

Abstract
Current medical countermeasures against organophosphate (OP) nerve agents are effective in reducing mortality, but do not sufficiently protect the CNS from delayed brain damage and persistent neurological symptoms. In this study, we examined the efficacy of neuregulin-1 (NRG-1) in protecting against delayed neuronal cell death following acute intoxication with the OP diisopropylfluorophosphate (DFP). Adult male Sprague Dawley rats were pretreated with pyridostigmine (0.1 mg/kg BW, i.m.) and atropine methylnitrate (20 mg/kg BW, i.m.) prior to DFP (9 mg/kg BW, i.p.) intoxication to increase survival and reduce peripheral signs of cholinergic toxicity but not prevent DFP-induced seizures or delayed neuronal injury. Pretreatment with NRG-1 did not protect against seizures in rats exposed to DFP. However, neuronal injury was significantly reduced in most brain regions by pretreatment with NRG-1 isoforms NRG-EGF (3.2 μg/kg BW, i.a) or NRG-GGF2 (48 μg/kg BW, i.a.) as determined by FluroJade-B labeling in multiple brain regions at 24 h post-DFP injection. NRG-1 also blocked apoptosis and oxidative stress-mediated protein damage in the brains of DFP-intoxicated rats. Administration of NRG-1 at 1 h after DFP injection similarly provided significant neuroprotection against delayed neuronal injury. These findings identify NRG-1 as a promising adjuvant therapy to current medical countermeasures for enhancing neuroprotection against acute OP intoxication.
Keywords: apoptosis, delayed neurotoxicity, diisopropylflurophosphate (DFP), neuroprotection, organophosphate, oxidative stress, rat model
Introduction
Organophosphates (OPs) are a class of phosphorus-containing organic chemicals that disrupt cholinergic neurotransmission by inhibiting acetylcholinesterase (AChE), an enzyme that catalyzes the hydrolysis of the neurotransmitter acetylcholine (ACh). OP nerve agents are the most toxic and rapidly acting of the known chemical warfare agents (Jett, 2007). Iraq briefly used OP nerve agents during the Iran-Iraq Gulf War of 1981-1987 (Newmark, 2004), and in 1995, a Japanese doomsday cult used sarin in a terrorist attack on the Tokyo subway system that killed twelve and sent more than 5,000 people to hospitals (Okumura et al., 2005; Yanagisawa et al., 2006; Hoffman et al., 2007). A year earlier, the cult killed seven people in a sarin attack in the Japanese city of Matsumoto (Okudera, 2002). As a result of escalating concerns regarding terrorist use of OP nerve agents following the World Trade Center attacks of September 11, 2001 in New York City, the development of therapeutic strategies for protection against chemical and biological warfare agents has become an important area of research.
Individuals that survive the acute intoxication with OP nerve agents often manifest long-term, chronic neurological symptoms (Okudera, 2002; Yanagisawa et al., 2006; Hoffman et al., 2007; Golomb, 2008). The current therapies for nerve agent exposure (atropine, oximes, reversible AChE inhibitors and benzodiazepines) are useful for increasing survival and reducing peripheral signs of cholinergic toxicity in exposure victims, but are not effective in preventing neuronal injury and long-term neurological damage. Therefore, new and more effective medical countermeasures against OP nerve agents must be developed to improve emergency treatment and protection of civilians, first responders and military personnel following acute intoxication with OP neurotoxins.
The OP diisopropylflurophosphate (DFP) is structurally and toxicologically similar to the OP nerve agents and thus is used as an OP nerve agent simulant in experimental animal models (Li et al., 2011). In this study, we employed the DFP neurotoxin exposure model to examine the neuroprotective efficacy of neuregulin-1 (NRG-1) against OP-induced neurotoxicity. NRG-1 belongs to a family of multipotent growth factors that have been shown to be neuroprotective and improve neurological function in rodent ischemic stroke models (Shyu et al., 2004; Xu et al., 2004; Xu et al., 2005; Guo et al., 2006; Xu et al., 2006; Li et al., 2007; Li et al., 2009; Iaci et al., 2010). The neuropathological sequelae of acute OP poisoning are similar to those observed in other acute CNS injuries, such as stroke, brain trauma and status epilepticus (Lemercier et al., 1983; McLeod et al., 1984; McDonough et al., 1987; Petras, 1994; Deshpande et al., 2010). Our findings demonstrate that NRG-1 prevents neuronal injury and apoptosis when administered with atropine and pyridostigmine immediately prior to DFP exposure. The therapeutic window for NRG-1 neuroprotection is at least 1 h when administered post-DFP exposure. These data suggest the clinical potential for NRG-1 for protecting against brain injury following acute OP intoxication.
Methods
Animals and DFP exposures
All animals used in these studies were treated humanely and with regard for alleviation of suffering and pain and all protocols involving animals were approved by the IACUC of Morehouse School of Medicine, Oregon Health & Science University and University of California, Davis (UCD) prior to the initiation of experimentation. Adult male Sprague-Dawley rats (280-320g; Harlan Laboratories, USA) were housed individually in standard plastic cages in a temperature-controlled room (22 ± 2°C) on a 12 h reverse light-dark cycle. Food and water were provided ad libitum.
Animals were anesthetized with 2% isoflurane (30% oxygen, 70% nitrous oxide) and then injected i.p. with DFP (D0879, Sigma Chemical Co., St. Louis, MO) at 9 mg/kg BW diluted in sterile distilled water. DFP was always prepared fresh within 5 min before administration. In the delayed neuronal injury studies, rats were injected i.m. with pyridostigmine bromide (PB; P1339, TCI America, Portland, OR) at 0.1 mg/kg BW in saline and with atropine methylnitrate (AMN; A0755, TCI America) at 20 mg/kg BW in saline 30 or 10 min prior to DFP injection, respectively. AMN and PB do not readily cross the blood brain barrier, so these drugs are centrally inactive (Shih et al., 1991) but effectively block peripheral OP neurotoxicity, thereby reducing mortality and facilitating detection of seizure symptoms (Kim et al., 1999). NRG-1 was administered inter-arterially 5-10 min prior to DFP injection or 1 or 4 hr after DFP injection. In all studies, anesthesia was stopped immediately following injection of DFP.
Intra-Arterial Administration of NRG-1
The left common carotid artery (CCA) was exposed in anesthetized animals through a midline incision and was carefully dissected free from surrounding nerves and fascia (Li et al., 2007). The occipital artery and superior thyroid branches of the external carotid artery (ECA) were isolated and electrocoagulated. The ECA was dissected further distally. The internal carotid artery (ICA) was isolated and carefully separated from the adjacent vagus nerve, and the pterygopalatine artery was ligated close to its origin with a 6-0 silk suture.
