

Summary
Discovery of the T-helper 17 (Th17) subset heralded a major shift in T-cell biology and immune regulation. In addition to defining a new arm of the adaptive immune response, studies of the Th17 pathway have led to a greater appreciation of the developmental flexibility, or plasticity, that is a feature of T-cell developmental programs. Since the initial finding that differentiation of Th17 cells is promoted by transforming growth factor-β (TGFβ), it became clear that Th17 cell development overlapped that of induced regulatory T (iTreg) cells. Subsequent findings established that Th17 cells are also unusually flexible in their late developmental programming, demonstrating substantial overlap with conventional Th1 cells through mechanisms that are just beginning to be understood but would appear to have important implications for immunoregulation at homeostasis and in immune-mediated diseases. Herein we examine the developmental and functional features of Th17 cells in relation to iTreg cells, Th1 cells, and Th22 cells, as a basis for understanding the contributions of this pathway to host defense, immune homeostasis, and immune-mediated disease.
Keywords: Th17 cells, Th1 cells, Treg cells, Th22 cells, T-cell development, plasticity, cytokines
Introduction
Naive CD4+ T cells are antigen-specific and multipotent. They continuously circulate between blood and lymph, passaging between the two circulatory systems through secondary lymphoid tissues that are specialized to promote their interaction with antigen-presenting cells (APCs) bearing antigens collected from tissues. Activation of a naive CD4+ T cell by recognition of processed antigenic peptides presented in complex with major histocompatibility complex (MHC) class II molecules on APCs initiates a program of differentiation in T-cell zones that is coordinated with clonal expansion to generate progeny with the potential for divergent functional specialization contingent on the integration of multiple signals from the local microenvironment. The type and intensity of the signals the T cell receives are largely determined by the dose of antigen and its intrinsic properties to activate APCs and other innate immune cells via pattern-recognition receptors (PRRs). This occurs rapidly and is communicated to activated T cells largely through cytokines, although non-cytokine mediators are also important. As the central cell coordinating immune responses, the linkage of distinct CD4+ T-cell developmental programs to environmental cues from innate immune cells serves to appropriately and efficiently match adaptive immunity to homeostatic, anti-inflammatory or host-protective, pro-inflammatory immune responses, as dictated by recognition of antigens derived from the extended metagenome of the host and its commensal microbiota or microbial pathogens, respectively.
Originally conceived in the context of a bipolar framework, the Th1-Th2 hypothesis, an increasingly complex view CD4+ T-cell lineage specification has emerged wherein at least four major subsets can be defined on the basis of distinct developmental and functional programming of naive precursors in secondary lymphoid tissues: Th1, Th2, Th17, and induced regulatory T (iTreg) cells. A case can be made for follicular helper T cells (Tfh) as an independent subset specialized for germinal center responses (1), although it remains to be determined whether Tfh cells represent instead a developmental branch of each of the other subsets. Now, precipitated initially by findings that linked early Th17 and iTreg development and followed by demonstration of the transition of Th17 cells to Th1-like progeny, the long-entrenched view that T-helper subsets are stable has given way to an appreciation of considerable plasticity (2, 3). In this review, we highlight the special case of the Th17 lineage, which, along with the developmentally linked iTreg lineage, is at the top of the hierarchy of programming plasticity among the established CD4+ T-cell lineages.
T-helper subsets: specialization driven by the need for a diversified antimicrobial defense
The evolutionary forces driving diversification and functional specialization of CD4+ T-cell subsets are based in the central role played by these cells in coordinating host defense against different classes of pathogens (exemplified by the Th1, Th2, and Th17 effector lineages), or in the case of the microbiome, suppressing untoward reactivity to beneficial symbionts (exemplified by the iTreg lineage). For each subset, the cellular targets that are ‘helped’ and the mediators by which help is provided reflect clearance strategies that are tailored to each class of pathogen. Both innate and adaptive immune cells are targets for regulation by each subset, and a distinct pattern of cytokines produced by each is largely responsible for coordinating the pattern of response.
Th1 cells are charged with augmenting the clearance of intracellular pathogens (e.g. intracellular bacteria and viruses) that have evolved strategies to survive or replicate within macrophages. Th1 cells produce interferon-γ (IFNγ), a potent activator of intracellular killing by macrophages, and also promote B-cell class switching to induce opsonizing immunoglobulin G (IgG) antibody isotypes optimized for killing of intracellular pathogens and may augment CD8+ T-cell responses, with which they share functional features. Th2 cells coordinate expulsion of parasitic helminthes by producing interleukin-4 (IL-4), IL-13, and IL-5 to orchestrate B-cell class switching to IgE, which arms mast cells and basophils for granule release, enhancing mucus production and by promoting the development and recruitment of eosinophils. Th17 cells orchestrate the clearance of extracellular bacteria and fungi, indirectly inducing and activating neutrophils through production of the signature cytokines, IL-17A and IL-17F. Th17 cells also promote B-cell class switching to opsonizing IgG antibodies (4). iTreg cells produce TGF-β, and in addition to suppressing the development of effector T-cell responses, appear to promote B-cell class switching to IgA in mucosal tissues and maintenance of IgA-producing plasmacytes in the intestine as a mechanism to sequester commensal bacteria within the intestinal lumen (5).
A common theme in the lineage specification of the different CD4+ T-cell subsets is the integration of actions of cytokine-induced signal transducer and activator of transcription (STAT) factors and ‘master regulators’ that guide each developmental program. The cytokines that drive CD4+ T-lineage specification are primarily derived from innate immune cells that recognize microbe-associated molecular patterns (MAMPs) to promote pro-inflammatory anti-microbial responses, or recognizing damage-associated molecular patterns (DAMPs) generated in response to non-infectious tissue injury. In the case of Th1 development, sequential actions of STAT1 induced by IFNγ and STAT4 induced by IL-12 promote expression of T-bet, a central transcription factor for Th1 programming. Th2 programming is induced by STAT6 downstream of IL-4 signaling, which upregulates GATA3, the master regulator of Th2 programming. Stability of these developmental programs is enforced through cell–extrinsic feedback loops exerted by the production of cytokines that also induce differentiation (e.g. IFNγ for Th1 and IL-4 for Th2) as well as cell-intrinsic transcriptional networks. Feedforward and feedback loops that reinforce one developmental program while suppressing the other lead to relatively stable, heritable Th1 and Th2 phenotypes (6).
In contrast to Th1 and Th2, Th17 development demonstrates an unusual tendency for developmental flexibility or plasticity (2, 3). While Th17 development is potently inhibited by the Th1 and Th2 cytokines IFNγ and IL-4 (7) as well as other STAT1-activating cytokines (e.g. type I interferons and IL-27) (7, 8), it shares with iTreg cells a developmental requirement for TGF-β (9–11), leading to early overlap in its transcriptional programming with iTreg cells. This appears to be an adaptation of the shared role of Th17 and iTreg cells in constraining the microbiota, particularly in the intestines (12). Late Th17 development is similarly flexible; recent reports have shown that cells committed to the Th17 developmental program can deviate to Th1-like progeny in response to either IL-23 or IL-12 signaling, enabling the Th17 program to shift in response to the prevailing cytokine milieu (2). Similarly, iTregs, but not thymically derived natural Tregs (nTregs), also retain substantial developmental plasticity, challenging the view engendered by years of studies of Th1 and Th2 biology that T-cell differentiation programs diverge rapidly and become fixed.
