et al., 2006; Park et al., 2005; Veldhoen et al., 2006). Th17 development is inhibited by the Th1 cytokine and interferon (IFN)-γ, as well other cytokines that activate STAT1 (e.g., type I interferons and IL-27), by the Th2 cytokine IL-4 (Batten et al., 2006; Harrington et al., 2005; Park et al., 2005; Villarino et al., 2003) and is constrained by IL-2 signaling via STAT5 (Laurence et al., 2007). TGF-β, which is required for Th17 development, has suppressive effects on Th1 development (Gorham et al., 1998), through inhibitory effects on STAT4 and T-bet signaling, and also inhibits Th2 development (Gorelik et al., 2000; Lin et al., 2005). STAT3 plays a central role in Th17 development, coupling early IL-6 signaling to induction of IL-21, an autocrine factor implicated in the progression of Th17 development (Harris et al., 2007; Korn et al., 2007; Mathur et al., 2007; Nurieva et al., 2007; Yang et al., 2007; Zhou et al., 2007). IL-21, also acting in part through STAT3 signaling, can induce expression of the transcription factors RORγt and RORα, which appear to be essential for Th17-lineage specification (Ivanov et al., 2006; Yang et al., 2008).
Despite the developmental signals that distinguish Th17 and Th1 differentiation, there are interesting parallels between the two developmental programs. IL-23R is induced in developing Th17 cells and IL-12Rβ2 is induced in developing Th1 cells to pair with the constitutively expressed IL-12Rβ1 chain to confer responsiveness to IL-23 and IL-12, respectively. The IL-12Rβ1 chain binds a common subunit of the IL-23 and IL-12 heterodimers—IL-12p40 (or IL-12β)—that pairs with IL-23p19 (or IL-23α) or IL-12p35 (or IL-12α), respectively (Aggarwal et al., 2003; Oppmann et al., 2000; Parham et al., 2002). Activation of the IL-23 or IL-12 receptors drives late events downstream of early Th17 or Th1 lineage commitment (Harrington et al., 2005; Mangan et al., 2006). Whereas the IL-12 receptor potently activates STAT4 in Th1 cells, the IL-23 receptor predominantly activates STAT3, but also recruits some STAT4 (Mathur et al., 2007; Oppmann et al., 2000; Parham et al., 2002; Yang et al., 2007).
The long-held association of autoimmune models such as experimental autoimmune encephalomyelitis (EAE), collagen-induced arthritis (CIA), and inflammatory bowel disease (IBD) with Th1-mediated pathogenesis has been revised in view of the requirement for IL-23, but not IL-12, for disease in these models (Cua et al., 2003; Langrish et al., 2005; Murphy et al., 2003). This has given rise to a new Th17-mediated pathogenesis paradigm (McGeachy and Cua, 2008). However, early reports that linked IL-23 to induction of IL-17 expression by memory CD4+ T cells also demonstrated induction of IFN-γ expression (Oppmann et al., 2000), and IFN-γ-producing T cells are typically found in association with IL-17-producing T cells in the context of both infectious and autoimmune inflammation. Interestingly, whereas mice lacking key components of the Th1 pathway (e.g., IL-12p35, IFN-γ, IFNγR, IL-12Rβ2, and STAT1) retain susceptibility to autoimmunity (Becher et al., 2002; Bettelli et al., 2004; Ferber et al., 1996; Gran et al., 2002; Willenborg et al., 1996; Zhang et al., 2003), deficiency of STAT4 and T-bet can preclude disease (Bettelli et al., 2004; Chitnis et al., 2001; Neurath et al., 2002), suggesting either that STAT4 and T-bet have important roles in both Th1 and Th17 inflammation or that cooperation of Th1 and Th17 responses is necessary for immune pathogenesis in certain settings.
Although IL-23 is dispensable for Th17 commitment (Mangan et al., 2006; Veldhoen et al., 2006), its requirement for Th17-associated immune protection or autoimmunity is undisputed, and it is speculated to enhance and/or maintain the Th17 phenotype (Veldhoen et al., 2006; Yang et al., 2007; Zhou et al., 2007). Indeed, Th17 cells differentiated by TGF-β and IL-6 in the absence of IL-23 have been shown to have a protective rather than pathogenic role in a model of EAE (McGeachy et al., 2007), consistent with an essential role for IL-23 in Th17 effector function. Nevertheless, the precise effects of IL-23 on Th17 lineage stability remain unclear (McGeachy and Cua, 2008; Weaver et al., 2007).
To explore the role of IL-23 in late Th17 development, we have examined the fate and phenotype of Th17 cells generated and maintained over the long term under defined cytokine conditions. Using an IL-17F reporter mouse to identify developing Th17 cells committed to expression of both IL-17F and IL-17A, we found that TGF-β was essential for maintenance of IL-17 expression, whereas IL-23 was not. Propagation of committed Th17 precursors in the presence of IL-23 without TGF-β resulted in progressive extinction of IL-17F and, to a lesser extent, IL-17A and promoted the emergence of IFN-γ-producing cells that lacked IL-17 expression. Remarkably, Th17 cells maintained over the long term by TGF-β demonstrated persistent responsiveness to IL-12 despite relatively low expression of IL-12Rβ2. Stimulation of Th17 cells with IL-12 induced a rapid, STAT4-and T-bet-dependent transition marked by extinction of RORγt, RORα, IL-17A, and IL-17F and an induction of a Th1-like gene-expression signature. Th17-committed cells generated in the presence or absence of IL-23 induced rapid development of colitis when transferred into immunodeficient recipients and showed transition of a subset of cells to IFN-γ expression. Blockade of IL-23 ablated development of colitis, but did not prevent persistence of IL-17-expressing cells nor transition to a subset of IFN-γ producers, whereas blockade of IL-23 and IL-12 ablated development of colitis and markedly diminished persistence of transferred Th17 cells. These findings support substantial developmental plasticity of the Th17 lineage and identify a mechanism for latent Th1-like responsiveness of Th17 cells that provides a basis for understanding the relationship between Th17 and Th1 mediated immunity.
RESULTS
TGF-β Is Required for Maintenance of IL-17 Expression by Th17 Cells
Although IL-23 is required for optimal expression of IL-17 under conditions of limiting TGF-β during Th17 development (Zhou et al., 2007), it is dispensable for Th17 commitment and its role in late Th17 development remains incompletely defined. To examine the possible requirement for IL-23 in maintenance of the Th17 phenotype, we propagated Th17 cells generated in the absence of IL-23 under defined cytokine conditions in vitro (Figure 1). Th17 cells maintained in the continued presence of exogenous TGF-β demonstrated an increase in the frequency of IL-17A+ progeny that was comparable in the presence or absence of exogenous IL-6 or IL-23. Neutralization of possible endogenous IL-6 had no detectable effect on the maintenance of IL-17 expression, consistent with its being nonessential for late events in Th17 development sustained by IL-21 (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007, and data not shown). In all cultures maintained with TGF-β, a subset of IL-17A+ cells coexpressed IFN-γ, despite the absence of IL-12 and irrespective of the addition (or absence) of IL-23 or IL-6. Notably, whereas the frequency of IL-17A+ cells decreased under conditions of IL-23 stimulation in the absence of TGF-β, the frequency of cells that expressed IFN-γ, without coexpression of IL-17A, markedly increased.
Figure 1. Requirement for TGF-β in the Maintenance of IL-17 Expression by Th17-Polarized Cells.

(A) FACS-sorted naive CD4+ T cells from OT-II TCR transgenic mice were cultured with irradiated Il12b−/− (IL-12p40-deficient) splenic feeder cells and 5 μg/ml OVAp for 7 days under Th17-polarizing conditions (TGF-β, 5 ng/ml; IL-6, 20 ng/ml; anti-IFN-γ, 10 μg/ml; anti-IL-4, 10 μg/ml), then stained intracellularly for IL-17A and IFN-γ after PMA/ionomycin activation for 5 hr in the presence of monensin (1° culture; left panel). Cells were harvested and restimulated with irradiated Il12b−/− splenic feeder cells in the presence of indicated cytokine(s) at the same doses or, in the case of IL-23 and IL-12, at 1 ng/ml, with anti-IFN-γ, anti-IL-4, and OVAp for an additional two rounds (7 days each) and stained intracelluarly for IL-17A and IFN-γ after PMA-ionomycin activation (post 3° culture; right panels).