NRG-1 isoforms are generated from one gene by alternative mRNA splicing, and most of them are synthesized as part of a larger transmembrane precursor. The NRG-1 transmembrane precursors consist of either an immunoglobulin (Ig)-like or cysteine-rich domain, an EGF-like domain, a transmembrane domain and a cytoplasmic tail (Holmes et al., 1992; Fischbach and Rosen, 1997; Falls, 2003; Talmage and Role, 2004). The EGF-like domain of NRG-1 is common to all isoforms and appears to be sufficient for activation of erbB receptors and downstream signal transduction pathways (Holmes et al., 1992). Using a Hamilton syringe, NRG-1 was administered via the ECA as a single bolus of either NRG-1β EGF-like domain (NRG-EGF; R&D Systems, Minneapolis, Minnesota) at 3.2 μg/kg in PBS with 1% BSA or NRG-1β full-length extracellular Ig-domain isoform (Iaci et al., 2010) (NRG-GGF2; generously provided by Acorda Therapeutics, Hawthorne, NY) at 48μg/kg (in 1% BSA/PBS). The doses were selected based on previous studies of the neuroprotective efficacy of intra-arterial administration of these NRG-1 preparations in rat stroke models (Xu et al., 2006; Li et al., 2007). The dose of NRG-GGF2 was 15 times greater than NRG-EGF because the molecular weight of NRG-GGF2 is 6 times that NRG-EGF. We, therefore, increased the ug/kg dose six times to produce molar equivalents. We also increased the dose of NRG-GGF2 another 2.5 times to account for the binding of its immunoglobulin-like domain to extracellular matrix molecules in the brain (Loeb and Fischbach, 1995). All surgical procedures were performed using sterile/aseptic techniques in accordance with IACUC guidelines. Rectal temperature was maintained between 36.5°C and 37.0°C with a Homeothermic Blanket Control Unit (Harvard Apparatus) during anesthesia.
Carotid catheterization and post-DFP administration of NRG-1
No neuroprotective effect of isoflurane was seen in the NRG-1 pre-treatment studies; however, approximately one-third of the animals showed some neuroprotection with isoflurane alone in post-treatment studies (data not shown). To mitigate the protective effect of isoflurane in the post-treatment studies, we used pre-implanted catheters to administer NRG-1 interaterially. In collaboration with Charles River Laboratories, we developed a modified version of Charles River's commercially available carotid catheterized rats in which the catheter was cranially directed to deliver drugs into the cerebral vasculature. The carotid artery was isolated and a catheter filled with lock solution (50% glycerol, 500U/ml heparin in PBS) was inserted into the external carotid artery in a superior direction towards the carotid bifurcation. Arterial catheters exited through the skin on the back of the neck for stability. The surgical procedure was optimized to ensure proper directionality of the catheter in the carotid artery and that the catheter remained patent. Between the PB and DFP injections, animals were anesthetized with isoflurane and catheters were placed into the external carotid artery. Animals were awake 2-3 min after isoflurane was discontinued and they were allowed to recover prior to NRG-1 administration. To examine the efficacy of NRG-1 post-DFP exposure, rats were restrained with a plastic constrainer and a single bolus dose of vehicle or NRG-EGF (3.2 or 16 μg/kg) was administered into the catheter 1 or 4 h after DFP intoxication.
Histochemistry and immunohistochemistry
At 24 h post DFP injection, rats were deeply anesthetized with 5% isoflurane and perfused transcardially with saline followed by cold 4% PFA solution in PBS for 30 min. Brains were quickly removed and cryoprotected in 30% sucrose. A subset of rats whose brains were used for TUNEL labeling and phospho-Akt were perfused with saline prior to harvest and then snap frozen in dry ice. For FluoroJade B (FJB) labeling, coronal sections of 20 μm thickness were cryosectioned from the entire brain of each animal. For TUNEL labeling and phospho-Akt immunostaining, the thickness is 10um. Sections were mounted on slides which were stored at −80°C until further processed. FJB (AG310, Millipore, Billerica, MA) labeling and TUNEL labeling using the in situ death detection kit from Roche Molecular Biochemicals (Indianapolis, IN) were performed as previously described (Li et al., 2011). Immunohistochemical localization of NeuN was as previously described using a mouse anti-NeuN mAb (1:200, MAB314, Millipore). To examine Akt phosphorylation, animals were euthanized 60 min (n=3) after NRG-EGF administration and immunohistochemistry was performed using rabbit monoclonal phospho-Akt primary antibodies (1:200 dilution; cat# 4060; Cell Signaling Technology, Danvers, MA). The sections were incubated with the phospho-Akt antibody at 4°C overnight, followed by DL488 conjugated goat anti mouse secondary antibody (1:400 dilution; 115-485-146, Jackson ImmunoResearch Laboratory, West Grove, PA) for 1 h at room temperature. Negative controls were treated with secondary antibody but without primary antibodies.
Quantification of FJB-positive cells and pAkt-immunopositive cells
Every fifth section obtained from coronal sectioning of the entire brain of each rat was labeled with FJB or phospho-Akt. A Zeiss microscope equipped with CCD camera (Carl Zeiss Microimaging Inc, Thornwood, NY) was used to capture digital images of 5 sections at the same level (as determined using a brain atlas) at 200× magnification. The number of FJB and phospho-Akt-positive cells was determined using Image Pro Plus software (Media Cybernetics, Inc., Bethesda, MD) by an individual who was blinded to the experimental treatments. Only profiles of neuronal somas were counted and FJB-positive fragments were excluded. A mean value of FJB and phospho-Akt -positive cells per unit area within the brain regions was obtained for each individual rat. Data were expressed as mean ± SEM. These mean values from each individual rat were used as the statistical unit of measure for analysis by one-way ANOVA to determine statistically significant treatment effects.
Evaluation of Seizure Activity
The intensity of seizures in the delayed neuronal injury model was evaluated using a 5-point ranking system specifically designed to measure seizure activity in animals pre-treated with peripheral antidotes (De Sarro et al., 1993; Kim et al., 1999). In this test, 0 was used to indicate no response; 1, myoclonic jerks of the contralateral forelimb; 2, mouth and facial movements (i.e. facial myoclonus, clonus of the jaw and vibrissae) and head nodding with or without mild forelimb clonus; 3, severe forelimb clonus; 4, rearing and severe forelimb clonus; 5, rearing and falling. The animals were observed for a period of 1 h after drug administrations.