Developmental plasticity in the Th17 lineage: overlapping Th17-iTreg and Th17-Th1 programs
In contradistinction to Th1 and Th2 differentiation, Th17 differentiation is linked to alternative developmental programs both early and late during lineage specification (2) (Fig. 1). Due to the shared requirement for TGFβ signaling in the differentiation of both iTreg and Th17 cells, the lineage-defining transcription factors forkhead box protein 3 (Foxp3) and receptor retinoic acid-related orphan receptor γt (RORγt) are transiently co-expressed early in specification of iTreg and Th17 cells. Th17 cells show further overlap with the Th1 pathway following their commitment to the Th17 lineage, conditioned by the cytokine balance in the microenvironment when they are re-activated by antigen. Recent studies indicate that the transition of Th17 precursors to progeny with Th1-like features represents an adaptation of the Th17 lineage that has important implications for host defense but also impacts certain immune-mediated diseases and anti-tumor responses. The Th1 transition of Th17 cells would appear to resolve longstanding controversies concerning the pathogenic potential of conventional Th1 cells.
Fig. 1. Overlap of the Th17, iTreg, and Th1 axes of differentiation.

Developmentally, the Th17 lineage overlaps the iTreg pathway early and Th1 pathway late. Shown on top are the key inductive factors for each subset (iTreg, Th17 and Th1), with the lineage-specifying STATs and master regulators indicated. The factors identified in red on the bottom indicate those factor that deviate the Th17 developmental program towards iTreg early (RA and IL-2) or Th1 late (IL-23 or IL-12).
Early Th17-iTreg axis
Prior to its discovery as a co-factor for Th17 cell differentiation, TGFβ1 was identified as an essential cytokine in the development and maintenance of iTreg cells from naive precursors (9–11). In addition to the immunosuppressive actions, it exerts through the induction of iTreg cells, TGFβ was also known to suppress of Th1 and Th2 responses, in part through mechanisms that repressed STAT4 and T-bet, and GATA3, respectively (13). The essential role for TGFβ1 in immune homeostasis is evident in the rapid-onset, multi-organ Th1-mediated disease in TGFβ1-deficient mice. The importance of role TGFβ’s actions on T cells is supported by similar disease that occurs in mice with deficiency of TGFβRII or expression of a dominant-negative TGFβRII targeted to T cells (14). The independent finding by our group and others that TGFβ is an important inductive cytokine for Th17 cells was therefore quite unanticipated (9–11) and marked a shift in our appreciation of the developmental flexibility in T-helper cell differentiation. No cytokine had been shown previously to promote the development of two distinct subsets, and TGFβ’s participation in the development of mature CD4 T cells that were either anti-inflammatory (iTreg) or highly pro-inflammatory (Th17) seemed counterintuitive. On the other hand, the finding that the pro-inflammatory cytokine IL-6 modulated TGFβ’s promotion of iTreg development in favor of Th17 development provided an elegant mechanism by which to match immune homeostasis or anti-pathogenic clearance to extracellular bacteria at barrier sites, such as the intestines, where the greatest number of iTregs and Th17 cells in the body reside, driven by the life-long interplay with the intestinal microbiota (12).
Discovery that the liganded nuclear RORγt is the lineage-defining transcription factor of Th17 cells set the stage for mechanistic insights into the developmental overlap of early Th17 and iTreg differentiation (15). Both Foxp3 and RORγt are induced by TGFβ signaling in antigen-activated naive CD4+ T cells, and although T cells that co-express both transcription factors have been identified in vivo and ex vivo in both mouse and human, the preponderance of data indicate that co-expression of these factors tends to be metastable and typically resolves to dominant expression of one or the other contingent on coordinate signaling by additional factors that favor Th17 versus iTreg specification.
In studies that mapped a physical interaction between Foxp3 and the ROR family member RORα, it was found that a motif encoded by exon 2 of Foxp3 (LQALL, similar to the LxxLL motif of other ROR co-activators and repressors) binds the carboxy-terminal AF2 domain of RORα and was essential for its repression (16). These results were extended to studies of Th17 cell development (17, 18), wherein similar Exon2-dependent repression of RORγt by Foxp3 was found to be critically dependent on TGFβ dose: high doses of TGFβ repressed RORγt function via increased Foxp3 and favored iTreg differentiation, whereas low doses of TGFβ cooperated with IL-6 to overcome Foxp3-mediated repression of RORγt, extinguish Foxp3 expression, and drive Th17 differentiation. Notably, whereas Foxp3 appears to play a direct role in repression of RORγt, the converse does not appear to be the case. That is, while IL-6 activation of STAT3 is required for repression of Foxp3, RORγt is not (19). Thus, Th17-promoting cytokines that activate STAT3, including IL-6, IL-21, and IL-23, override the Foxp3-mediated repression of RORγt in naive T cells exposed to TGFβ to induce Th17 cell differentiation by a mechanism that remains to be defined.
Although studies in mice and humans have identified conditions under which Th17 cells can transition into iTreg cells (20), it is not clear that this occurs to an appreciable extent in Th17 cells that have downmodulated Foxp3. In contrast, Foxp3+ iTregs that have downmodulated RORγt do retain the capacity to transdifferentiate into Th17 cells under pro-inflammatory conditions that produce STAT3-inducing cytokines such as IL-6 or IL-23 (19, 21). This is in contrast to Foxp3+ Tregs that develop intrathymically (so-called nTregs), which are resistant to a similar Th17 transition. The basis for latent plasticity of iTregs but not nTregs reflects differential epigenetic modification of the Foxp3 locus induced during differentiation of the closely related lineages in the periphery or thymus, respectively (22). In the thymus, nTregs undergo demethylation of an upstream cis-regulatory element (Foxp3 CNS2) that is bound by Foxp3 in a Runx1- and Cbf-β-dependent manner to establish a positive feedback loop that stabilizes Foxp expression. During iTreg development, this element fail is not demethylated, thereby preventing positive Foxp3 autoregulation. Although the mechanism by which Th17 cells resist reciprocal transition to Treg cells extinction of Foxp3 is not well understood, a positive feedback loop wherein RORγt transactivates its own expression does not appear to exist.
While IL-6 acts to promote Th17 differentiation in the presence of TGFβ, factors that shift the balance in favor of Foxp3 expression to antagonize Th17 differentiation have also been identified. The vitamin A metabolite retinoic acid (RA), which is produced by intestinal, but not extraintestinal DCs, is a potent non-cytokine cofactor for iTreg development (23, 24). RA signaling through nuclear RAR receptors expressed by naive CD4+ T cells blocks the inhibitory effect of IL-6 on Foxp3 induction, thereby accentuating Foxp3-mediated antagonism of RORγt (25). Additionally, RA is reported to directly inhibit RORγt in CD4+ T cells (26). The antagonism of Th17 differentiation by acts in part through IL-2, a known inhibitor of Th17 differentiation (27), as antibody-mediated neutralization of IL-2 or use of IL-2-deficient CD4+ T cells blunts iTreg differentiation in favor of Th17 differentiation in the presence of TGFβ plus RA (24). Accordingly, the actions of RA were found to be partially STAT5-dependent; RA induced substantially less Foxp3 and failed to inhibit IL-17 induction in STAT5-deficient T cells (26). Importantly, many DNA binding sites targeted by STAT3 in Th17 lineage gene loci can also bind STAT5, providing a mechanism for competitive antagonism of these trans-factors downstream of IL-6 and IL-2 family cytokine receptors (28). STAT3 and STAT5 bind seven shared sites in the Il17a-Il17f gene locus (28). While binding of STAT3 correlated with permissive histone modifications, STAT5 binding correlated with repressive histone modifications. Thus, RA-dependent amplification of IL-2-induced STAT5 could directly interfere with Th17 programming by STAT3 at multiple gene targets, including Rorc, as a mechanism to promote iTreg differentiation at the expense of Th17 differentiation (28). Collectively, then, modulation of a shared TGFβ-induced transcription program by cytokines that affect the dynamic balance between STAT3 and STAT5 levels during the early stages of Th17 and iTreg cell differentiation plays a central role in specifying Th17-iTreg fate.