(B) IfngThy1.1/Thy1.1 OT-II naive CD4+ T cells were cultured with irradiated Il12b−/− splenic feeder cells under Th17-polarizing conditions for 6 days and a fraction of the recovered cells were stained intracellularly for IL-17A and IFN-γ after PMA-ionomycin activation (left panel). A second fraction of recovered CD4+ T cells were activated for 5 hr with OVAp and Il12b−/− feeder cells, and Thy1.1+ (IFN-γ+) cells were depleted by magnetic sorting. Thy1.1 depletion was confirmed by surface staining for Thy1.1 (middle panel), and the isolated Thy1.1- cells were cultured with Il12b−/− feeder cells and OVAp and either TGF-β plus IL-6 or IL-23 in the presence of anti-IFNγ plus anti-IL-4 for three additional rounds (right panels), then stained intracellulary for IL-17A and IFN-γ after PMA-ionomycin activation. Numbers in each quadrant indicate percentages of total CD4+ T cells. All data are representative of at least three independent experiments.
Because a minor fraction of IFN-γ+ cells was generated under Th17-polarizing conditions in the absence of IL-12 and IL-23 (Figure 1A), we postulated that IL-23 might act on this small subset as a source for outgrowth of IFN-γ+ progeny under conditions in which TGF-β was deficient. To address this, we used naive CD4+ T cells from IFN-γ reporter mice (IfngThy1.1/Thy1.1) that permit specific depletion of IFN-γ-producing cells via the Thy1.1 reporter molecule (Harrington et al., 2008). Th17 cells were polarized as before, except that the small fraction of IFN-γ+ (Thy1.1+) cells was depleted prior to further propagation with either TGF-β and IL-6 or IL-23 (Figure 1B). Similar to previous results, the fraction of IFN-γ-producing cells was significantly increased by IL-23 stimulation in the absence of continued TGF-β, whereas IL-17A-producing cells were comparably amplified in the presence of TGF-β—as were the subset of IL-17A+IFN-γ+ cells. Collectively, these results suggest that TGF-β is a critical factor for both differentiation and maintenance of IL-17-producing effector cells; Th17 cells do not stably express IL-17A in the absence of continuous TGF-β signaling. In contrast, IL-23 appears to be dispensable for both the induction and maintenance of Th17 cells, but instead is associated with transition to progeny that extinguishes IL-17 expression and enhances IFN-γ expression under conditions of low or absent TGF-β. Thus, developing Th17 cells can give rise to “Th1-like” progeny in the absence of ongoing TGF-β signaling. Importantly, the converse is not true; Th1-polarized cells are resistant to transition to a Th17 cytokine-expression pattern when propagated under Th17 conditions (Figure S1 available online and Harrington et al., 2005). Accordingly, Th17 cells appear to have greater plasticity with respect to their cytokine expression during late stages of development.
Th17 Cells Respond to IL-12 to Rapidly Extinguish IL-17 and Promote IFN-γ Expression
Th17 cells upregulate IL-23R as part of their developmental program to confer responsiveness to IL-23 (Harrington et al., 2005; Mangan et al., 2006; Oppmann et al., 2000; Zhou et al., 2007). In contrast, developing Th1 cells upregulate IL-12Rβ2 to confer IL-12 responsiveness, and it is thought that the reciprocal expression of IL-23R and IL-12Rβ2, both of which pair with constitutively expressed IL-12Rβ1, is central to differential development of Th17 and Th1 cells, respectively (Harrington et al., 2005; Mangan et al., 2006; Zhou et al., 2007). Given the observed deviation of Th17 cells toward enhanced IFN-γ expression in the presence of IL-23, we examined the possible effect of IL-12 on Th17 cells (Figure 2). Naive CD4+ T cells polarized with Th17-inductive cytokines and antigen-presenting cells (APCs) deficient in expression of IL-12 and IL-23 (Il12b−/−) were divided into fractions that were maintained for one to three more cycles of stimulation with addition of the indicated exogenous cytokines. Consistent with previous results, TGF-β was required to maintain high frequencies of IL-17-expressing cells. Even in the absence of added cytokines, antigen stimulation alone (OVAp) induced progressive decline in IL-17A expression and enhanced IFN-γ expression in the absence of exogenous TGF-β. IL-23 signaling in the absence of TGF-β significantly and consistently increased the frequency of IFN-γ+ cells and decreased IL-17A+ cells relative to the absence of TGF-β alone.
Figure 2. Th17 Cells Respond to IL-12 to Rapidly Upregulate IFN-γ and Extinguish IL-17.

OT-II CD4+ T cells were cultured with irradiated Il12b−/− splenic feeder cells under Th17-polarizing condition for 7 days (1° culture, top panel), and a fraction of recovered T cells were restimulated and analyzed for intracellular IL-17A and IFN-γ as in Figure 1. The remaining T cells were cultured again with irradiated Il12b−/− splenic feeder cells in the presence of indicated cytokine(s), anti-IFNγ, anti-IL-4, and OVAp as in Figure 1 for 7 days (2° culture, middle panel), and a fraction was reanalyzed for intracellular cytokine expression. The remaining recovered cells from secondary cultures were recultured under identical cytokine conditions for a third and fourth round (3° and 4° cultures, bottom panel), then analyzed for intracellular IL-17A and IFN-γ after PMA-ionomycin activation as before. Numbers in quadrants indicate percentage of total CD4+ cells in each. Data are representative of at least three experiments.
Remarkably, a large majority of Th17 cells restimulated with IL-12 in the absence of TGF-β produced IFN-γ (~72%) and IL-17A expression was markedly extinguished, such that a small minority of cells produced IL-17A (~8%) after only a single round of culture. With continued maintenance in IL-12, most cells became IFN-γ single-producers (~85%) and IL-17A was nearly completely extinguished (<1%). The addition of TGF-β blunted the effect of IL-12, but there was progressive deviation of the cytokine phenotype toward enrichment of IFN-γ producers and loss of IL-17 producers nonetheless, such that IFN-γ expression became dominant despite continued TGF-β availability. This was in contrast to the maintenance of a Th17 phenotype by combined addition of TGF-β and IL-23 and was observed despite the neutralization of IFN-γ. Thus, although Th17 cells are characterized by their upregulation of IL-23R and relatively low IL-12Rβ2, the latter is sufficient to confer marked, rapid deviation of cytokine phenotype in response to IL-12 when unopposed by TGF-β.
IFN-γ-Producing Effectors Arise from IL-17-Producing Precursors
In the foregoing experiments, an appreciable fraction of T cells generated under Th17-polarizing conditions did not express either IL-17A or IFN-γ, thereby raising the possibility that cells that developed into IFN-γ producers might emerge from uncommitted (IFN-γ− IL-17A−) precursors. Furthermore, because Th17 cells can express IL-17A and IL-17F, we sought to examine the relationship of IL-17F and IL-17A expression and the possibility that cells that deviated to an IL-17A− IFN-γ+ phenotype might retain expression of IL-17F. We therefore generated IL-17F reporter mice in which the Thy1.1 gene was introduced via gene targeting into the endogenous Il17f gene, immediately distal to the stop codon in exon 3 and downstream of an internal ribosome entry site (IRES); these mice are hereafter referred to as Il17fThy1.1/Thy1.1 mice (Figure S2). In these mice, Thy1.1 reporter expression is regulated by the endogenous Il17f locus and expression of the endogenous Il17f gene is not disrupted, thus providing a reporter model with high fidelity and a fully functional Il17f gene that enables identification and isolation of IL-17F+ cells. Importantly, the IL-17F reporter is expressed early in Th17 development and by a greater number of Th17 polarized cells than IL-17A (Figure S2 and data not shown); the large majority of cells that express IL-17A are a subset of cells that express IL-17F, thereby establishing that an appreciable fraction of developing Th17 cells express only IL-17F, and validating IL-17F as a preferable marker for identification of early committed Th17 cells (Figure 3A and Figure S2).
Figure 3. IFN-γ-Producing Effectors Arise from IL-17-Producing Precursors.

(A) FACS-sorted naive CD4+ T cells from Il17fThy1.1/Thy1.1 reporter mice were cultured with irradiated Il12b−/− splenic feeder cells under Th17-polarizing conditions for 6 days as in Figure 1. A fraction of recovered cells were analyzed for expression of CD4, Thy1.1 (IL-17F), and intracellular cytokine IL-17A and IFN-γ after PMA-ionomycin stimulation for 5 hr in the presence of monensin. The flow-cytometric plots are gated on CD4+ cells.