Acetylcholinesterase (AChE) activity
Blood was collected in heparinized tubes by cardiac puncture immediately after euthanasia, which was immediately followed by harvesting of brains with rapid dissection of brain regions on ice. All samples were snap frozen on dry ice and stored at -80°C. AChE activity was determined using the standard Ellman assay (Ellman et al., 1961) with 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) and acetylthiocholine iodide (ASChI) as substrate as previously described (Li et al., 2011). Pseudocholinesterase activity was inhibited by adding 100 μM tetraisopropyl pyrophosphoramide (iso-OMPA) to the assay. Data from brain samples were normalized using protein concentration as determined using the BCA assay according to the manufacturer's directions (Pierce, Rockford, IL). AChE activity in blood samples was normalized according to blood volume after correcting for addition of heparin.
Determination of ATPase activity and detection of nitrotyrosine of the ATPase beta subunit
Cortical brain tissues were prepared as previously described (Ross-Inta et al., 2010) with the following modifications. For ATPase activity assays, tissues were homogenized in 3-4 volumes of buffer (20 mM HEPES, pH 7.4 supplemented with kinase, phosphatase, and proteolytic inhibitors) per g tissue (wet weight), incubated on ice for 10-15 min, re-homogenized, and then snap frozen in liquid nitrogen. The ATPase activity assay was performed as previously described (Fujisawa et al., 2009). Each sample contained 3 μg of protein, 140 μl of Reaction Buffer (1.5 mM phospho(enol) pyruvate, 6.3 units/ml pyruvate kinase, 4.5 units/ml lactic dehydrogenase, 0.25 mM NADH, 45 mM MgCl2 and 45 mM HEPES, pH 7.5). The reaction was initiated by adding 2 mM ATP followed 5 min later by the addition of oligomycin (5 μg/ml). Reaction rates at 37°C were assessed at 340 nm in an Infinite M200 (Tecan) microplate reader. The oligomycin-sensitive rate was expressed as nmol ATP hydrolyzed/min/mg protein.
Protein extracts for western blots were obtained by snap freezing brains in liquid nitrogen prior to homogenization in a Dounce homogenizer with 20 downward strokes of a tight fitting pestle in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 0.5% IGEPAL, 0.1% SDS, 0.012% deoxycholate, 0.5% Triton X-100, pH 7.4) containing protease and phosphatase inhibitors. Homogenates were transferred to centrifuge tubes and rotated overnight at 4°C followed by centrifugation at 16,000 × g at 4°C. RIPA-soluble protein fractions were quantified with the BCA Protein Assay Kit. The protein extracts were concentrated and partly delipidated by acetone precipitation, through the addition of four volumes of -20°C acetone to each homogenate. Acetone-containing mixtures were vortexed and placed at -20°C for 24 h. Samples were then centrifuged at 16,000 × g for 10 min at 4°C. After pouring off the supernatant, the pellet was resuspended and washed two more times with acetone at -20°C, spinning each wash at 16,000 × g for 10 min at 4°C. After removing the supernatant from the final wash, the samples were placed in a SpeedVac for 15 min to remove residual acetone. Proteins were denatured in SDS-PAGE sample buffer (BioRad) plus 2-mercaptoethanol at 100°C for 5 min, loaded (5 μg) and run on an SDS-PAGE gel, transferred to PVDF membranes, then probed with mouse monoclonal antibodies reactive to nitrotyrosine (Millipore, Billerica, MA) and β-ATPase (BD Transduction Laboratories, San Jose, CA). Membranes were visualized with ECL chemiluminescent reagents on a Kodak 2000MM Imager. The loaded protein amounts were plotted against the densitometry readings to ensure that the chemiluminescence response was within a linear range of the protein range. The experiments were run in duplicate or triplicates and repeated two times in independent experiments. Data were expressed as mean ± SEM. The data were evaluated by using the t-test (StatSimple v2.0.5; Nidus Technologies, Toronto, Canada) setting p < 0.05 as statistically significant.
Plasma and brain pharmacokinetics of NRG-1 in the absence or presence of DFP
Pharmacokinetics (pK) studies were performed at the NIH CounterACT Preclinical Development Facility, SRI International (Menlo, CA). Animals were administered 5 μg/kg NRG-EGF i.a. in the absence or presence of DFP (9 mg/kg, i.p) with PB and AMN pretreatment as described above. At 10 min after the atropine injection, NRG-EGF was injected into the carotid artery. Approximately 10 min (5-15 min range) after NRG-EGF injection, DFP was administered. NRG-1 levels were evaluated in plasma at 10, 20, 40, 60, 90 min and 2, 4, 6, 12, 24 h and in whole brain lysates at 20 min, 1 h and 4 h post-DFP injection (n=3 per time point). Venous blood samples were placed on wet ice (for no more than 15 min) and processed to plasma by centrifugation for 6-8 min at 2-8°C. Plasma samples were transferred to cryovials and stored at -80°C until analysis. Whole brains were removed immediately after euthanasia, rinsed quickly with media (RPMI 1640 culture media containing protease inhibitors) and then placed in a tube with fresh media. Whole brains were homogenized and sonicated in lysis buffer containing a protease inhibitor cocktail immediately after collection. Homogenates were stored at -80°C until analysis. NRG-EGF concentrations were determined in plasma and brain tissue homogenates by an ELISA method using a polyclonal goat anti-human NRG-1-β1 EGF-domain Ab (AF-396-NA; R&D Systems) as both the capture and detecting antibody. After blocking with 5% normal goat serum, NRG-EGF standards (NRG-1β EGF-like domain, R&D Systems; 20 to 10,250 pg/ml) and samples were added to the wells. For plates containing plasma samples, NRG-EGF was diluted in dilution buffer containing 10% normal rat plasma. For plates containing lysate samples, NRG-EGF was diluted in dilution buffer containing 10% lysis buffer. For quality control, normal rat plasma (100%) and rat brain lysates (100%) were spiked with 10 ng/ml NRG-EGF and used fresh or stored frozen until analysis. Both fresh and frozen stored spiked samples were analyzed (at 1:10 dilution in dilution buffer) in parallel with the test samples in the ELISA. The detector Ab was biotinylated goat anti-human NRG1-β1 EGF domain (cat# AF-396-NA; R&D Systems, biotinylated using a using NHS-PEO4-Biotin (Pierce, Rockford, IL)). After washing, streptavidin-HRP conjugate (diluted 1:10,000) then TMB substrate (Sigma-Aldrich, St. Louis, MO) was added to each well. TMB stop solution was added to each well and the plate was read with a spectrophotometer (450 nm, with wavelength subtraction at 570 nm). The optical density of each well was measured by a Power Wave HT 340 microplate reader with KC4™ software, version 3.4 (BioTek Instruments, Winooski, VT). The software calculated the delta OD values for each well (450 OD-570 OD) to remove background OD. The mean OD value of the buffer blank on each plate was subtracted from all other OD values on the plate (standards, controls, and samples) to obtain the net OD values for further calculations. Standards were plotted with the concentration of NRG-EGF on the x-axis and mean net OD values on the y-axis. The standard curve was calculated using four-parameter regression analysis, and the resulting equation was used to calculate the concentration of NRG-EGF in all other samples on the plate. Mean and standard error were calculated for the plasma and brain drug concentrations. Pharmacokinetic parameters were calculated with WinNonlin (version 5.2) using noncompartmental modeling of the plasma and brain drug levels versus time data. The following parameters were determined: maximum concentration (Cmax), time to maximum concentration (Tmax), elimination half-life (t1/2) and clearance.