Late Th17-Th1 axis
The proximal cytokines governing Th1 and Th2 effector cell development are not only distinct from those governing Th17 cell development, but they also potently suppress Th17 development (7, 29). Thus, both type I and II interferons (IFNα/β and IFNγ, respectively) and IL-4 inhibit the development of Th17 cells from naive precursors. In our own studies leading to the definition of Th17 cells as products of a differentiation pathway distinct from Th1 and Th2, a key clue that Th17 cells did not derive from the Th1 pathway, as had been hypothesized, was the finding that neutralization of the Th1-promoting cytokine IFNγ was necessary to enable development of IL-17 producing cells (7). However, in the earliest reports that linked IL-17 production by memory CD4+ T cells to IL-23, IFNγ was also elicited, and in all studies that have identified Th17 cells in vivo, whether in the context of immune-mediated inflammation or at homeostasis, IFNγ-producing T cells accompany IL-17-producing T cells in the same microenvironment, suggesting crosstalk between the Th17 and Th1 lineages.
Resolution of this apparent paradox came with studies that identified late developmental plasticity in the Th17 lineage wherein Th17 precursors were found to give rise to Th1-like progeny (2). Employing an IL-17F reporter mouse model, which permitted the identification and isolation of pure populations of Th17 cells committed to both IL-17F and IL-17A expression, we found that repeated stimulation of isolated Th17 cells under non-polarizing conditions ex vivo produced progeny with expression of IL-17A and IFNγ, either singly or in combination (IL-17F tended to be lost even when a subset retained IL-17A expression) (30). Addition of exogenous TGFβ led to sustained expression of high IL-17A and IL-17F; neither IL-6 nor IL-23 sustained pure IL-17A and IL-17F expression. Indeed, IL-23 promoted progressive diversification of the Th17 precursor population, producing a subset of progeny that extinguished expression of IL-17A and IL-17F, and expressed IFNγ instead. Addition of exogenous IL-12 rapidly and irreversibly extinguished expression of the Il17a/f locus in purified Th17 cells, which transitioned into IFNγ-producing cells indistinguishable from classical Th1 cells by gene expression profiling. Importantly, Th17 cells that transitioned under conditions of selective IL-23 exposure had a gene expression profile distinct from those exposed to IL-12, despite many shared features of conventional Th1 cells. The Th17 to Th1 transition induced by IL-23 and IL-12 was dependent on STAT4 and T-bet, as absence of these transcription factors prevented the transition. Notably, addition of TGFβ strongly inhibited the transition induced by IL-23 but had limited effect on the IL-12-mediated transition, consistent with greater activation of STAT4 downstream of IL-12 (30).
Independent findings from other groups that examined cloned human lines (31) or Th17 cells purified by a cytokine capture technique (32) led to similar conclusions. In the latter study, it was found that Th17 cells could also transition to Th2 progeny, a finding that is finding some traction in human studies of asthma (33, 34). Notably, Th17 cells isolated from the memory pool of mice were resistant to a Th1 transition ex vivo, raising the important possibility that aspects of Th17 programming yet to be identified might stabilize the Th17 program so as to confer long-lived Th17 memory (32). Importantly, studies of human CD4+ T-cell lines derived from patients with Crohn’s disease closely paralleled subsequent findings in mice (31), suggesting relevance of these findings to human disease, and consistent with subsequent genome-wide association studies (GWAS) that have identified overlapping linkage to Th17 and Th1 pathway components (35).
Studies that have explored Th17-Th1 transitions in models of chronic immune-mediated disease have confirmed the plasticity identified in vitro and have implicated a pathogenic role for the Th1-like cells derived from Th17 precursors by the actions of IL-23 (36). This is in accord with the landmark studies that identified a pathogenic link between IL-23, but not IL-12, in disease models of multiple sclerosis [experimental autoimmune encephalomyelitis (EAE)] and rheumatoid arthritis [collagen-induced arthritis (CIA)] (37, 38). In our own studies, Th17 cells derived ex vivo from IL-17F reporter mice induced rapid and severe colitis following transfer into immunodeficient recipients. Recovered T cells from diseased tissue and secondary lymphoid tissues demonstrated IL-23-dependent phenotypic divergence similar to that observed in vitro (30). Similarly, CD45RBhi cells lacking IL-23R failed to transition to IFNγ/IL-17A double producers and did not trigger colitis (39). In Th17 transfer models of EAE and type I diabetes, IL-23–dependent transition of transferred Th17 cells into Th1-like progeny has been linked to disease development (36, 40), although a pathologic role for Th1 cytokines has been found to vary depending on the model. In a study complementary to the foregoing transfer studies, it was found using transgenic fate reporter mice that nearly all IFNγ-producing CD4+ T cells recovered from the spinal cords of mice with EAE had expressed IL-17A during their development providing strong support for the development of Th1-like cells from Th17 precursors (36). In this model, both the Th17 to Th1 conversion and disease pathogenesis required IL-23, in accord with previous reports.
An interesting feature of the late developmental divergence of Th17 cells that deserves further comment derives from recent studies of Th17 function in anti-tumor immunity. In a melanoma model that examined tumor eradication by transfers of effector T cells derived from TCR transgenic CD4+ T cells specific for the melanoma antigen tyrosinase-related protein 1 (TRP-1), Th17 but not classical Th1 cells were found to have potent anti-tumor activity (41). Although IL-17 proved contributory (42), tumor clearance was dependent on the expression of T-bet and IFNγ by Th17 cells that converted to Th1-like cells post-transfer, similar to results in immune-mediated inflammatory disease models (43). Comparative transcriptome profiling of Th17 and Th1 cells recovered following transfers into tumor-bearing mice revealed the acquisition of many Th1 gene signatures by the Th17 cells, in keeping with their phenotypic shift but also revealed heightened expression of signature genes that confer heightened self-renewal and multipotency in hematopoietic stem cells (HSCs). This observation supports the view that Th17 cells serve as multipotent, self-renewing precursors capable of differentiating into Th1-like effector like progeny (Th17/Th1) (43) and are advantageous for tumor eradication at least in part because of their superior self-renewal properties compared to conventional Th1 cells (44). These findings further point to a multipotent capacity of Th17 cells that is retained even as their capacity for iTreg development is extinguished. Thus, both in vitro and in vivo, Th17 precursors give rise to progeny that retain or extinguish IL-17A expression, or gain IFNγ expression — with or without co-expression of IL-17. Because Th1-like cells that arise from Th17 precursors lose expression of genes involved in self-renewal and do not appear to revert back to Th17 cells, they are more terminally differentiated and survive less well than the Th17 cells from which they arise. It is thus speculated that the superior self-renewing potential of Th17 cells relative to their Th1 progeny might provide a long-lived pool of cells that can similarly contribute to superior tumoricidal actions and chronic immune disease (43).
A TGFβ-independent path to Th17 cell differentiation and pathogenesis?
From the outset, the role of TGFβ in Th17 development and function has generated controversy. This has continued unabated and remains incompletely resolved, although recent findings have begun to offer solutions. Central to this controversy have been disparate findings as to whether Th17 cells do or do not require TGFβ signaling to differentiate, initially fueled by species differences between mice and humans. This is further complicated by recent studies supporting the existence of at least two functional subclasses of Th17 cells distinguished by their development in the presence or absence of TGFβ, and reports that Th17 cells can produce their own TGFβ, including TGFβ1 and TGFβ3, which would appear to exert distinct programming functions. Finally, complexity inherent in the study of TGFβ family regulation in vivo, where abundant tissue stores of latent TGFβ isoforms and multiple pathways to their activation make definitive studies difficult, have made this issue challenging.