(B) Cells from (A) were collected and Thy1.1+ (IL-17F+) cells isolated by magnetic sorting. The purity of sorted Thy1.1+ cells was confirmed by surface staining of Thy1.1. Numbers in the histograms are the fraction of CD4+ T cells in the indicated gates.
(C and D) Thy1.1+ cells were restimulated with irradiated Il12b−/− splenic feeder cells in the presence of indicated cytokine(s), anti-IFNγ, anti-IL-4, and anti-CD3 as in Figure 1 for a second (C) and fourth round (D) and were stained for CD4, Thy1.1 (IL-17F), and intracellular IL-17A and IFN-γ after PMA-ionomycin activation for 5 hr. Numbers in each quadrant indicate the percentage of total CD4+ T cells. Data are representative of at least three independent experiments.
To examine the fate of committed Th17 cells, we activated naive CD4+ T cells from Il17fThy1.1/Thy1.1 mice with anti-CD3 and Il12b−/− (IL12p40-deficient) splenocytes under Th17-polarizing conditions and isolated Thy1.1+ (IL-17F+) cells by magnetic or FACS sorting (Figure 3B and Figure S3). Recovered IL-17F+ cells were restimulated under distinct cytokine conditions, and expression of IL-17A, IL-17F (Thy1.1) and IFN-γ was assessed after secondary and quaternary cultures (Figures 3C and 3D and Figure S3). In agreement with previous results, cells that expressed IFN-γ, without coexpression of IL-17A or IL-17F, were found to emerge from IL-17F+ Th17 precursors in the presence of IL-12 or IL-23 and absence of TGF-β, with IL-12 deviating the cytokine-expression pattern more rapidly and strongly. Interestingly, a minor subset of cells that coexpressed IL-17A, IL-17F, and IFN-γ developed independently of a requirement for IL-12, IL-23, or IFN-γ signaling and were retained under conditions of continuous TGF-β availability (Figure 3 and Figure S3; data not shown). Notably, IL-17F and IL-17A became coordinately expressed after continued culture in TGF-β and were similarly extinguished by IL-12 and IL-23 signaling without TGF-β. In parallel studies using immobilized anti-CD3 and anti-CD28 to examine the effects of TGF-β, IL-12, and IL-23 on recovered IL-17F+ cells, comparable effects were seen, indicating that the effects on Th17-phenotype shifts are not dependent on indirect effects on APCs (data not shown). Furthermore, the emergence of distinct cytokine phenotypes from initial populations sorted for expression of IL-17F was not due to the outgrowth of a minor contaminating population of IL-17F− cells because intentional contamination of the starting Thy1.1+-sorted population with a fraction of Thy1.1− cells did not substantially alter the observed expression pattern (data not shown, and Figures 2 and 3).
Concordant with the functional effects of IL-12 on committed Th17 cells, we found detectable expression of IL-12Rβ2 mRNA in Th17 cells isolated from IL-17F reporter mice, albeit significantly reduced expression compared to that of Th1 cells (Figure S4). Importantly, the responsiveness of Th17 cells to IL-12 is not transient but is retained over the long term by Th17 cells maintained in the presence of TGF-β, consistent with retention of IL-12Rβ2 expression despite the absence of prior IL-12 or IL-23 signaling or expression of detectable T-bet (Figure S5 and data not shown). Taken together, these data establish that fully committed Th17 cells can convert to IFN-γ-producing cells via IL-12 and IL-23 signaling and that TGF-β maintains IL-17A and IL-17F expression and opposes IL-12- and IL-23-driven Th17 transition to IL-17− IFN-γ+ progeny.
Gene-Expression Signature of IL-12-Driven, IFN-γ-Producing T Cells that Develop from Th17 Precursors
Although the Th17 subset was originally defined as a distinct effector lineage on the basis of its development independently of the Th1 and Th2 lineages (Harrington et al., 2005; Park et al., 2005), the IFNγ+IL-17− phenotype of Th17 progeny generated by IL-12 and IL-23 signaling suggests shared features with classical Th1 cells. To further characterize this population and develop molecular signatures with which to extend comparative studies of IL-12- and IL-23-deviated Th17 cells and classical Th1 cells, we performed gene-expression profiling of Th17 and Th1 cells to identify differentially expressed genes. FACS-sorted naive CD4+ T cells from IfngThy1.1/Thy1.1 reporter mice were activated on Il12b−/− APCs under Th1- or Th17-polarizing conditions for 5 days, isolated and restimulated with PMA and ionomycin for 5 hr, and subsequently enriched for the IFN-γ+ fraction (in Th1 cells) or depleted of the IFN-γ+ fraction (in Th17 cells) by Thy1.1-based magnetic sorting. The sorted Th1 and Th17 cells were processed for microarray-based gene-expression analyses, as were naive CD4+ T cells that had been identically stimulated (Figure S6).
Genes in activated Th1 or Th17 cells that were increased at least 4-fold relative to activated naive cell precursors were determined and compared with one another (Figure S6A). The transcriptome of the Th17 population was quite distinct from that of Th1 cells. Of 1246 genes upregulated in either or both Th17 and Th1 cells, 674 were specifically elevated in Th17 cells and 336 were specifically elevated in Th1 cells. The remaining 236 genes were significantly elevated in both subsets relative to naive controls. Of the genes specifically upregulated in Th17 or Th1 cells, many showed marked expression differences between the subsets, as indicated by heat-map analysis (Figure S6B). We used RT-PCR analysis to validate a subset of the 100 genes most differentially expressed between the populations and examined selected genes for acute expression changes induced in Thy1.1+ (IL-17F+) Th17 cells derived from Il17fThy1.1/Thy1.1 mice restimulated under distinct cytokine conditions (Figure 4).
Figure 4. Divergence of Transcriptomes of Th17 Progeny Restimulated under Different Cytokine Conditions.

FACS-sorted naive CD4+ T cells from Il17fThy1.1/Thy1.1 mice were cultured with irradiated Il12b−/− splenic feeder cells under Th17-polarizing conditions for 6 days. Cells were collected and Thy1.1+ (IL-17F+) cells were isolated by magnetic sorting as in Figure 3. Isolated Thy1.1+ cells were restimulated with anti-CD3 and anti-CD28 in the presence of indicated cytokine(s) and processed for mRNA quantification of the indicated genes at 6 hr (Fasl and Gzma) or 18 hr after stimulation by real-time PCR. All data were normalized to 18S RNA and are expressed as fold differences (log2) relative to anti-CD3- and anti-CD28-stimulated cells. Data are representative of two or three experiments.
Expression of mRNA encoding lineage-associated transcription factors was remarkable for the rapid downregulation of the Th17 factors Rorc and Rora, and reciprocal upregulation of Th1 factor Tbx21 (T-bet) in committed Th17 cells stimulated by IL-12 without TGF-β (Figure 4, upper panel). This correlated well with corresponding downregulation of Il17a and Il17f, and upregulation of Ifng. Comparable effects were observed for IL-23 in cells polarized several rounds without TGF-β, consistent with the more modest, progressive effects of IL-23 compared to the rapid effects of IL-12 (Figure S7). Th1-specific cytocidal factors, exemplified by Fasl and Gzma, demonstrated similarly rapid IL-12-mediated induction in Th17 cells that was blunted by TGF-β (Figure 4, lower panel), as did several other Th1-specific granzyme family members (not shown). Notably, mRNA encoding IL-21, an autocrine factor for Th17 development (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007), was acutely upregulated by IL-12 in the absence of TGF-β. In contrast, the IL-10 family member Il22 was significantly enhanced by IL-23 but suppressed by IL-12, and its induction by IL-23 was blunted by TGF-β, as previously reported (Zheng et al., 2007). Il24, which was found to be the most differentially expressed by Th17 cells among IL-10 family members (Figure S6 and data not shown), was upregulated by IL-23 but suppressed by IL-12 in the absence of TGF-β, analogous to Il22. Interestingly, Ccr6, and its ligand, Ccl20, both lineage-specific markers for Th17 cells, were similarly inhibited by IL-12 and IL-23, whereas Ccr5, a Th1-specific chemokine receptor, was induced by IL-12 and IL-23. Collectively, these data support a rapid reprogramming of Th17 cells by IL-12, with induction of a number of genes that are characteristic of classical Th1 cells, suggesting that Th17 cells retain substantial developmental plasticity to permit deviation toward a more Th1-like phenotype contingent upon IL-12 signaling. Importantly, although IL-23 supported IFN-γ expression, it had effects on late Th17 development that were distinct from those of IL-12 (e.g., IL-22 and IL-24 induction) and that reflected unique features of IL-23-dependent programming related to its indirect effects on epithelial cells (Zheng et al., 2007; Zheng et al., 2008).