Results
Effect of NRG-1 on seizures and AChE activity following acute intoxication with DFP
To examine the effects of NRG-1 on delayed neuronal injury, we employed a DFP intoxication model in which animals are treated with PB and AMN prior to DFP exposure to reduce mortality and peripheral cholinergic symptoms (Kim et al., 1999; Li et al., 2011). In the absence of these supportive therapies, > 90% of DFP-exposed animals die within several hours of DFP injection. In contrast, treatment with PB and AMN increases the survival rate of DFP-exposed animals to approximately 90% but does not prevent DFP-induced seizures (see Figure 1). We also previously demonstrated using this model that acute DFP intoxication causes a regionally specific delayed neuronal injury in rat brain (Li et al., 2011). DFP-treated animals exhibit seizures, characterized by tonic-clonic seizures within 5 to 10 min after DFP injection. Tonic-clonic seizures generally subsided within 40-60 min after the DFP injection but DFP-treated animals continued to display mild clonic seizures for up to 4-5 h post-DFP exposure. In the current study, seizure activity is noted only during the first hour post-DFP exposure because we have observed that animals that do not have tonic-clonic seizures during that hour also do not exhibit injured neurons (data not shown). Pretreatment with either NRG-EGF or NRG-GGF2 had no effect on DFP-induced seizure activity (Figure 1). Analyses of AChE activity in the brain and blood 24 h after DFP administration indicated a reduction of AChE activity to about 25% of that observed in brains and blood from control animals and this inhibition was not significantly altered by pretreatment with NRG-1 (Figure 2A).
Figure 1. NRG-1 pretreatment does not alter seizure activity in rats acutely intoxicated with DFP.

Rats were pretreated with PB (0.1 mg/kg, i.m.) and AMN (20 mg/kg, i.m.) 30 or 10 min, respectively, prior to injection of DFP (9 mg/kg, i.p.) or an equal volume of vehicle (sterile water). NRG-1 (NRG-EGF, 3.2 μg/kg) or NRG-GGF2, 48 μg/kg) or vehicle (1% BSA in PBS) were administered via the carotid artery 5 min prior to DFP injection. Seizure severity was evaluated using a 5-point scale in which 0 indicates no response; 1, myoclonic jerks of the contralateral forelimb; 2, mouth and facial movements and head nodding with or without mild forelimb clonus; 3, severe forelimb clonus; 4, rearing and severe forelimb clonus; 5, rearing and falling.
Figure 2. NRG-1 pretreatment does not alter AChE activity in rats acutely intoxicated with DFP.

Rats were pretreated with PB (0.1 mg/kg, i.m.) and AMN (20 mg/kg, i.m.) 30 or 10 min, respectively, prior to injection of DFP (9 mg/kg, i.p.) or an equal volume of vehicle (sterile water). NRG-GGF2 (48 μg/kg) or vehicle (1% BSA in PBS) was administered via the carotid artery 5 min prior to DFP injection. The Ellman assay was used to measure AChE activity which was normalized to protein concentration in brain samples and to volume for whole blood samples. Data in panels A and B are expressed as mean ± SEM (N = 5 per treatment group). Statistically significant differences were identified using one way ANOVA with post hoc Tukey's test; ***p < 0.001 compared to controls.
Plasma and brain pharmacokinetics of NRG-EGF
The lack of effect of NRG-1 on seizure and AChE activity raised questions as to whether it was accessing the brain. To address this question, we investigated the pharmacokinetics of NRG-EGF and its ability to cross the blood-brain barrier after a single i.a. administration to control rats and rats challenged with DFP. As indicated in Table 1, the plasma Cmax values after i.a. administration were 2050 and 2330 ng/ml for the control and DFP groups, respectively, and the Tmax was 0.17 h (10 min). The plasma t1/2 was calculated as 0.14 h (8 min) and NRG-EGF was undetectable after 20 min. NRG-EGF was not detected in brains of animals not injected with NRG-EGF, but it was present in brains of animals treated with exogenous NRG-EGF at 20 min post administration and remained at a constant level for up to 4 h post-NRG-EGF injection. Acute intoxication with DFP did not significantly alter brain levels of NRG-EGF following i.a. injection of NRG-EGF, nor did it alter brain and plasma PK parameters. In the brain, the Cmax was 25.0-30.2 pg/mg; the Tmax did not vary significantly from 20 min to 4 h post-NRG-1 injection. These findings are consistent with two previous studies demonstrating that NRG1 EGF-like domain and full length NRG1 both enter the brain within 10-15 min and are stable once in the brain (Kastin et al., 2004; Rosler et al., 2011).
Table 1. Pharmacokinetics of NRG-1 in brain and plasma.
| Brain | Brain w/DFP | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| control | 20 min | 60 min | 4 hr | 20 min | 60 min | 4 hr | |
|
|
|||||||
| NRG-1 (pg/m g protein) | nd | 24.9 | 24.9 | 24.2 | 30.2 | 19.4 | 17.3 |
| Std. Error (±) | - | 1.3 | 1.6 | 2.6 | 10.4 | 0.5 | 1.8 |
|
| |||||||
| Plasma | Plasma | Plasma w/DFP | |||||
|
| |||||||
| 10 min | 10 min | 20 min | 40 min | 10 min | 20 min | 40 min | |
|
| |||||||
| NRG-1 (pg/ml plasma) | nd | 2050.0 | 718.3 | nd | 2330.0 | 1019.3 | nd |
| Std. Error (±) | - | 343.1 | 361.8 | - | 143.8 | 94.6 | - |
nd = not detected
n=3 per time point
NRG-1 protected against the delayed neuronal injury observed in the brains of animals acutely intoxicated with DFP
FJB staining was used to assess neuronal damage in brains collected 24 h after DFP injection. Consistent with our previous findings (Li et al., 2011), acute DFP intoxication caused extensive neuronal cell death in multiple brain regions. Pretreatment with either the EGF-like domain (NRG-EGF) or the full-length extracellular domain (NRG-GGF2) of NRG-1 significantly inhibited DFP-mediated neuronal injury as shown in representative photomicrographs of FJB labeling in the CA1 and dentate gyrus of the hippocampus (Figure 3). NRG-1 pretreatment resulted in approximately a 90% reduction in FJB labeling in numerous cortical regions (Figure 4), the CA1, CA3 and dentate gyrus of the hippocampus (Figure 5A). However, NRG-1 only partially attenuated FJB labeling in the insular, piriform, entorhinal, perirhinal and ectorhinal cortices. The amygdala and thalamus displayed nucleus-specific effects of NRG-1 following DFP intoxication. NRG-1 treatment did not protect neurons in the basal and lateral amygdaloid nuclei, however, neuronal injury was reduced in the medial nuclei by NRG-GGF2 and the central amygdaloid nuclei by both NRG-1 isoforms (Figure 5B). In the thalamus, NRG-1 reduced neuronal injury only in the dorsolateral nucleus (Figure 5C). Neuroprotection by NRG-1 was also found in midbrain structures including the septum, nucleus accumbens and substantia nigra (Figure 5D). We did not observe a difference in neuroprotection by NRG1 between the ipsilateral and contralateral brain hemispheres.