Following the initial reports that identified TGFβ as a cofactor for mouse Th17 differentiation (9, 11, 45), two independent groups reported that TGFβ is dispensable for differentiation of human Th17 cells (46, 47). Later, refuting the dispensability of TGFβ in human Th17 differentiation, three studies reinstated TGFβ as an important Th17 differentiation factor, albeit less robust than in murine T cells and more dependent on the pro-inflammatory mediator IL-1β and IL-23 (48–50). In fact, in no studies have the high frequencies of IL-17-producers that can be induced from naive mouse CD4+ T-cell precursors been replicated in human T cells. The basis for this species difference remains unresolved, although it is worth noting that the ‘naive’ CD4+ T-cell precursors used in human studies, whether isolated from peripheral blood on the basis of CD45RA/O isoform surface markers or isolated from cord blood, may not be entirely comparable to their murine counterparts. Thus, it is unclear to what degree the species differences reflect the T-cell populations studied or inherent differences in Th17 developmental programming.
The dispensability of TGFβ in Th17 differentiation resurfaced again, this time in the mouse, when it was reported that there can be two pathways of Th17 differentiation: a TGFβ-dependent pathway that gives rise to ‘non-pathogenic’ Th17 cells and a TGFβ-independent pathway that gives rise to ‘pathogenic’ Th17 cells (51). Naive precursors polarized in the presence of IL-6, IL-1β, and IL-23, but absence of TGFβ signaling, induced a population of so-called Th17 (23) cells that induced EAE upon passive transfer into normal mice. In contrast, naive cells polarized under identical conditions but with exogenous TGFβ1 and not IL-23 [so-called Th17(β) cells], failed to induce EAE following transfers, despite expressing considerably higher amounts of IL-17A. A large number of genes were differentially expressed between the two Th17 subclasses: conventional Th17(β) cells expressed higher levels of Il9, Il10, and Ccl20, whereas TGFβ-independent Th17(23) cells expressed higher levels of Il2, Il33, and Il18r1. Not surprisingly, while RORγt was induced in both populations, T-bet was selectively upregulated in Th17(23) cells, as were other Th1 pathway genes — in accord with previous reports, including our own, that highlighted the antagonistic effects of TGFβ on the IL-23-induced, STAT4- and T-bet-dependent transition of Th17 cells into Th1-like cells (2).
While this study hinted at possible subclasses of Th17 cells distinguished by early differentiation in the presence or absence of TGFβ1, it is unclear under what conditions the latter might occur physiologically. TGFβ1 is produced by a variety of immune cells and is widely expressed in tissues, at least in latent form. It is also normally present in blood; normal human subjects have over 2 ng/ml TGFβ1 in their plasma (52). It is therefore difficult to conceive of a tissue environment completely devoid of its expression. Indeed, it has been shown that in addition to its production by Treg cells, effector T cells are themselves an important physiologic source of TGFβ1, with Th17 cells producing higher amounts than either Th1 or Th2 cells. Thus, Th17 cells are an autocrine source for this developmental co-factor (53). Indeed, T-cell-specific deletion of TGFβ1 was found to obviate Th17 development and EAE disease (53), as was T-cell-specific expression of a dominant-negative TGFβRII transgene (54). These findings suggest that Th17 cells fail to develop in vivo without the availability of TGFβ signaling to promote Th17 induction; without Th17 development, Th17-driven disease does not occur.
The preponderance of published data collectively support a bi-phasic model wherein TGFβ co-signaling with IL-6 is essential early for robust Th17 induction, whereas TGFβ late blunts IL-23-driven STAT4 signaling to maintain a multipotent Th17 state. Th17 cells sustained by ongoing TGFβ signaling are poised to deviate down a program of Th1-like development contingent on locally increased IL-23 and decreased TGFβ1 (30) (Fig. 2). This model is consistent with the finding that neutralization of TGFβ at the local site of Th17 priming inhibited EAE development, whereas global neutralization of TGFβ enhanced EAE severity, presumably by shifting the late Th17-Th1 transition in favor of IL-23 dominance (55). Similarly, this is consistent with an early study showing that Th17 cells recovered from draining lymph nodes of mice primed for induction of EAE were non-pathogenic upon transfers to normal mice if cultured ex vivo with TGFβ1 plus IL-6, whereas cells cultured with IL-23 transferred disease (56).
Fig. 2. TGFβ and IL-6 gradients regulate early Th17, iTreg, and Th22 commitment.
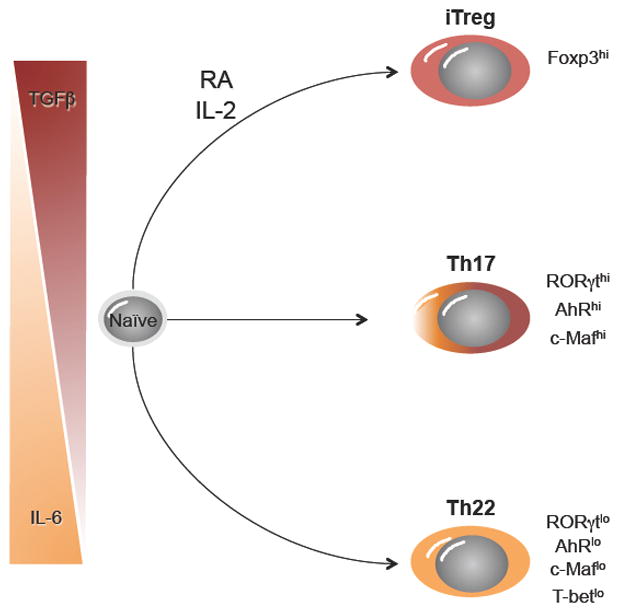
The differentiation of naive CD4+ T cells can proceed down one of three pathways under the influence of differential TGFβ and IL-6 cytokine gradients. Expression of key transcription factors for each cell type is indicated. RA, retinoic acid.
In a new wrinkle on the TGFβ-Th17 story, it was recently reported that Th17 cells produce, in addition to TGFβ1, TGFβ3 (57). In contrast to TGFβ1, which antagonizes the development of pathogenic Th17 cells, TGFβ3 purportedly promotes pathogenic features of Th17 cells in the EAE model. Induced downstream of IL-23 and T-bet in Th17 cells differentiated in the presence of TGFβ1, it was proposed that TGFβ3 might promote a pathogenic Th17 phenotype by mechanisms yet to be defined. Interestingly, differentiation of naive CD4+ T cells with IL-6 plus TGFβ3 induced a gene expression profile that was distinct from that induced by differentiation with IL-6 plus TGFβ1, and resembled that of previously reported Th17(23) cells (51). Also similar to Th17(23) cells, Th17 cells differentiated with TGFβ3 induced EAE in a passive transfer model. Quite remarkably, disease induced by Th17 cells differentiated with TGFβ3 was independent of T-bet, suggesting that although T-bet is required for optimal TGFβ3 expression, there might exist a TGFβ3-dependent, T-bet-independent mechanism that can drive neuroinflammation. Because TGFβ1 and TGFβ3 are ligands for the same TGFβ receptor complex, it is unclear how they might induce distinct developmental programs, although divergent downstream signals generated by the two ligands were identified. An important caveat is that no careful dose titration comparisons were performed with the two ligands, leaving open the possibility that the signaling and Th17 programming differences observed might reflect quantitative rather qualitative effects. In view of the well documented dose-dependent effects of TGFβ1 on Th17 development, both early and late, aspects of the TGFβ3 programming observed could reflect its weaker agonism of the TGFβ receptor. Indeed, as discussed below, IL-6 programming of naive CD4+ T cells in the presence of low or absent TGFβ1 induces a similar phenotype, although its function in EAE pathogenesis has not been investigated.
Th22; a third branch of the Th17 family tree with ties to pathogenic Th17 cells?