Conversion of Th17 Cells Is STAT4 and T-bet Dependent
STAT4 and T-bet are important factors in the IL-12-dependent development of Th1 cells, but are dispensable for the development of Th17 cells (Harrington et al., 2005; Park et al., 2005). However, conversion of Th17 precursors to effectors with features similar to Th1 cells by IL-12—a potent activator of STAT4—implicated this factor in IL-12-dependent silencing of IL-17A and IL-17F and upregulation of IFN-γ. Because IL-23 also activates STAT4, albeit less potently than IL-12 (Mathur et al., 2007; Oppmann et al., 2000), it was possible that STAT4 activation by IL-23 might also contribute to the phenotype shift induced by IL-23. Given the important role of T-bet in Ifng gene expression, we wished to test whether T-bet might also contribute to the observed phenotype shift initiated by IL-12 or IL-23 signaling.
Naive CD4+ T cells isolated from wild-type (WT), Stat4−/−, and Tbx21−/− (T-bet-deficient) mice were differentiated in vitro with WT or Il12b−/− (IL-12p40-deficient) APCs under Th17-polarizing conditions. As shown in Figure 5A, T cell deficiency of STAT4 or T-bet had no significant effect on the development of IL-17-producing Th17 cells, nor did the absence of IL-23 or IL-12, as previously reported (Harrington et al., 2005; Mangan et al., 2006). However, T cell deficiency of either STAT4 or T-bet significantly blunted IL-12-driven upregulation of IFN-γ (Figure 5B). Notably, whereas STAT4-deficient T cells demonstrated comparable expression of IL-17A under all conditions of restimulation, T-bet-deficient T cells showed a significant decrement in the frequencies of IL-17A+ cells after restimulation with IL-12, suggesting that the extinction of IL-17 expression is at least partially dependent on STAT4 via a T-bet-independent mechanism (Figure 5B, right panel). Thus, the complete transition of IL-12-stimulated Th17 precursors is both STAT4 and T-bet dependent, but the requirements for STAT4 and T-bet in IFN-γ upregulation and IL-17 silencing are not the same.
Figure 5. Transition of Th17 Precursors into IFN-γ-Producing Cells Is Stat4 and T-bet Dependent.

(A) CD4+ T cells from WT (BALB/c), Stat4−/−, or Tbx21−/− mice were cultured with either WT or Il12b−/− splenic feeder cells and anti-CD3 under Th17-polarizing conditions for 7 days, and then a fraction was analyzed for CD4 and intracellular IL-17A and IFN-γ after PMA-ionomycin stimulation for 5 hr.
(B) Recovered CD4+ T cells were restimulated with either irradiated WT or Il12b−/− splenic feeder cells in the presence of indicated cytokine(s), anti-IFNγ, anti-IL-4, and anti-CD3 for an additional 5 days and analyzed as in (A).
(C and D) Naive CD4+ T cells from BALB or B6 WT, BALB.Stat4−/−, or B6.Tbx21−/− mice were cultured as in (A) for one round, a fraction was analyzed for CD4 and intracellular cytokines (IL-17A and IFN-γ) as in (A), and the remainder was recultured for three additional rounds under the indicated cytokine conditions as in (B). After the third round of culture, recovered BALB.WT and BALB.Stat4−/− CD4+ T cells ([C], left and right panels) or B6.WT and B6.Tbx21−/− ([D], left and right panels) were activated with PMA-ionomycin and analyzed for CD4 and intracellular IL-17A and IFN-γ. Numbers in quadrants indicate percentages of total CD4+ T cells in each. Data are representative of at least two independent experiments.
Consistent with a role for STAT4 and T-bet in the transition of Th17 precursors by IL-12, the IL-23-driven transition, although less robust in comparison to that induced by IL-12, was similarly STAT4 and T-bet dependent (Figures 5C and 5D). Accordingly, after three rounds of IL-23-driven restimulation of Th17 precursors, the frequencies of IFN-γ+IL-17A− cells were significantly decreased in STAT4- and T-bet-deficient T cells compared to WT controls. In contrast to STAT4-dependent effects of IL-12 on suppression of IL-17 expression, IL-23-driven effects on IL-17 expression appeared more complex, perhaps reflecting more modest STAT4 recruitment by IL-23 and/or its recruitment of STAT3 and STAT1. Finally, it is notable that Th17 cells differentiated in the absence of IL-12, IL-23, IFN-γ, STAT4, or T-bet retain a modest, but significant and reproducible, subset of cells that coexpress IL-17A and IFN-γ, implicating an alternative pathway to IFN-γ expression yet to be defined.
Th17 Precursors Give Rise to IFN-γ-Expressing Cells in a Transfer Model of Colitis
Although IL-17 and IFN-γ production by individual CD4+ T cells tend to be mutually exclusive, both phenotypes have typically been found in autoimmune- and infectious-disease models associated with Th17-mediated immunity. The foregoing in vitro studies identified differential effects of TGF-β, IL-23, and IL-12 on late-stage development of committed Th17 cells and suggested that the IFN-γ-producing T cells found in these models might arise from Th17 precursors. To explore this possibility, we examined the fate and disease-inducing potential of committed Th17 precursors in a transfer model of colitis (Figure 6). Naive CD4+ T cells from Il17fThy1.1/Thy1.1 mice were polarized ex vivo under Th17 conditions, in the absence or presence of IL-23, then FACS sorted for Thy1.1 (IL-17F) expression (Figure S8) and transferred into congenic, RAG-deficient recipients. CD4+ CD45RBhi and CD45RBlo fractions were also isolated from Il17fThy1.1/Thy1.1 reporter mice and were cotransferred as a negative control; alternatively, CD45RBhi cells were transferred alone into RAG-deficient recipients as a positive control, for the development of colitis (Powrie et al., 1994; Powrie et al., 1993).
Figure 6. A Subset of Th17 Cells Extinguish IL-17 and Transition to Single-Positive IFN-γ Cells in Transfer Model of Colitis.

(A and B) FACS-sorted CD4+CD45RBhi T cells (4 ×105) or CD4+CD45RBlo (2 ×105) T cells isolated from pooled spleen and LNs of Il17fThy1.1/Thy1.1 mice were transferred into congenic Rag1−/− mice as indicated. Th17-polarized T cells were generated from naive Il17fThy1.1/Thy1.1 CD4+ T cells with Il12b−/− APCs and anti-CD3 (2.5 μg/ml) as in Figure 1, in the absence (Th17 − IL-23) or presence (Th17 + IL-23) of 10 ng/ml IL-23. Thy1.1− (IL-17F− ) and Thy1.1+ (IL-17F+) cells were isolated by FACS sorting (see Figure S8), and 3 ×105 cells were transferred as indicated. As shown in (A), 8 weeks after transfer, mice were sacrificed and intestinal tissues were collected for histological processing and analysis. Shown are representative sections of colon from indicated experimental groups; all magnifications represent 40×. (B) shows the incidence and severity of colitis 4 (upper panel) or 8 (lower panel) weeks after transfer of the indicated cell populations. n represents the number of recipients in each group.
(C and D) IL-17A, Thy1.1 (IL-17F), and IFN-γ expression of CD4+ T cells isolated from MLNs of indicated recipient mice 4 weeks (upper two panels) or 8 weeks (lower panel) after cell transfer. Recovered cells were stimulated ex vivo with PMA-ionomycin and analyzed for CD4, Thy1.1, and intracellular IL-17A and IFN-γ. Numbers in each quadrant indicate percentages of total CD4+ T cells (C). Cumulative data for frequencies of cytokine-positive CD4+ T cells, as analyzed as in (C), are shown in (D). Data are the means ± SEM for tissues and groups indicated. All data are representative of results from five similar experiments.