Figure 3. NRG-1 protects against DFP-induced delayed neurotoxicity.
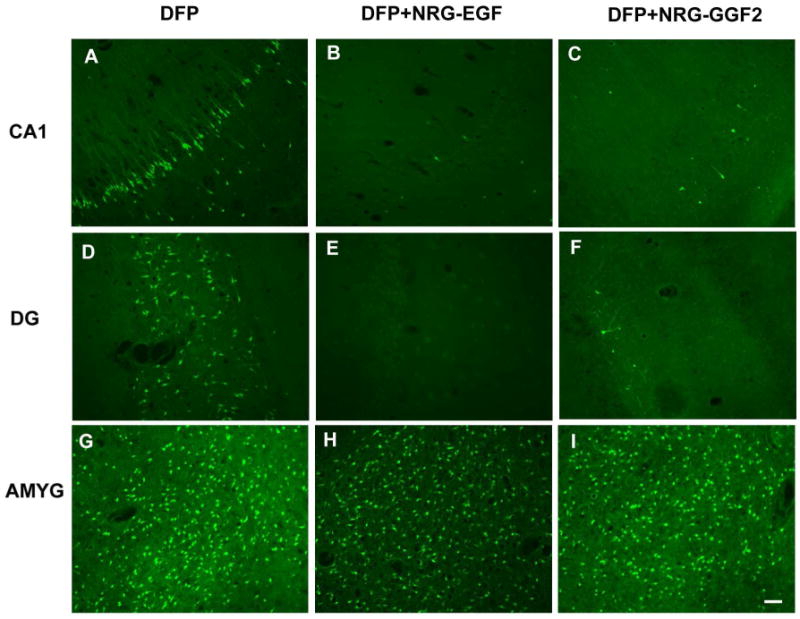
Rats were pretreated with PB and AMN prior to DFP (9 mg/kg, i.p.). NRG-1 (NRG-EGF, 3.2 μg/kg or NRG-GGF2, 48 μg/kg) or vehicle was administered via carotid artery 5 min prior to DFP exposure. Brains collected at 24 h post-DFP injection were labeled with Fluoro-Jade B (FJB). Representative photomicrographs of FJB labeling are shown from the CA1 region of the hippocampus (CA1; panels A,B,C), hippocampal dentate gyrus (DG; panels D,E,F) and amygdala basal nucleus (AMYG; panels G,H,I) of vehicle (A,D,G), NRG-EGF (B,E,H) and NRG-GGF2 (C,F,I) treated rats. Scale bar = 100 μm.
Figure 4. NRG-1 pretreatment protects against DFP-induced neuronal injury in the cerebral cortex.

Rats were pretreated with PB and AMN prior to DFP (9 mg/kg, i.p.). NRG-1 (NRG-EGF, 3.2 μg/kg or NRG-GGF2, 48 μg/kg, i.a.) or vehicle was administered via carotid artery 5 min prior to DFP exposure. Brains collected at 24 h post-DFP injection were labeled with FJB. The number of FJB-positive cells was quantified from digital images of coronal sections (200× magnification) in specific brain regions at the same level across animals as determined using a brain atlas. Data are expressed as the mean ± SEM (N = 7 animals per treatment group). Statistically significant differences were identified using one way ANOVA with post hoc Tukey's test; *p <0.05, **p <0.01 and ***p <0.001 compared to controls. The brain regions analyzed were the auditory (AUD), cingulate (CING), motor (MOT), retrosplenial (RS), somatosensory (SS), visual (VIS), piriform (PIR), insular (INS), entorhinal (ENT) and ectorhinal (ECT) cortices.
Figure 5. Neuroprotection by NRG-1 against DFP neurotoxicity varies across brain regions.
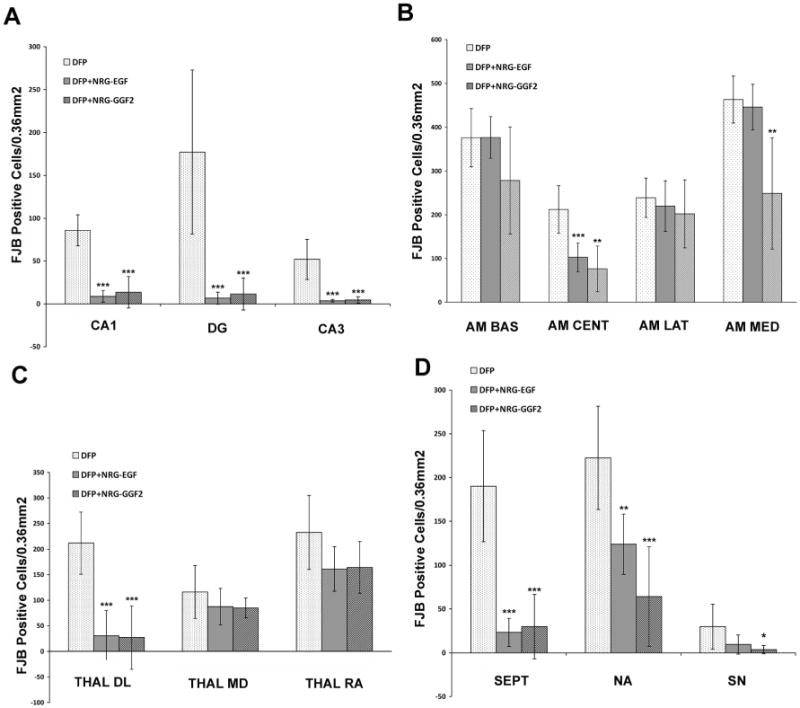
Rats were pretreated with PB and AMN prior to DFP (9 mg/kg, i.p.). NRG-1 (NRG-EGF, 3.2 μg/kg or NRG-GGF2, 48 μg/kg, i.a.) or vehicle was administered via carotid artery 5 min prior to DFP exposure. Brains collected at 24 h post-DFP injection were labeled with FJB. The number of FJB-positive neuronal cells was quantified from digital images of coronal sections (200× magnification) in specific brain regions at the same level across animals as determined using a brain atlas. Data are expressed as the mean ± SEM (N = 7 animals per treatment group). Statistically significant differences were identified using one way ANOVA with post hoc Tukey's test; *p <0.05, **p <0.01 and ***p <0.001 compared to controls. The brain areas examined were the CA1 (CA1), dentate gyrus (DG) and CA3 (CA3) regions of the hippocampus (A); the basal (AM BAS), central (AM CENT), medial (AM MED) and lateral (AM LAT) nuclei of the amygdala (B); the dorsolateral nucleus (THAL DL), mediodorsal (THAL MD) and reunion area (THAL RA) of the thalamus (C) and the septum (SEPT), nucleus accumbens (NA) and substantia nigra (SN) (D).