IL-22 is a cytokine of the IL-10 family that is often regarded as a signature product of Th17 cells (58, 59). A critical mediator in the coordination of host defense at barrier sites such as the skin and respiratory and intestinal tracts, IL-22 produced by immune cells activates epithelial cells for heightened anti-bacterial resistance (60, 61) and is a good fit with the Th17 repertoire of host protective functions. However, in humans, IL-22 was initially identified as a Th1 subset-derived cytokine (62), and a subset of human CD4+ T cells referred to as Th22 cells that are characterized by production of IL-22 in the absence of IL-17 has been described (63). The allegiance of IL-22 to a specific CD4+ T-cell lineage would therefore appear to be ambiguous and is currently under debate.
The initial report that detailed conditions for the development of IL-22-producing effectors from naive murine CD4+ T cells identified reciprocal effects of TGFβ1 on the induction of IL-22 and IL-17A by cells polarized ex vivo in the presence of IL-6 and reported that IL-23 induced IL-22 (59). Naive cells activated with IL-6 in combination with anti-IL-4 and anti-IFNγ in the absence of exogenous TGFβ1 expressed high IL-22 and minimal IL-17A, whereas cells activated with IL-6 in the presence of exogenous TGFβ1 — that is, conventional Th17-polarizing conditions — produced high IL-17A and minimal IL-22. Thus, while promoting optimal Th17 differentiation, TGFβ1 potently inhibits induction of IL-22, suggesting that optimal IL-22 production is compromised is canonical Th17 cells. As in conventional Th17-polarizing conditions, antibody mediated blockade of IFNγ and IL-4 indicated that the effects of TGFβ to inhibit IL-22 were not simply due to suppression of Th1 and Th2 programming but rather must inhibit aspects of IL-6-induced programming. It was subsequently reported that the repression of IL-22 by TGFβ1 was mediated by c-Maf, a component of the conventional Th17 transcription factor network induced by TGFβ signaling (64).
The subsequent identification of a human ‘Th22’ subset that produced IL-22 without IL-17 co-production (63, 65, 66), suggested parallels with mouse ‘Th17’ cells induced in the absence of TGFβ1. Human Th22 cells were designated as a distinct subset on the basis of their unique cytokine profile and minimal expression of RORγt (63). However, in recent studies by our group (67), it was found that murine effectors differentiated by IL-6 in the absence of TGFβ1 similarly expressed high levels of IL-22 and low levels of RORγt. Notably, in comparing the transcriptome of these ‘Th22’ cells with conventional Th17 cells, more than 600 genes were differentially expressed and the two transcriptomes bore a striking resemblance to those previously reported for ‘pathogenic’ Th17(23) cells and ‘non-pathogenic’ Th17(β) cells (51). Prominent among the genes expressed at higher levels by the ‘Th22’ cells, were IL-23R, STAT4, and T-bet — Th1 lineage genes that are likely linked to the pathogenic potential of Th17(23) cells. AhR, a liganded transcription factor that is required for IL-22 production from conventional Th17 cells (68), was expressed at much lower levels in Th22 cells, despite their higher production of IL-22. Interestingly, while both the Th22 and Th17 cells expressed significant levels of TGFβ3, this isoform was higher in the Th22 subset, suggesting that TGFβ1 represses TGFβ3 expression (RB, unpublished observation).
In functional studies of these two subsets, we found that when transferred into IL-22-deficient mice challenged with the enteropathogenic bacterium Citrobacter rodentium, the ‘Th22’ cells conferred complete protection from fatal infection, whereas conventional Th17 cells did not (67). This identified an important role for these cells in supporting intestinal barrier function under pathogen-induced inflammatory conditions. In light of the repression of IL-22 by TGFβ (59), these findings suggest that mechanisms to curb activation of TGFβ, which is abundant in latent form in the intestines, might be required to promote Th22 induction or enhanced IL-22 production by Th17 cells.
Notably, despite marked reduction of AhR expression by Th22 cells relative to Th17 cells, optimal expression of IL-22 and protection from infection by the Th22 subset was dependent on co-expression of AhR and T-bet, supporting a previous study that found heightened production of IL-22 by Th17 cells retrovirally transduced with T-bet (69). Collectively, these results indicate that irrespective of the term one chooses to call them (Th22, Th17/Th22, or pathogenic Th17), these cells are phenotypically and functionally distinct from conventional Th17 cells and represent a branch of the Th17 family tree that is favored by IL-6 and IL-23 signaling under conditions of low or absent TGFβ signaling (Fig. 2). Thus, despite clear developmental ties between Th22 and Th17 cells, each would appear to constitute a distinct functional population. This suggests an early overlapping developmental diversity between iTreg, Th17, and Th22 that is modulated by competing gradients of TGFβ and IL-6.
These findings raise new questions regarding the possible pathogenic potential of Th22 cells and IL-22. Although a non-redundant cytokine required for host protection against bacterial and fungal pathogens in the intestinal and respiratory tracts (60, 61, 67, 70), IL-22 can also contribute to pathogenic inflammation in these tissues (71–73). Elevations of IL-22 in the involved tissues of patients with inflammatory bowel disease, psoriasis, and rheumatoid arthritis might contribute to these diseases (74–78), although this remains to be determined. Finally, given the strong similarities between Th22 and Th17 cells that induce EAE, these findings raise questions regarding the possible pathogenic potential of Th22 cells in neuroinflammation.
A new perspective on Th17-mediated immune regulation and pathogenicity
In view of the substantial experimental support for the Th17-Th1 model in immune-mediated disease and the essential role for IL-23 in driving pathogenic diversion of Th17 cells, a longstanding question has been: what attributes of Th17 that are dependent on IL-23 are essential for their pathogenicity? While important negative data have come from studies that pre-date discovery of the Th17 pathway, a new candidate has been advanced by recent reports.
Whereas expression of the Th1 trans-factors STAT4 and T-bet by deviated Th17 cells appear essential to EAE disease development (79), other Th1 lineage factors are not. In particular, mice with deficiencies of IFNγ, IFNγRI, and IL-12Rβ2 remain susceptible to disease (80–84). Similarly, mice lacking the signature Th17 cytokines IL-17A and IL-17F, as well as IL-22 and IL-21 are not completely protected from disease (56, 85, 86). Thus, core Th17 cytokines do not appear to be essential, and pathogenicity linked to STAT4 and T-bet is neither due to IFNγ nor dependent on IL-12-mediated effects. To the contrary, at least in some studies deficiency of IL-12 and IL-12 signaling heightened disease susceptibility, as did deficiency of IFNγ (87). Thus, activation of STAT4 and T-bet by IL-23, but not IL-12, must be driving other factors important for EAE development, the nature of which has remained elusive.
Two recent reports have made major inroads into this issue, independently identifying GM-CSF as a T-cell cytokine induced by IL-23 that is indispensible for Th17-mediated EAE pathogenesis (88, 89). Although GM-CSF was identified as a product of Th17 cells in early studies (90), its role in EAE pathogenesis had not been explored. GM-CSF, or CSF2, is a member of the common β-chain (βc) receptor family of hematopoietic cytokines, which includes IL-3 and IL-5. It promotes the development of granulocytes and macrophages from myeloid progenitors in bone marrow. In addition to its hematopoietic actions, GM-CSF has pro-inflammatory effects on macrophages and DCs, acting to promote maturation and recruitment as well as promoting heightened antigen presentation functions and release of pro-inflammatory mediators by these cells. Although GM-CSF can be produced by non-T cells (e.g. myeloid and stromal cells), in both reports development of EAE was contingent on the expression of GM-CSF by transferred of Th17 cells (88, 89).