Transfer of Th17-committed cells induced significant disease that was comparable to that induced by CD45RBhi cells at 8 weeks after transfer, whether the Th17 population was generated in the absence or presence of IL-23 (Figures 6A and 6B). Importantly, however, disease onset was considerably more rapid in mice receiving IL-17F+ Th17 populations compared to CD45RBhi controls (Figure 6B and Figure S9). The Th17 cells pre-treated with IL-23 induced significantly higher disease scores than Th17 cells not pretreated with IL-23 at 4 weeks after transfer (p < 0.05), although this did not achieve statistical significance at 8 weeks. Notably, disease was also induced by transfer of Th17-polarized cells sorted for lack of IL-17F expression, and although the disease severity was significantly reduced at early time points, this pool of cells retained the capacity to develop into pathogenic effectors.
Concordant with the onset of significant colitis histologically, flow-cytometric analyses of T cells recovered from diseased mice and activated ex vivo showed similar cytokine-expression phenotypes, whether isolated from recipients of Thy1.1+ Th17 cells induced with or without IL-23 or from recipients of CD45RBhi cells (Figures 6C and 6D). Although essentially all transferred Th17 cells initially expressed IL-17F (Thy1.1+) (Figure S8), there was marked, progressive extinction of IL-17F relative to IL-17A over the course of colitis development. Thus, although approximately half of the transferred cells expressed IL-17F alone (IL-17-F+IL-17A−), and all IL-17A+ cells coexpressed IL-17F (IL-17F+IL-17A+), few IL-17F+ cells were recovered, and the large majority of IL-17F+ cells remaining were a subset of IL-17A+ cells. Further, there was significant induction of IFN-γ expression in transferred T cells (Figures 6C and 6D), despite negligible or undetectable IFN-γ+ cells in the sorted fractions prior to transfer (Figure S8). Neither the observed IL-17F extinction or IFN-γ enhancement appeared to be due to preferential enrichment of IL-17+ cells in the inflamed intestines, given that T cells recovered from the colons of diseased mice showed a similar cytokine phenotype to those of the mesenteric lymph nodes (MLNs) (data not shown). Finally, the cytokine-phenotype shift observed in the transferred T cells was not accompanied by expression of IL-10 because few (<0.4%) of CD4+ T cells expressed detectable IL-10 prior to transfer or after recovery (Figure S9 and data not shown). Thus, as in vitro, committed Th17 cells can give rise to progeny that extinguished IL-17 expression and upregulated IFN-γ expression, although, interestingly, the majority of recovered cells expressed neither. These results further establish that Th17 cells induced by TGF-β and IL-6, either in the absence and presence of IL-23, are colitogenic after transfer into immunodeficient recipients and give rise to a subset of IFN-γ+ progeny that emerges as a substantial component of the effector T cell response during the development of immunopathology.
To determine whether IL-12 or IL-23 was required for the phenotype shift observed in mice that received transfers of committed Th17 precursors, we examined the effects of neutralizing antibodies to IL-23R or IL-12p40 on the development of colitis and cytokine phenotype of transferred T cells generated from Il17fThy1.1/Thy1.1 mice. Administration of either anti-IL-23R or anti-IL-12p40 significantly ameliorated disease development induced by transfers of sorted IL-17F+ Th17 populations 4 weeks after transfer (p < 0.01; Figure 7A), regardless of whether the transferred T cells were pretreated with IL-23. Blockade of IL-23 signaling resulted in a significant increase in ratio of IFN-γ- to IL-17A-producing T cells (Figure 7B) because of a higher fraction of IFN-γ+ T cells (~2-fold increase) among the CD4+ T cells recovered; there was no significant change in the fraction of IL-17A+. However, the total numbers of CD4+ T cells numbers were substantially reduced (>90%). Thus, absolute numbers of IL-17+ and IFN-γ+ cells were markedly reduced in the absence of IL-23. Notably, anti-IL-12p40 administration caused an even more profound loss of recoverable T cells, obviating assessment of the effects on combined blockade of IL-23 and IL-12 signaling on late-stage phenotype conversion and suggesting that survival of transferred Th17 cells was not sustained in the absence of IL-23 or IL-12 signaling.
Figure 7. Effects of IL-23 and IL-12 Blockade on Th17 Precursors in Transfer Model of Colitis.
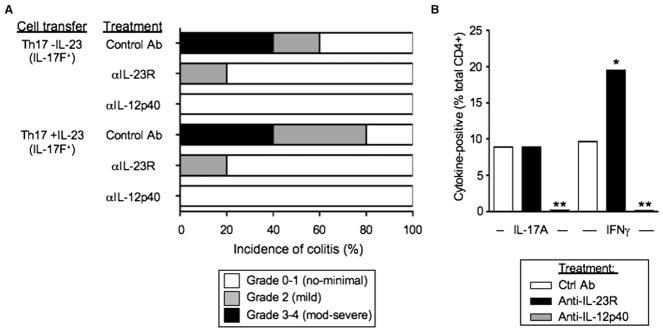
(A) Th17-polarized T cells were generated from naive Il17fThy1.1/Thy1.1 CD4+ T cells with Il12b−/− APCs and anti-CD3 (5 μg/ml) as in Figure 1, in the absence (Th17 -IL-23) or presence (Th17 +IL-23) of 5 ng/ml IL-23. Thy1.1+ (IL-17F+) cells were isolated by FACS sorting and 3 × 105 cells were transferred into congenic Rag1−/− mice as indicated. Recipients were administered 100 μg of anti-IL-23R, anti-IL-12p40, or isotype control via i.p. injection in saline 1 day prior and 1 day after T cell transfers and once weekly thereafter (five total doses). Mice were sacrificed 4 weeks after transfer and analyzed histologically and scored for development of colitis.
(B) Mice that received Th17 -IL-23 (IL-17F+) T cells and treated as in (A) were sacrificed 4 weeks after transfer, and cells recovered from MLNs were stimulated ex vivo with PMA ionomycin and analyzed for CD4 and intracellular IL-17A and IFN-γ by FACS. Data are the means from five mice in each treatment group. *p < 0.05 and **p < 0.01 versus control treatment group. Data are representative of two similar experiments.
DISCUSSION
A prominent feature of Th17 immunity induced in both infectious and autoimmune settings is the development of a substantial component of IFN-γ-producing cells, some of which coexpress IL-17, but a majority of which don’t. The recognition early that IL-23 induction of IL-17 expression in the memory pool of CD4+ T cells was accompanied by low amounts of IFN-γ expression (Oppmann et al., 2000), together with the recognition that IL-23 and IL-12 shared the common IL-12p40 (IL-12β) subunit and their receptors used the common IL-12Rβ1 subunit, gave rise to speculation that IL-17-producing effectors developed as a late branch of the Th1 lineage (Bettelli and Kuchroo, 2005; Mathur et al., 2006; McKenzie et al., 2006). In this study, we provide evidence that the opposite is true. Using IL-17F reporter mice for isolation of committed IL-17-producing effectors, we found that Th17 precursors coexpressed IFN-γ independently of IL-23 and IL-12—or STAT4—signaling, but also responded to IL-23 and IL-12 to give rise to progeny that shifted their phenotype toward enhanced IFN-γ expression and IL-17A and IL-17F extinction, contingent upon limited or absent TGF-β. Thus, Th17 cells demonstrate substantial developmental plasticity after their commitment to the Th17 program, and TGF-β appears to be critical for the maintenance of IL-17 expression, at least in part by opposing the STAT4-mediated effects of IL-23—and particularly IL-12—to extinguish expression of IL-17F and IL-17A and promote expression of IFN-γ. A role for TGF-β in both the induction and maintenance of IL-17A and IL-17F expression draws a further parallel in shared functions for this cytokine in the developmental programs of both Th17 and induced Treg cells (Weaver et al., 2006).