NRG-1 blocked DFP-induced apoptosis and rescued NeuN immunoreactivity
TUNEL staining of brains collected 24 h after injection with DFP revealed TUNEL-positive cells in all of the 23 brain regions examined. NRG-1 pretreatment effectively abolished TUNEL labeling in a similar regional pattern as FJB labeling as illustrated in representative photomicrographs of the lateral dorsal thalamic nuclei (Figure 6C versus Figure 6D). As previously shown (Li et al., 2011), acute DFP intoxication reduces the relative intensity of NeuN immunoreactivity in injured neurons as illustrated in the dorsal lateral thalamic nuclei (Figure 6E). These injured areas showed high numbers of FJB labeling (Figure 6A), which co-localizes with the low or no NeuN expressing cells (Figure 6E). Neighboring neurons that were not injured showed relatively higher levels of NeuN immunoreactivity. NRG-1 pretreatment rescued NeuN immunoreactivity (Figure 6F).
Figure 6. NRG-1 blocks DFP-induced apoptosis and rescues NeuN immunopositive cells.

Rats were treated with PB, AMN and either vehicle or NRG-EGF (3.2 μg/kg, i.a.) before injection with DFP (9 mg/kg, i.p.). As illustrated in representative photomicrographs of the lateral dorsal thalamus, in control animals that did not receive NRG-1, at 24 h post-DFP injection there are many FJB-labeled cells (A), numerous TUNEL positive cells (B) and reduced NeuN immunoreactivity in injured neurons (C) with relatively higher levels of NeuN immunoreactivity in adjacent uninjured neurons (arrows). However, NRG-1 treatment significantly attenuated FJB labeling (B), abolished TUNEL staining (D) and rescued NeuN immunoreactivity (F). These figures are representative of brains from NRG-EGF treated animals, however similar results were seen when NRG-GGF2 was used. Scale bar = 100 μm.
NRG-1 pretreatment reduced DFP-induced oxidative stress
Acute DFP intoxication caused oxidative damage in the cortex detectable at both 24 h (Figure 7A) and 72 h (Figure 7B) post DFP injection by western blot analyses using Ab that specifically recognizes nitrated tyrosine (nTyr) residues. Reprobing these blots with Ab specific for the mitochondrial ATPase beta subunit confirmed that the 55 kDa nTyr immunoreactive band corresponds to this ATPase subunit (Figure 7A and B). The level of nitrated tyrosine per beta subunit was increased by 2-fold in brains from DFP-treated rats and this effect was completely prevented in animals pretreated with NRG-EGF (Figure 7A, B, C). It has been demonstrated that nitration of critical tyrosine residues in this protein is accompanied by loss of ATPase activity resulting in decreased ATP production (Fujisawa et al., 2009; Haynes et al., 2010). As predicted, ATPase activity was reduced by DFP (by 40%) and this loss of activity was completely abrogated by pretreatment with NRG-EGF (Figure 7D).
Figure 7. Acute DFP intoxication causes oxidative stress that is reduced by NRG-1 pretreatment.

Rats were treated with PB, AMN and either vehicle or NRG-EGF (3.2 μg/kg, i.a.) prior to injection with DFP (9 mg/kg, i.p.). Homogenates of cortical brain tissues collected at 24 (A) or 72 h (B) post-DFP injection were separated by SDS PAGE and probed with Ab that reacts with nitrotyrosine (nTyr) or the ATPase beta subunit. Densitometric analyses (C) indicate that DFP intoxication increased the normalized level of nitrated tyrosine and this effect was attenuated by pretreatment with NRG-EGF. ATPase activity was reduced by DFP and pretreatment with NRG-EGF protected ATPase activity (D). Experiments were run in duplicate or triplicate and repeated two times in independent experiments. Data were expressed as the mean ± SEM. Statistically significant differences were identified using the t-test (StatSimple v2.0.5; Nidus Technologies, Toronto, Canada); *p < 0.05 relative to control.
Administration of NRG-1 increase Akt phosphorylation
NRG-1 has been previously shown to protect neurons by activating the CDK5/Akt/PI3 kinase pathway (Li et al., 2003; Croslan et al., 2008). In this study, NRG-EGF dramatically and significantly increased immunoreactivity for phospho-Akt in the brains of DFP-intoxicated animals at 60 min after DFP administration. The pattern of Akt phosphorylation coincided with brain regions where NRG-1 blocked FJB labeling as demonstrated by the representative figure from the somatosensory cortex (Figures 8A-C). Akt phosphorylation slightly increased following DFP intoxication alone, but NRG-1 stimulated Akt phosphorylation by 5-fold (Figure 8D). NRG-EGF did not increase phospho-Akt levels in brain regions where it did not protect neurons from DFP intoxication, such as the amygdala (data not shown).
Figure 8. NRG-1 upregulates phospho-Akt immunoreactivity.

Rats were treated with PB, AMN and either vehicle or NRG-EGF (3.2 μg/kg, i.a.) before injection with DFP (9 mg/kg, i.p.). Animals were euthanized 60 min after NRG-EGF administration, and brain sections from these animals were immunostained using antibodies that specifically recognized the phosphorylated form of Akt. As illustrated in representative photomicrographs from the somatosensory cortex, in control animals that did not receive NRG-EGF, there are few p-Akt -labeled cells (CTRL; A). Levels of phospho-Akt increase slightly at 60 min after DFP intoxication (DFP; B), but the percentage of phospho-Akt positive cells increases significantly 60 min after NRG-1 administration (DFP+NRG; C). The number of phospho-Akt positive neuronal cells in the somatosensory cortex increased 5-fold following NRG-EGF administration (D). Statistically significant differences were identified using one way ANOVA with post hoc Tukey's test (n = 3); ***p <0.001 compared to controls. Scale bar = 100 μm.