One of the actions of GM-CSF appears to be the upregulation of IL-23 by resident microglia and myeloid cells that infiltrate the inflamed CNS (89). This points to a positive feedback loop by which Th17 cell expansion and deviation to Th1-like functions are amplified. Interestingly, it was found that in contrast to IL-23, which promoted GM-CSF production by myeloid cells, IFNγ and IL-12 suppressed its production (88). Nevertheless, like Th17 cells, Th1 cells required expression of GM-CSF to induce neuroinflammaton. Although not formally tested, this suggests that the important ‘Th1’ cells that are targeted for IL-23-induced GM-CSF production are derived from the Th17 lineage. Importantly, however, although T-bet is essential for pathogenicity of Th17-derived effectors, its absence did not impair GM-CSF expression (88), which was shown to be RORγt dependent in one of the studies (88) but not the other (89). Thus, while these studies identify an important new mechanism by which Th17 cells drive chronic inflammation, questions related to the role of T-bet-dependent actions in Th17-mediated inflammatory disease remain open. It also remains to be determined whether the non-redundant role for GM-CSF in neuroinflammation in the EAE model extends to other immune-mediated diseases.
Orchestration of transcription factor networks in CD4+ T-cell fate decisions; towards an understanding of lineage specification and stability or plasticity in T-helper cells
Transcription factor (TF) networks underpin both divergence and stabilization of CD4+ T-cell phenotypes, acting through key trans-factor nodes to establish expression or repression of large sets of genes. In view of the prominent early and late developmental plasticity that are features of the Th17 pathway, it is perhaps not surprising that the range of extrinsic and intrinsic factors involved in this pathway is proving to be quite diverse.
An important consideration in establishment or repression of TF networks relates to the functional activation and stability of individual factors. For example, individual STAT family members, which link distinct cytokine signals to divergent gene transcription networks for each CD4+ T-cell subset, are active for only minutes to hours. For the Th17 pathway, cytosolic phosphorylation and dimerization of the lineage-specifying STAT3 by JAK family kinases activated by receptors binding IL-6, IL-21, or IL-23 leads to rapid translocation to the nucleus to bind target cis-regulatory elements (CREs). STAT3 actions are rapidly terminated at several levels: proximally through inhibition of JAK kinase activity by cytokine signaling-induced SOCS3 in a negative feedback loop and distally by inhibition and degradation by PIAS family proteins, as well as de-phosphorylation by tyrosine phosphatases. Accordingly, as for each of the STATs, phospho-STAT3 acts transiently and can only influence TF networks during initiation of programming, as occurs early in Th17 lineage specification, or by sequential, episodic activation, contingent on persistence of cytokines in the microenvironment. In the special case of Th17 cells, in which sequential activation of STAT3 can occur through different cytokine receptors (i.e. IL-6, IL-21, and IL-23), this could be a unique strategy to achieve on-going STAT signaling.
In contrast, other lineage–specific TFs are stably expressed and thus are more likely to contribute to lineage stabilization and maintenance. Key among these are the lineage-defining TFs, or master regulators, which have been identified for each major lineage and are defined as necessary and sufficient for lineage specification (i.e. T-bet/Th1, GATA3/Th2, RORγt/Th17, and Foxp3/iTreg). Also included here are TFs that can be expressed by more than one lineage and appear to have more universal roles in establishing lineage-specific TF networks (e.g. IRF4). Models of lineage specification and stability must account for the integration of multiple TFs, whether transiently or stably expressed, and whether lineage-specific or shared between lineages.
Rapid advances in identification of the factors involved and the interplay of key regulatory nodes that define stability or plasticity of the networks established are beginning to provide a mechanistic basis for the tendency of Th17 differentiation to be more flexible and less stable than that of the Th1 or Th2 cell pathways. Here, we consider emerging data on the links between environmental sensors (e.g. TCR/CD28 and cytokine receptors) and the TFs they induce or activate, and cell-autonomous TFs activated downstream of these sensors that establish lineage-specific regulatory networks. Although there remain major gaps in our understanding, existing data have begun to provide testable models and are being accelerated by the implementation of systems biology and network theory approaches.
Chromatin landscape pioneers: AP1-IRF complexes and the STATS — once and future activators of enhancers
Although key lineage-specifying TFs for each of the T-helper subsets are now known, identification of specific functions in the promotion and maintenance of distinct gene expression programs and transcription networks has been elusive. Like other metazoan differentiation programs, CD4+ T-cell differentiation requires the coordinated expression of key trans-factors and epigenetic modifications of the CREs to which they bind to influence gene expression or repression and to establish interacting networks that confer lineage stability or heritability despite episodic rounds of active cell division. In developing Th17 cells, as for other CD4+ T-cell subsets, a central unanswered question has concerned the division of labor between factors that recruit chromatin remodeling complexes to generate accessible sites and factors that bind these sites to enhance (or repress) transcription and to what extent these are separable. A flurry of recent studies has begun to solve this puzzle: one group of studies has identified the cooperative binding of members of the AP1 and IRF transcription factor families that appears to initiate remodeling of CREs in immune gene targets across CD4+ T-cell lineages (91–93); another has identified a predominant role for STATs over master regulators to activate lineage-specific enhancer functions at these CREs (94).
Of the trans-factors that are expressed by Th17 cells, four have been found to be indispensable for robust Th17 differentiation: STAT3, RORγt, IRF4, and BATF (95–99). In coincident reports from three groups, a basis for previous findings that IRF4- and BATF-deficient CD4+ T cells have impaired Th17 differentiation was identified. It was discovered that IRF4 cooperates with heterodimers of BATF and the JUN members of the AP1 family (JunB and c-Jun) to bind a newly identified AP1-IRF composite elements (AICEs) (91–93). IRF4 is a member of the interferon regulatory factor (IRF) transcription factor family, expression of which, along with IRF8, is largely restricted to the immune system. Previously characterized as an important TF in B cells, macrophages, and DCs, cells in which it binds target sequences cooperatively with Ets family TFs (PU.1 and Spi-B), termed Ets-IRF composite elements or EICEs (92), the new findings extend the binding partners with which IRF4 and IRF8 can interact to contribute to immune cell lineage specification.
BATF, a member of the ATF subset of the AP1 family, was shown previously to act proximal to RORγt in Th17 differentiation, and targeted a modified AP1 consensus site in several signature Th17 genes, including sites in the Il17a-Il17f locus previously identified as DNase I hypersensitivity sites that bore permissive epigenetic marks only in Th17 cells (97, 100). Notably, although Rorc expression was impaired in BATF-deficient cells polarized under Th17-inducing conditions, Th17 differentiation was not rescued by enforced expression of RORγt, establishing that failure of Th17 differentiation could not be explained simply on the basis of impaired Rorc expression.
The binding of IRF4-BATF-JUN complexes at AICEs throughout the T-cell genome proves to be highly important to establishment of transcriptional networks in developing Th17 cells, where it binds in association with the lineage-specifying factors STAT3 and RORγt (91). Remarkably, it was found that CREs targeted by IRF4-BATF-JUN complexes in Th17 cells were comparably occupied in CD4+ T cells activated under non-polarizing cytokine conditions (Th0), indicating that binding of these IRF-AP1 complexes to CREs in developing Th17 cells precedes binding of STAT3 and RORγt. Accordingly, deficiency of either IRF4 or BATF broadly compromised recruitment of STAT3 and RORγt to CREs under conditions of Th17 polarization. Thus, IRF4-BATF-JUN complexes, which are activated independently of lineage-specifying cytokine signaling downstream of TCR signaling, act as pioneer factors that would appear to generate a permissive chromatin landscape required for subsequent binding of lineage-specifying factors. This provides the first global mechanistic link coupling TCR and lineage-specifying cytokine signaling on the genome-wide scale and could have implications for integration of other signals emanating from the TCR that might act as pioneer factors. Specifically, it has long been known that AP1 family members cooperate with members of the nuclear factor of activated T cells (NFAT) family to induce T-cell gene downstream of TCR signaling, and in some sites reside adjacent to IRF consensus sequences (93). It is not unlikely that NFAT-AP1 complexes might interact with IRF4 or IRF8 to similarly pioneer chromatin accessibility at distinct CREs of immune response genes in T cells, or might do so independently.