Although differential expression of IL-23R and IL-12Rβ2 has been thought to distinguish Th17 and Th1 developmental programs, we found that whereas IL-12Rβ2 expression is substantially diminished in Th17 cells, it remains fully functional. Even after long-term maintenance of Th17 cells in the absence of IL-12 or IFN-γ, there was rapid phenotype deviation in response to IL-12, indicating mechanisms for induction and retention of IL-12Rβ2 that are not extinguished by TGF-β. TCR signaling alone can induce low levels of IL-12Rβ2 in human T cells in anticipation of STAT4 signaling (Letimier et al., 2007), consistent with our results and similarly to effects of IL-12 on human Th17 clones (Annunziato et al., 2007). Thus, the balance of TGF-β and IL-12 or IL-23 controls the late-stage phenotype of Th17 cells. Activation of STAT4 by IL-12 and IL-23 was important for the induction of Th17 progeny that expressed increased IFN-γ in the absence of IL-17 coexpression, and the more potent effects of IL-12 in the induction of the IL-17 to IFN-γ transition are consistent with stronger activation of STAT4 by the IL-12 receptor (Oppmann et al., 2000; Parham et al., 2002). Although STAT4 was essential for IL-12-mediated suppression of IL-17A and IL-17F and for IFN-γ induction, the effects of T-bet induction appeared more limited to the enhancement of IFN-γ; deficiency of T-bet did not preclude the suppression of IL-17 by IL-12 or IL-23, although the reduced expression of T-bet induced by IL-23 in the absence of TGF-β complicates this conclusion.
Importantly, although both IL-23 and IL-12 promoted increased IFN-γ expression and deviation from IL-17 expression in Th17 precursors, the programs induced were not the same. Both kinetics of deviation and transcriptomes of the cells induced differed. Thus, despite the fact that both IL-12 and IL-23 showed similarly rapid effects on the suppression of a number of “signature” Th17 factors (e.g., CCR6, CCL20, and AhR), the suppression of IL-17A and IL-17F and upregulation of T-bet and IFN-γ occurred within hours in response to IL-12, but required days in response to IL-23. Furthermore, IL-23-induced effects were more potently opposed by TGF-β signaling and IL-23 typically failed to completely extinguish IL-17A. In support of previous studies (Kolls and Linden, 2004; McGeachy et al., 2007; Zheng et al., 2007), we found that IL-23 is unique in its promotion of IL-22 expression; compared to IL-23, IL-12 and TGF-β both markedly blunted induction of IL-22. Interestingly, chronic IL-23 signaling showed differential effects on Rorc and Rora expression; the former was suppressed in association with upregulation of Tbx21, whereas Rora expression was retained.
The transcriptome of Th17 cells deviated by IL-12 signaling was largely indistinguishable from classical Th1 cells. In the absence of tempering TGF-β activity, IL-12 rapidly suppressed the Th17 transcription factors Rorc and Rora, but upregulated Tbx21, and promoted expression of the cytocidal factors Fasl and multiple granzymes (e.g., Gzma) among other genes that are highly expressed by conventional Th1 cells, but not Th17 cells. This extends the importance of IL-12 as a potent cytokine for promoting an effector program well adapted for eradication of intracellular pathogens and implies that the “master regulators” of Th17 differentiation—RORγt and RORα (Ivanov et al., 2006; Yang et al., 2008)—can be overridden by cell-extrinsic factors.
Prior to discovery of the Th17 lineage and a nonredundant role for IL-23 in several models of autoimmunity, immune pathogenesis in these models was ascribed to dysregulated Th1 responses (reviewed by McGeachy and Cua, 2008; Weaver et al., 2007). A simple Th1 correlation was complicated by the persistence of disease development in studies using mice deficient for IFN-γ, IFN-γR, STAT1, or IL-12p35 (Becher et al., 2002; Bettelli et al., 2004; Ferber et al., 1996; Gran et al., 2002; Willenborg et al., 1996) and was largely dispelled with the discovery that mice deficient for IL-23 (IL23a−/−), but not IL-12 (Il12a−/−), were resistant to autoimmunity (Cua et al., 2003). Accordingly, T cell-transfer studies demonstrated more potent disease induction by Th17 cells compared to Th1 cells (Elson et al., 2007; Langrish et al., 2005), and successful treatment with blocking antibodies specific for IL-23 established that Th17 cells were the principal effectors in autoimmune pathogenesis in several disease models (Chen et al., 2006; Elson et al., 2007). Nevertheless, mice deficient in STAT4 or T-bet, which are dispensable for Th17 development, are resistant to autoimmunity in these models (Bettelli et al., 2004; Chitnis et al., 2001; Neurath et al., 2002), suggesting that these signaling factors might play a Th17-independent role. The findings herein link STAT4 and T-bet to a late shift in the Th17 developmental program, thereby offering an explanation for these results, and may explain the observed predominance of Th17 cells early and “Th1” cells late in the CNS in EAE (Dardalhon et al., 2008). Although studies in additional autoinflammatory models will be needed, our colitis studies establish that IFN-γ cells can emerge from Th17 precursors in the context of T cell-driven inflammation, and although precise mechanisms remain to be established because of the loss of Th17 cell survival in the absence of IL-23 and IL-12, we speculate that in the absence of STAT4 and T-bet, aspects of the late events in Th17 development critical for maintenance of ongoing inflammation are impaired. This is highlighted by the balance of IL-17A+IFN-γ− and IL-17A− IFNγ+ cells in colitic mice and the lack of colitis development after adoptive transfers of CD45RBhi T cells deficient in T-bet expression (Neurath et al., 2002).
An important facet of our findings concerns the phenotype of Th17 cells maintained in TGF-β. In contrast to a recent report that demonstrated an IL-10-dependent protective effect of Th17 cells induced with TGF-β and IL-6 in the absence of IL-23 (McGeachy et al., 2007), we found minimal induction of IL-10 expression in cells polarized with TGF-β and IL-6, even after prolonged culture under these Th17-inducing conditions in the absence of IL-23, and Th17 cells induced under these conditions were pathogenic, not protective upon adoptive transfers. Accordingly, the transfer of TGF-β-and-IL-6-polarized Th17 cells derived from IL-17F reporter mice into immunodeficient hosts induced colitis similarly to that of CD45RBhi transfers (Powrie et al., 1996; Powrie et al., 1994), implying that the IL-10-associated regulatory function attributed to Th17 cells induced without IL-23 may be model specific. Indeed, our finding that TGF-β is a maintenance factor for IL-17A and IL-17F expression by Th17 cells might also contribute to the anticolitic effect of TGF-β neutralization previously attributed to blockade of regulatory cell function in the CD45RBhi model (Powrie et al., 1996). However, CD45RBhi cells that are poorly responsive to TGF-β by virtue of expression of a dominant-negative transgenic TGF-β receptor (dnTβRII) induced colitis after adoptive transfers into Rag1−/− mice (Izcue et al., 2008), albeit with reduced disease severity. It is unclear whether this might be due to default Th1 development, which can also drive colitis (Iqbal et al., 2002), whether TGF-β signaling in these cells remains adequate for some TGF-β-dependent functions, or whether other factors are involved. Additional studies to define the cytokine phenotypes and the origins of colitiogenic T cells that emerge under conditions of reduced TGF-β actions in this model will be needed to resolve this.
A notable feature of Th17 biology revealed by the Il17fThy1.1/Thy1.1 reporter mouse concerns the relative expression patterns of IL-17A and IL-17F. IL-17F expression was dominant early in Th17 development, such that a substantial fraction of Th17 cells expressed IL-17F alone and the majority of IL-17A-expressing cells were a subset of the larger IL-17F pool, consistent with early developmental predominance of IL-17F homodimer (IL-17F-F) and IL-17A-F heterodimer expression and limited IL-17A homodimer expression (Liang et al., 2007). TGF-β promoted the enhanced expression of both cytokines in vitro, inducing progressive convergence of IL-17A and IL-17F expression; thus essentially all cells maintained coexpression of both cytokines with continuing TGF-β stimulation. Interestingly, IL-23 restimulation in the absence of TGF-β appeared to induce more pronounced extinction of IL-17F than IL-17A, resulting in a predominance of IL-17A expression by cells that retained IL-17. Given the higher binding affinity of IL-17A-A for the IL-17RA receptor (Hymowitz et al., 2001), and its more potent proinflammatory effects compared to IL-17A-F or IL-17F-F in some settings (Chang and Dong, 2007; Liang et al., 2007), this may represent an important effect of IL-23 in promoting Th17 inflammation and is consistent with the relative retention of IL-17A compared to IL-17F in our colitis studies.