Neuroprotection by NRG-1 administered post-DFP exposure
To examine the efficacy of NRG-1 post-DFP exposure, NRG-1 (3.2 or 16 μg/kg) or vehicle was administered into a pre-implanted arterial catheter 1 or 4 h after DFP intoxication. At a dose of 3.2 μg/kg, no neuroprotective effects of NRG-1 were seen when administered 1 h post-DFP exposure. However, at a 5 times greater dose of 16μg/kg, NRG-1 prevented DFP-mediated neuronal injury in both brain hemispheres when administered 1 h following DFP exposure. The pattern of neuroprotection when NRG-1 was administered 1 h after DFP injection was similar to that observed with NRG-1 pre-treatment. Figure 9 is a representative image demonstrating FJB labeling in the CA1 region of the hippocampus in DFP-exposed animals treated with vehicle (Fig. 9A) versus DFP-exposed animals receiving NRG-1 (Fig. 9B) 1 h post-DFP exposure. There was ∼90% reduction in FJB labeling in the CA1 of the hippocampus (Fig. 9C), cingulate cortex (Fig. 9D) and most other brain regions examined. While pretreatment with NRG-1 did not protect against neuronal cell death in the amygdala, treatment with NRG-1 1 h after DFP injection effectively blocked neuronal injury in the amygdala of a subpopulation of animals (Fig 9E). No neuroprotection was observed when NRG-1 was administered 4 h after DFP injection (data not shown).
Figure 9. NRG-1 is neuroprotective when administered 1 h post-DFP injection.
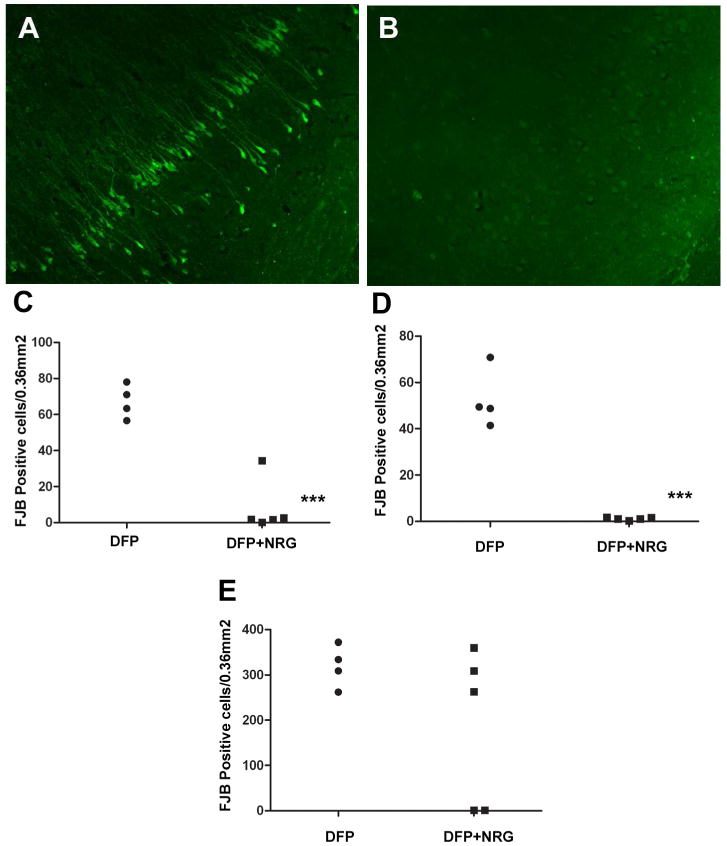
Rats were pretreated with PB and AMN prior to injection with DFP (9 mg/kg, i.p.) or vehicle (control). Between PB and DFP injections, catheters were placed into the external carotid artery. Animals were awake 2-3 min after isoflurane was discontinued and allowed to recover prior to NRG-1 administration. One h after DFP injection, animals were restrained and a single bolus dose of vehicle or NRG-1 (16 μg/kg) was administered via the catheter. Animals were euthanized 24 h post-DFP injection and neuronal injury quantified using FJB. Representative photomicrographs of FJB labeling in the CA1 region of the hippocampus from animals intoxicated with DFP in the absence (A; N = 4) or presence (B; N = 5) of NRG-1 treatment indicate that administration of NRG-1 1 h after DFP injection provides significant protection against DFP-induced delayed neuronal injury. These findings are confirmed by quantitative analyses of the number of FJB-labeled in cells the CA1 region of the hippocampus (C), cingulate cortex (D) and amygdala medial nucleus (E). Statistically significant differences were identified using a t- test; ***p<0.001. Scale bar = 100 μm.
Discussion
The results of this study demonstrate the efficacy of NRG-1 in protecting against delayed neuronal injury in a rat model of acute DFP intoxication. In initial studies, nearly all animals displayed severe seizures and died within 30 min after acute DFP intoxication without PB and AMN. To examine the effects of NRG-1 on delayed neuronal injury, we used a model in which rats are treated with PB and AMN prior to DFP intoxication to increase survival and reduce peripheral signs of cholinergic toxicity (Kim et al., 1999; Kadriu et al., 2009; Deshpande et al., 2010; Li et al., 2011). In our delayed neuronal injury model, more than 90% of the animals survived and delayed neuronal injury was observed 4 to 72 h after DFP exposure in specific brain regions (Li et al., 2011). When animals were pretreated with NRG-1, DFP-induced neuronal injury was blocked in most brain regions as determined by FJB labeling. Previous studies from our laboratory demonstrated that the FJB labeled cells observed following DFP intoxication were neurons (Li et al., 2011). Similar results were obtained using either the EGF-like domain fragment of NRG-1 (NRG-EGF) or the full-length extracellular domain NRG-1 isoform (NRG-GGF2). NRG-1 pretreatment blocked neuronal injury in the hippocampus, thalamus and several cortical regions. However, the neuroprotective effect of NRG-1 varied across brain regions, as evidenced by partial attenuation of DFP-induced neuronal injury in some but not all loci in the amygdala. The reason for the regional specificity of NRG-1 neuroprotection is unclear. Plausible explanations include, but are not limited to, factors such as differential access of exogenously administered NRG-1 to various brain regions, region-specific mechanisms of neuronal death, and regional differences in expression of receptors for NRG-1 or levels of endogenous NRG-1.