In a complementary study that globally mapped the activity of lineage-specific enhancers in the context of Th1 and Th2 differentiation, binding of the p300 acetyl transferase complex was validated as a reliable indicator of active enhancers and its recruitment to CREs was found to be strongly dependent on the actions of lineage-specific STATs (94). In the case of STAT6 actions in Th2 cells, it was found that while only ~30% of Th2-specific enhancers demonstrated detectable binding of STAT6, in cells deficient for STAT6, there was loss of ~80% of Th2-specific p300 sites. Thus, activation of lineage-specific enhancers in Th2 cells was strongly dependent on STAT6 actions, whether by direct binding or activation of other p300-recruiting trans-factors induced by STAT6. Notably, the Th2 master regulator, GATA3, bound a minority of the sites bound by STAT6 (~35%), and enforced expression of GATA3 in STAT6-deficient cells induced reactivation of only ~50% of the lineage-specific enhancers. In Th1 cells, developmental programming of which involves sequential STAT1 and STAT4 activation by IFNγ and IL-12, respectively (101), activation of Th1-specific enhancers was also found to be highly dependent on the STATs; T-bet had a markedly lower impact on enhancer activation, with only ~15% of the total number of active enhancers requiring T-bet expression. Further, while ~50% of the lineage-specific enhancers in Th1 cells bound either STAT1 or STAT4, the remainder required binding of both STAT1 and STAT4, suggesting cooperativity at these CREs. Collectively, these findings establish that activation of STATs downstream of lineage-specifying cytokines represents the dominant mechanism for T-lineage-specific enhancer activation and therefore the establishment of lineage-specific gene expression networks.
Although Th17 cells were not evaluated in this study nor were requirements for binding of IRF4-BATF-JUN complexes for STAT recruitment to AICEs, collectively the findings of this and the foregoing studies support a model in which STAT3 recruitment to activating CREs in Th17 cells requires the sequential actions of IRF4-BATF-JUN pioneer complexes that enable enhancer activating STAT3-dependent recruitment of p300 complexes and RORγt. Given that targeting of p300-CBP family acetyl transferases to nucleosomes is mediated by binding of the acetylated histone lysines by their bromodomains, it is possible that IRF4-BATF-JUN complexes initiate the enhancer activation cascade by initial recruitment of other acetyl transferases, via a mechanism to be determined.
Although highly expressed in Th1, Th2, and Th17 cells, only Th17 cell development was defective in BATF-deficient T cells (97). This is in contrast to IRF4-deficient T cells, which, in addition to demonstrating a Th17 developmental deficit, also have impaired Th2 development (98). Like BATF, IRF4 (and IRF8) is highly expressed in naive CD4+ T cells and in each of the effector lineages (reviewed in 98, authors’ unpublished findings). In view of the widespread binding of IRF4-BATF-JUN complexes in Th0 cells (91), questions are raised as to why deficits of these factors would curtail certain T-helper differentiation programs (e.g. Th17) and not others (e.g. Th1). Because there is substantial overlap in the thousands of CREs targeted in the different T-helper subsets (94), whether targeted for expression or repression, the lack of a requirement for IRF4-BATF-JUN complexes in Th1 cells, for example, cannot be ascribed to a distinct set of binding targets. Certainly, it is possible that other AP1-IRF complexes might compensate. Thus, although the numbers differed between studies (92, 93), there was agreement that a substantial fraction of the sites that bound BATF did not co-bind IRF4 (although IRF8 binding was not examined). Conversely, depending on the study, at less than 20%–50% IRF4 sites was BATF binding detected (92, 93), implicating other partners for the IRFs. While some of this variance likely reflects the dynamic and transient nature of these complexes, it is likely that other factors are also operative. Alternatively, insofar as lineage-specific were found to be the major mediators of p300 recruitment (94), it is conceivable that STATs might go it alone at some sites and recruit p300 complexes without requirement for IRF4-BATF-JUN complexes, or instead might cooperate with pioneering factors of completely different composition. Could cooperativity of STAT1 and STAT4 at a large fraction of the Th1 enhance landscape bypass a need for other pioneers? Finally, are different rules for establishment of lineage-dependent regulatory networks at play here? In this regard, it is notable that c-Maf was found to be a global repressor in Th17 cells, where it is induced by both STAT3 and IRF4, and acts to repress BATF (91). Because c-Maf is expressed in Th2 and Th17 cells (64, 91, 102), but not in Th1 cells, are there altered thresholds for the pioneering actions of AP1-IRF complexes when c-Maf is not a principal component of the network, making a role for AP1-IRF complexes superfluous? These questions remain open, but are likely to be answered soon.
What role the old masters?
A central discovery that emerges from these studies is the surprisingly limited role for the master regulators T-bet, GATA3, and RORγt in directly initiating the establishment of active enhancer networks in Th1, Th2, and Th17 lineages. These results are echoed in a similar recent study of Foxp3 actions in Treg cells (103). The fact remains that, although indispensable, the STATs cannot go it alone. Thus, genetic deficiency, knockdown, or pharmacologic inhibition of the respective master regulators undermines the lineage-specific network to which each contributes even when STAT activation is intact. This begs the question of the ultimate function of the ‘masters.’
Perhaps one answer lies in timing. As noted above, TFs that serve to pioneer CREs (e.g. IRF4-BATF-JUN complexes) and the STATs that further remodel and activate (or repress) their enhancer function act transiently, activated rapidly downstream of TCR or CD28 and a lineage-specifying cytokine receptor, respectively, and inactivated nearly as rapidly. For the networks induced to expand and remain stable, especially in the face of the burst of cell cycling that is induced in antigen activated naive T cells, someone must remain behind to ‘mind the store’, ensuring that the networks are stabilized at the same time they are being built out. Is this the role for the master regulators, which are more stably expressed and are retained in dividing cells? And does variance in the expression stability of these stability factors in turn underlie the relative stability or flexibility of the different lineages? Although the final answers are not yet in, these new data are certainly consistent with such a model and provide glimpses into the unique strategies of each lineage to accomplish its mandate to orchestrate distinct immune responses.
The Th2 lineage would appear to be the simplest network, relying on one STAT and one master that potently induces its own expression to generate a robust positive feedback loop. Although less robust in directly inducing its own expression, T-bet does appear to enhance its own expression in Th1 cells (as well as in Th1-like cells that emerge from Th17 precursors), and, like GATA3 in Th2 cells, which supports positive feedback through reiterative cycles of IL-4 production and STAT6 signaling, T-bet indirectly promotes its sustained expression through positive feedback of the major cytokine it induces (e.g. IFNγ) to promote episodic STAT1 signaling in Th1 cells. Accordingly, both Th2 and Th1 lineages have evolved robust positive feedback loops to create highly stable lineage-specifying programs that integrate robust trans-factor and cytokine networks.
This is in distinct contrast to the Th17 lineage, where RORγt appears to lack similar assets for stabilizing its own expression and the network it underpins. RORγt does not appear to enhance its own expression, and although it does promote expression of IL-21, which can act as an autocrine cytokine for STAT3 activation, it also promotes expression of the IL-23 receptor, which though also a STAT3 activator, activates STAT4 that in turn induces T-bet, which directly suppresses RORγt expression (100). Indeed, once the balance tips in favor of a T-bet network, largely in a setting of reduced or absent ongoing TGFβ repression of STAT4 activation, RORγt expression is irreversibly lost as is much of the RORγt-sustained Th17 network (100). Collectively, this might explain the brittle nature of the Th17 phenotype and strong propensity for plasticity that is not shared by the Th1 or Th2 lineages.