Why has the Th17 lineage evolved to enable a shift from dominant IL-17 to dominant IFN-γ expression? Given that the selective pressures shaping the emergence of the Th17 lineage have been driven by pathogenic challenges, important evolutionary benefit must be derived from this developmental transition. Several evolutionary benefits are envisioned and are not mutually exclusive. First, given the potent tissue destructive capacity of the “rapid response,” neutrophilic inflammation driven by IL-17 cytokines, transition to a less tissue destructive, monocytic pattern of inflammation promoted by IFN-γ, i.e., transition from an “acute” to “chronic” inflammation pattern, could represent an evolutionary adaptation to limit tissue injury during late-stage clearance of certain infectious agents. In this regard, IFN-γ inhibits Th17 development, and the transition to IFN-γ production might play a role in negative feedback on Th17 development as a mechanism to regulate exaggerated IL-17 production while retaining an active protective response. Alternatively, the IL-17-to-IFN-γ transition may represent a strategy by which the T cell response can adapt to modulation of survival strategies employed by pathogens that can shift to an intracellular survival mode to evade host mechanisms of extracellular eradication. Association of Th17 responses with a number of extracellular bacterial and fungal pathogens has been established, although few pathogens induce T cell IL-17 responses without a corresponding induction of IFN-γ. Even pathogens that are dependent upon IFN-γ for host protection, such as Mycobacterium tuberculosis, can elicit early IL-17 responses that transition to IFN-γ-dominated responses (Khader et al., 2007). Although this has been attributed in part to a role for Th17 cells in recruiting Th1 cells in rechallenge responses, we propose that much of the Th1 “recruitment” attributed to Th17 cells reflects transition of primed Th17 effectors to IFN-γ-producing progeny; the transition of antigen-specific, antipathogenic Th17 cells to IFN-γ producers with distinct functional properties might permit the adaptive T cell response to efficiently coordinate pathogen clearance in response to changing cytokine cues from innate immune cells as the pathogen adapts to evade first-line host clearance mechanisms. Finally, the enrichment of Th17 cells at host-environment interfaces, particularly the intestines, in which there is abundant TGF-β and an ongoing homeostatic response to the commensal flora, may facilitate rapid responses to epithelial barrier breach to accentuate clearance of extracellular organisms while facilitating clearance of organisms ingested by newly recruited myeloid cells that can produce IL-12 and shift the T cell response to enhance intracellular killing of ingested microbes. In a sense then, Th17 cells represent the ultimate adaptive immune cells, representing a “two-for-one” effector T cell lineage capable of adjusting its effector program as needed to match requirements for efficient orchestration of early and late immune-clearance mechanisms and balancing a need to retain antipathogen responses while curbing the host tissue destruction wrought by unchecked neutrophilic responses.
EXPERIMENTAL PROCEDURES
Mice
The following mice were purchased from the Jackson Laboratories and/or bred at our facility: BALB/cByJ (BALB/c), C57BL/6J (B6), B6.OT-II TCR transgenic mice (OT-II), BALB/c.129S2-Stat4tm1Gru/J (STAT4-deficient), B6.129S6-Tbx21tm1Glm/J (T-bet-deficient), B6.129S1-Il12btm/Jm/J (IL-12p40-deficient), and B6 Rag1−/−. The generation of IFNg-Thy1.1 (IfngThy1.1/Thy1.1) reporter mice was described previously (Harrington et al., 2008). BALB.Tbx21−/− mice used in some studies were a kind gift from D. Bullard (UAB). All animals were bred and maintained in accordance with institutional animal care and use committee regulations.
Generation of Il17fThy1.1/Thy1.1 Reporter Mice
The BAC clone RP24-305203, which contains the Il17f and Il17a genes, was purchased from Children’s Hospital Oakland Research Institute (CHORI). The Il17f gene and flanking sequence were retrieved from the BAC clone into a pBluescript plasmid containing a TK cassette by recombineering-based gap repair (Harrington et al., 2008). A targeting construct containing an EMCV IRES element, Thy1.1 coding sequence, and LoxP-flanked neomycin resistance cassette was flanked with miniarms of homology to exon 3 Il17f genomic sequence and integrated between the Il17f stop codon and 3′ untranslated sequence (Figure S2), as previously described for similar targeting of the Ifng gene (Harrington et al., 2008). The Il17F-Thy1.1 targeting construct was linearized with NotI and electroporated into Bruce4 mouse embryonic stem cells, as previously described (Harrington et al., 2008). Drug-resistant clones were selected for homologous recombination by Southern-blot analysis and microinjected into albino C57BL/6-blastocysts at the UAB Transgenic Mouse Facility, and founder lines were established from chimeric mice as described (Harrington et al., 2008 and see Figure S2).
CD4+ T Cell Preparation and Culture
CD4+ T cells were purified from pooled spleen and lymph nodes by mouse CD4 Dynabeads and then DETACHaBEAD mouse CD4+, according to the manufacturer’s directions (Invitrogen). Naive CD4+ T cells were isolated by FACS sorting for the CD25loCD62Lhi fraction. Unless otherwise indicated, naive CD4+ T cells were cultured at a ratio of 1:5 with irradiated splenic feeder cells for 7 days in RPMI containing 10% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 μM sodium pyruvate, 1× non-essential amino acids, 2.5 μM β-mercaptoehanol, and 2 μM L-glutamine (R-10). OT-II TCR transgenic CD4+ cells were activated with 5 μg/ml OVA peptide (OVAp), whereas nontransgenic cells were stimulated with 2.5 μg/ml anti-CD3 (clone 145-11), supplemented with 5 ng/ml rhTGF-β1 (R&D Systems), 20 ng/ml rmIL-6 (R&D Systems), 10 μg/ml anti-IFN-γ (clone XMG1.2), and 10 μg/ml anti-IL-4 (clone 11B11). In restimulation cultures, viable T cells were recovered on day 7 of previous cultures and activated with fresh splenocytes, cytokines, and antibody mixtures as indicated. Unless otherwise indicated, rmIL-23 or rmIL-12 (R&D Systems) were added at 1 ng/ml. Thy1.1+ cells of Il17fThy1.1/Thy1.1 or IfngThy1.1/Thy1.1 reporter mice were isolated or depleted by magnetic sorting according to the manufacturer’s instructions (Miltenyi) or by FACS sorting, as indicated.
Flow-Cytometric Analyses
CD4+ T cells were collected and, where indicated, stimulated with PMA (50 ng/ml; Sigma) and ionomycin (750 ng/ml; Calbiochem) for 5 hr in the presence of Golgi Plug (BD). Intracellular staining was performed as previously described (Maynard et al., 2007). We used LIVE/DEAD Fixable Green Dead Cell Stain (Invitrogen) to exclude dead cells in flow-cytometric analyses. Fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ (XMG1.2), phycoerythrin (PE)-conjugated anti-CD90.1 (OX-7), anti-IL-17 (TC11-18H10), and peridinin chlorophyll protein (PerCP)-conjugated anti-CD90.1 (OX-7) were purchased from BD Biosciences; Alexa-Fluor-647-conjugated anti-IL-17A (17B7), anti-IL-17F (18F10), allophycocyanin (APC)-conjugated anti-IL-10 (JES5-16S3), and PE-Cy7-conjugated anti-CD4 (GK1.5) were purchased from eBioscience. Samples were acquired on an LSRII instrument (BD Biosciences), and data were analyzed with CellQuest Pro (BD Biosciences) or FlowJo software (Tree Star).