An additional consideration in the Th17-RORγt network concerns the fact that, unlike GATA3 or T-bet, RORγt is a liganded nuclear receptor that remains an orphan. Although exogenous anatagonists of RORγt’s activity have been discovered recently (104, 105) and although its natural ligand(s) would appear to derive from cholesterol biosynthesis pathway, it remains undiscovered. It is therefore formally possible that endogenous or environmental antagonists of its function exist. While this makes RORγt an excellent drug target, it also might render RORγt transcriptional functions susceptible to changing environmental circumstances that further contribute to the intrinsic instability or plasticity of the Th17 lineage. Is it possible that, akin to the aryl hydrocarbon receptor (AhR) for which endogenous and environmental ligands contribute to dynamic regulation of this trans-factor’s regulation of Il22 expression, ligands derived from the diet or the microbiota in the intestines, where Th17 cells reside in greatest abundance might modulate RORγt activity? Although purely speculative, the fact that RORγt is unique among the master regulators in being a liganded receptor opens the possibility for an additional layer of regulation and therefore plasticity, and could hold some surprises going forward.
Conclusion and perspectives
The different CD4+ T-cell lineages have adapted common as well as unique developmental strategies that are tailored to their specialized roles in host defense. This is nowhere better exemplified than the Th17 pathway, which has evolved a remarkable capability to adapt to changing environmental circumstances to match phenotype with function. Beginning with overlapping developmental programming shared with iTreg, and now Th22, and ending with terminal differentiation shared with Th1, the Th17 pathway encompasses its own microcosm of CD4+ T-cell lineage diversity. The basis for this unusual proclivity for developmental flexibility is likely the product of the strategic deployment of this subset in barrier tissues, particularly the lower intestinal tract, where the immune system must carefully balance the need to nurture a diverse, host-beneficial microbiome while restraining its entry into host tissues (12).
As evidenced from the growing range of immune-mediated diseases that are linked to the Th17 pathway, the advantages of this strategy for immune homeostasis and host defense hold considerable risk, the breadth of which is still being defined as Th17 driven inflammation that results from dysregulated responses to the microbiome has left the gut and appears destined to impact disease in many distant body tissues, including the brain (12). While much progress in understanding the basis for Th17 developmental plasticity has been made, much remains to be learned. What is the nature of memory (or memories) in the Th17 lineage? Do stable IL-17 producing T cells exist, and if so, what is their function? How do Th1-like cells that develop from Th17 precursors differ in their transcriptional and functional programming from conventional Th1 cells, and to what end in host defense and disease pathogenesis? The recent breakthroughs in deciphering the transcriptional regulatory networks of CD4+ T-cell lineages, and Th17 in particular, illuminate the remarkable complexity embodied in these developmental programs but also provide considerable insights into the ‘logic’ of their circuitry that is likely to pay substantial dividends for decoding ties to disease and identifying nodes for targeted intervention. This promises new treatment modalities that could scarcely have been conceptualized at the time, not long ago, when this immune pathway was discovered.
Acknowledgments
The authors thank members of the Weaver lab for helpful comments and Gloria Gaskins for administrative assistance. This work was supported by from the NIH (CTW and RDH) and the Crohns and Colitis Foundation of America (RB and CTW).
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 2.Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsdoerffer M, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. 2010;107:14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci USA. 2009;106:19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 7.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 8.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 10.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 11.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 14.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 15.Ivanov, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 16.Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180:4785–4792. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- 17.Zhou L, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichiyama K, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283:17003–17008. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 19.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood. 2011;117:6532–6541. doi: 10.1182/blood-2010-11-317321. [DOI] [PubMed] [Google Scholar]
- 21.Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–2352. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 22.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 25.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 26.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Yang XP, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lexberg MH, et al. Th memory for interleukin-17 expression is stable in vivo. Eur J Immunol. 2008;38:2654–2664. doi: 10.1002/eji.200838541. [DOI] [PubMed] [Google Scholar]
- 33.Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 cells: new players in asthma pathogenesis. Allergy. 2011;66:989–998. doi: 10.1111/j.1398-9995.2011.02576.x. [DOI] [PubMed] [Google Scholar]
- 34.Cosmi L, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol. 2010;125:222–230. e221–224. doi: 10.1016/j.jaci.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 35.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirota K, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 38.Murphy CA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahern PP, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bending D, et al. Highly purified Th17 cells from BDC2. 5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119:565–572. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muranski P, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin-Orozco N, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muranski P, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luckey CJ, Weaver CT. Stem-cell-like qualities of immune memory; CD4(+) T cells join the party. Cell Stem Cell. 2012;10:107–108. doi: 10.1016/j.stem.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 46.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17- producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 47.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 48.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volpe E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 50.Yang L, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghoreschi K, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakefield LM, et al. Transforming growth factor-beta1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin Cancer Res. 1995;1:129–136. [PubMed] [Google Scholar]
- 53.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 55.Veldhoen M, Stockinger B. TGFβ1, a ‘Jack of all trades’: the link with pro-inflammatory IL-17-producing T cells. Trends Immunol. 2006;27:358–361. doi: 10.1016/j.it.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 56.McGeachy MJ, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 57.Lee Y, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang SC, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 60.Aujla SJ, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 62.Gurney AL. IL-22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Int Immunopharmacol. 2004;4:669–677. doi: 10.1016/j.intimp.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 63.Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10:857–863. doi: 10.1038/ni.1767. [DOI] [PubMed] [Google Scholar]
- 64.Rutz S, et al. Transcription factor c-Maf mediates the TGF-β-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. 2011;12:1238–1245. doi: 10.1038/ni.2134. [DOI] [PubMed] [Google Scholar]
- 65.Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol. 2010;31:354–361. doi: 10.1016/j.it.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 66.Eyerich S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Basu R, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012 doi: 10.1016/j.immuni.2012.08.024... [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stockinger B, Hirota K, Duarte J, Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol. 2011;23:99–105. doi: 10.1016/j.smim.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 69.Lazarevic V, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gessner MA, et al. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun. 2012;80:410–417. doi: 10.1128/IAI.05939-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kamanaka M, et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med. 2011;208:1027–1040. doi: 10.1084/jem.20102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 73.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andoh A, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 75.Ikeuchi H, et al. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum. 2005;52:1037–1046. doi: 10.1002/art.20965. [DOI] [PubMed] [Google Scholar]
- 76.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 77.Wolk K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol. 2007;178:5973–5981. doi: 10.4049/jimmunol.178.9.5973. [DOI] [PubMed] [Google Scholar]
- 78.Wolk K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 79.Yang Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. The J Exp Med. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferber IA, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 81.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 82.Zhang GX, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 83.Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110:493–497. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. The J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haak S, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kreymborg K, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–8104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
- 87.Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117:1119–1127. doi: 10.1172/JCI31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Codarri L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 89.El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23- induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mcgeachy MJ, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ciofani M, et al. A validated regulatory network for th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glasmacher E, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li P, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543–546. doi: 10.1038/nature11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vahedi G, et al. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ivanov, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schraml BU, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lohoff M, Mak TW. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat Rev Immunol. 2005;5:125–135. doi: 10.1038/nri1552. [DOI] [PubMed] [Google Scholar]
- 99.Hu CM, Jang SY, Fanzo JC, Pernis AB. Modulation of T cell cytokine production by interferon regulatory factor-4. J Biol Chem. 2002;277:49238–49246. doi: 10.1074/jbc.M205895200. [DOI] [PubMed] [Google Scholar]
- 100.Mukasa R, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schulz EG, Mariani L, Radbruch A, Höfer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 102.Ho IC, Lo D, Glimcher LH. c-maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J Exp Med. 1998;188:1859–1866. doi: 10.1084/jem.188.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Samstein RM, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huh JR, et al. Digoxin and its derivatives suppress T(H)17 cell differentiation by antagonizing RORγt activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Solt LA, et al. Suppression of T(H)17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]