Gene-Expression Analyses
mRNA was extracted from T cells with TRIZOL (Invitrogen) and then treated with DNA-free (Ambion). cDNA synthesis was performed with Superscript III first-strand synthesis system (Invitogen). Real-time PCR was performed on a Bio-Rad iCycler with primer pairs specific for cDNAs of Il12rb1, Il12rb2, Il23r, Rorc, Rora, Tbx21, Il17a, Il17f, Ifng, Ccr6, Ccr20, Ccr5, Il22, Il24, Il21, and Fasl and Gzma mRNA transcripts with SYBR GreenER qPCR supermix (Invitrogen). The following primer sequences were used: IL-12Rβ1, sense primer, 5′-TACAGTTCAGGCGCCGGAT-3′, antisense primer, 5′-AGAGTTAACCTGA GGTCCGCAG-3′; IL-12Rβ2, sense primer, 5′-CCTCTTAACAGCACGTCCTG G-3′, antisense primer, 5′-GGTCTCAGATCTCGCAGGTCA-3′; IL-23R, sense primer, 5′-GCCAAGAAGACCATTCCCGA-3′, antisense primer, 5′-TCAGTGCT ACAATCTTCTTCAGAGGACA-3′; Rorγt, sense primer, 5′-CAGCCAACATGTG GAAAAGCT-3′, antisense primer, 5′-GGGAAGGCGGCTTGGA-3′; Rorα, sense primer, 5′-TGACGCCCACCTACAACATC-3′, antisense primer, 5′-CATCCATA TAGGTGCTGAGGTCAT-3′; IL-17A, sense primer, 5′-TGAAGGCAGCAGCGA TCA-3′, antisense primer, 5′-GGAAGTCCTTGGCCTCAGTGT-3′; IL-17F, sense primer, 5′-CGCCATTCAGCAAGAAATCC-3′, antisense primer, 5′-CTCC AACCTGAAGGAATTAGAACAG-3′; IFN-γ, sense primer, 5′-ACAATGAACGCT ACACACTGCAT-3′, antisense primer, 5′-TGGCAGTAACAGCCAGAAACA-3′; CCR6, sense primer, 5′-CCTCTGTGCCCGGGTTTAC-3′, antisense primer, 5′-CATTATCATTTTCGACGGTCTCACT-3′; CCL20, sense primer, 5′-TCAACT CCTGGAGCTGAGAATG-3′, antisense primer, 5′-CCATGCCAAAGCAAGGAA GA-3′; CCR5, sense primer, 5′-GTTCCTGAAAGCGGCTGTAAA-3′, antisense primer, 5′-GCAGTCAGGCACATCCATAGAC-3′; IL-24, sense primer, 5′-CTGT GCAAACTCAGGATGACATC-3′, antisense primer, 5′-CGAGACATTCCGCAG AACCT-3′; FASL, sense primer, 5′-GGCTGGGTGCCATGCA-3′, antisense primer, 5′-GGCACTGCTGTCTACCCAGAA-3′; Granzyme A, sense primer, 5′-GGGTGGGAGAGCCACGAT-3′, antisense primer, 5′-GAAGAAAAAGGAGAGT AGCAAGAGATG-3′; IL-22, sense primer, 5′-TCCGAGGAG TCAGTGCTAA A-3′, antisense primer, 5′-AGAACGTCTTCCAGGGTGAA-3′; and IL-21, sense primer, 5′-AAGATTCCTGAGGATCCGAGAAG-3′, antisense primer, 5′-TGCAT TC GTGAGCGTCTATAGTG-3′. Reactions were run in triplicate and normalized to 18S rRNA.
Adoptive-Transfer Model of Colitis
FACS-sorted naive CD4+ T cells from Il17fThy1.1/Thy1.1 mice were cultured with Il12b−/− feeder cells under Th17-polarizing condition for 6 days, and viable cells were recovered (Lympholyte; Cedarlane). Selection for Thy1.1 expressing cells was performed by magnetic or FACS sorting, and isolated CD4+Thy1.1+ cells were washed and resuspended in PBS for adoptive transfer. A total of 3 ×105 Thy1.1+ cells were injected i.p. into age-matched B6.RAG1-deficient recipients. For CD45RB transfer controls, CD4+ T cells from pooled spleen and lymph nodes of Il17fThy1.1/Thy1.1 reporter mice were isolated as above, with Dynal Beads, and the recovered CD4+ T cells were stained with APC-conjugated anti-CD4 (RM4-5) and FITC-conjugated anti-CD45RB (16A) (BD Biosciences) sorted into CD4+CD45RB+ and CD4+CD45RB− fractions as previously described (Maynard et al., 2007). Sorted cells were washed and resuspended in PBS for adoptive transfers. B6.RAG1-deficient recipients were injected intraperitoneally with 4.0 ×105 cells. For treatment studies, recipient mice were injected i.p with 100 μg of anti-IL-23R (a gift from R. Kastelein, SP Biopharma), anti-IL-12p40 (C17.8), or isotype control in saline 1 day before and 1 day after T cell transfers and once weekly thereafter.
All recipient mice were monitored regularly for signs of disease, including weight loss, hunched appearance, pilo-erection of the fur coat, and loose stools. At 4 or 8 weeks after transfer, mice were sacrificed and spleen, MLNs, and colon were recovered. Colons were cut longitudinally, and ~2 cm lengths of tissue were obtained from the proximal, middle, and distal portions of the colon, fixed immediately in 10% formalin, and processed for histopathological analyses (Maynard et al., 2007). Samples were coded and scored by a pathologist in a blinded fashion as previously described (Maynard et al., 2007). Expression of Thy1.1 (IL-17F) and intracellular IL-17A and IFN-γ from recovered T cells was assessed by flow-cytometric analysis as above, after activation with PMA and ionomycin ex vivo.
Statistical Analysis
Statistical significance was calculated by unpaired Student’s t test, Mann-Whitney U or ANOVA as appropriate, with Prism software (GraphPad). All p values ≤ 0.05 are considered significant and are referred to as such in the text. Unless otherwise specified, all studies for which data are presented are representative of at least two similar studies.
Supplementary Material
Acknowledgments
The authors are grateful to D. Chaplin, T. Benveniste, and C. Klug, as well as members of the Weaver laboratory, for their helpful comments and suggestions. We gratefully acknowledge R. Kastelein for provision of anti-IL-23R. We also thank C. Song, M. Blake, and B.J. Parsons for expert technical assistance and G. Gaskins for editorial assistance and acknowledge the UAB Transgenic Mouse Facility for blastocyst injections, the UAB Digestive Diseases Research Developmental Center (DDRDC) for generation and phenotyping of gene-targeted mice, the UAB Epitope Recognition and Immunoreagent Core Facility for antibody preparations, and the UAB Comprehensive Cancer Center Gene Expression Shared Facility for gene-expression studies. This work was supported by grants from the NIH (C.T.W.) and Daichi-Sankyo Co. Ltd. (C.T.W.).
Footnotes
Note Added in Proof
During revision of the current manuscript, an article by Lexberg et al. used a cytometric IL-17A secretion assay to identify plasticity in Th17 cells similar to findings in our study (Eur. J. Immunol. 38, 2654–2664, 2008). Their study also identified resistance to phenotype deviation of IL-17A+ cells found in the CD4+CD62Llo memory pool, suggesting that mechanisms exist, as yet undefined, for stabilizing the cytokine phenotype of at least a subset of IL-17A+ cells.
ACCESSION NUMBERS
Gene-expression data have been deposited in the NCBI Gene Expression Omnibus with the accession number GSE 14026.
Supplemental Data include nine figures and can be found with this article online at http://www.immunity.com/supplemental/S1074-7613(08)00546-3.
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110:493–497. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Kuchroo VK. IL-12- and IL-23-induced T helper cell subsets: Birds of the same feather flock together. J Exp Med. 2005;201:169–171. doi: 10.1084/jem.20042279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17:435–440. doi: 10.1038/cr.2007.35. [DOI] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773–4777. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- Gorham JD, Guler ML, Fenoglio D, Gubler U, Murphy KM. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J Immunol. 1998;161:1664–1670. [PubMed] [Google Scholar]
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: Evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452:356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal N, Oliver JR, Wagner FH, Lazenby AS, Elson CO, Weaver CT. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J Exp Med. 2002;195:71–84. doi: 10.1084/jem.2001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Letimier FA, Passini N, Gasparian S, Bianchi E, Rogge L. Chromatin remodeling by the SWI/SNF-like BAF complex and STAT4 activation synergistically induce IL-12Rbeta2 expression during human Th1 cell differentiation. EMBO J. 2007;26:1292–1302. doi: 10.1038/sj.emboj.7601586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, Goldman SJ, Dunussi-Joannopoulos K, Williams CM, Wright JF, Fouser LA. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- Lin JT, Martin SL, Xia L, Gorham JD. TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: Differential involvement of Stat4 and T-bet. J Immunol. 2005;174:5950–5958. doi: 10.4049/jimmunol.174.10.5950. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Cua DJ. Th17 cell differentiation: The long and winding road. Immunity. 2008;28:445–453. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Villarino A, Hibbert L, Lieberman L, Wilson E, Mak T, Yoshida H, Kastelein RA, Saris C, Hunter CA. The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity. 2003;19:645–655. doi: 10.1016/s1074-7613(03)00300-5. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.