As an initial assessment of mechanisms associated with the neuroprotective effects of NRG-1, we examined NeuN immunoreactivity and TUNEL labeling in brains of DFP-exposed animals receiving vehicle versus NRG-1. NeuN is a neuronal marker and the loss of NeuN immunoreactivity is associated with neuronal damage following acute brain injuries, including brain trauma, stroke and exposure to DFP and soman (Sato et al., 2001; Unal-Cevik et al., 2004; Collombet et al., 2006; Kadriu et al., 2009; Li et al., 2011). Our findings showed that DFP reduced NeuN labeling in injured neurons, but treatment with NRG-1 rescued NeuN immunoreactivity. It has also been reported that DFP exposure in the presence of PB and AMN resulted in delayed neuronal apoptosis (Kim et al., 1999; Kadriu et al., 2009; Li et al., 2011). DFP-induced TUNEL labeling in neurons was completely blocked by NRG-1 pretreatment in a similar regional pattern as FJB labeling. An evolving hypothesis in the field is that oxidative stress mechanisms contribute to the toxic effects of acute intoxication by OP compounds, including DFP (Gupta et al., 2001; Soltaninejad and Abdollahi, 2009). Consistent with this possibility, we found that acute DFP intoxication in the presence of PB and AMN increased nitration of the ATPase beta subunit. As predicted by prior studies demonstrating that nitration of ATPase beta reduced its activity (Fujisawa et al., 2009; Haynes et al., 2010), we observed a concomitant decrease in ATPase activity. Pretreatment with NRG-1 blocked DFP-induced tyrosine nitration and maintained ATPase activity at control levels. Since the energy needs of neurons are mainly, if not exclusively, derived from oxidative phosphorylation, these findings have significant implications regarding the mechanism(s) by which acute OP intoxication causes delayed neuronal cell death. How NRG-1 protects against DFP-induced nitrative damage remains the subject for future investigations. However, collectively, these data indicate that NRG-1 prevents DFP-induced delayed neuronal damage by blocking apoptotic and oxidative stress mechanisms and these effects may be mechanistically linked. Both acute OP intoxication and ischemia are characterized by neuronal injury that is associated with common mechanisms and neuropathology (Lemercier et al., 1983; McLeod et al., 1984; McDonough et al., 1987; Petras, 1994; Deshpande et al., 2010). Thus, as observed in these studies of acute DFP intoxication, NRG-1 prevented delayed neuronal injury in rodent models of ischemic stroke and inhibited ischemia-induced apoptotic and oxidative stress mechanisms (Shyu et al., 2004; Xu et al., 2004; Xu et al., 2005; Guo et al., 2006; Li et al., 2007; Li et al., 2009).
To determine whether the effects of NRG-1 were due to direct activation of the NRG-erbB signaling pathway in the brain, we first confirmed that NRG-1 reached the brain. Our pharmacokinetic studies demonstrated that exogenous NRG-EGF enters the brain after i.a. administration. Brain levels of NRG-EGF remain constant from 20 min to 4 h post-NRG-EGF injection. These findings are consistent with previous studies showing that NRG-1 crosses the BBB and blood spinal cord barrier in mice (Kastin et al., 2004; Carlsson et al., 2011; Kato et al., 2011; Rosler et al., 2011). While brain levels remained constant, NRG-EGF had a very short half-life in plasma, and was undetectable more than 20 min after administration. Interestingly, acute DFP intoxication did not result in higher NRG-1 levels in brain or alter the plasma profile of this compound. The time frame in which we observed entry of exogenous NGF into the brain is similar to that recently reported for radiolabeled full length NRG-1 (similar to NRG-GGF2) injected i.v. and i.p. into mice (Rosler et al., 2011), and an earlier study that employed radiolabeled NRG-1 EGF-like domain (NRG-EGF) (Kastin et al., 2004), although full length NRG-1 appears to be more stable over the long term. The short plasma half-life of NRG-EGF might reduce the likelihood of unwanted side effect in other tissues. The BBB permeability, persistence in brain tissues and short plasma half-life together suggest that NRG-1 could be a promising candidate for safe treatment of neuronal injury after nerve agent exposure and other acute CNS injuries.
In a second approach to determining whether the effects of NRG-1 were due to direct activation of the NRG-erbB signaling pathway in the brain, we examined the activation of Akt following NRG-EGF administration. We and others have shown that NRG-1 protects neurons by activating the CDK5/Akt/PI3 kinase pathway (Li et al., 2003; Croslan et al., 2008). Here, we demonstrated that NRG1 significantly increased phosphorylation of Akt in the somatosensory cortex of DFP-intoxicated animals. Phosphorylation of Akt was seen in both neurons and non-neuronal cells (data not shown), consistent with our studies demonstrating that NRG1 is both neuroprotective and anti-inflammatory in stroke models (Xu et al., 2005; Croslan et al., 2008). These data are consistent with a previous report demonstrating that exogenous radiolabeled NRG-1 stimulated the phosphorylation/activation of the NRG-1 receptor erbB4 at 1 h after administration (Rosler et al., 2011). Collectively, these data demonstrate that NRG-1 can enter the brain and directly activate the erbB signaling pathway. However, this does not rule out the possibility that other mechanisms, such as regulation of CNS seizures and hypoxic injury, contribute to the neuroprotective effects of NRG-1.
We also investigated the neuroprotective efficacy of NRG-1 when administered after DFP exposure. Previous reports from rat stroke models showed that NRG-1 had a therapeutic window of at least 13.5 h for prevention of neuronal death (Xu et al., 2006) and up to 7 d for improvement of neurological function (Iaci et al., 2010) after the ischemic event. Studies of delayed neuronal injury in DFP-intoxicated rats pretreated with PB and AMN demonstrated that the earliest signs of FJB labeling occurred 4 h after DFP injection (Li et al., 2011). Therefore, we examined the efficacy of NRG-1 when administered 1 and 4 h after DFP intoxication. NRG-1 was neuroprotective when administered 1 h but not at 4 h post-DFP injection. Further studies are underway to investigate strategies for increasing the neuroprotective efficacy of NRG-1 at later post-exposure times, including combination drug therapies as have been shown to be effective in the rat stroke model (Li et al., 2007).
Our data clearly demonstrate that NRG-1 administered before or up to 1 h after exposure to an acutely intoxicating dose of DFP is extremely effective in blocking neuronal injury. This neuroprotective effect does not appear to be mediated by anti-convulsant effects of NRG-1 since neither the incidence nor severity of seizures was altered by NRG-1. Whether the reduction of delayed neuronal injury by NRG-1 translated into improved functional outcome as determined by behavioral measures is the focus of future studies. In recent clinical studies of the cardioprotective effects of NRG-1 in human patients, doses of NRG-EGF that showed efficacy for treating heart failure were similar to the doses used in our studies and were shown to be safe and tolerable (Gao et al., 2010; Jabbour et al., 2011). These results further indicate that NRG-1 could be useful as an adjuvant to existing antidotal treatments for treating individuals acutely intoxicated by OP nerve agents.
Highlights.
NRG-1 blocked DFP induced neuronal injury.
NRG-1 did not protect against seizures in rats exposed to DFP.
NRG-1 blocked apoptosis and oxidative stress-mediated protein damage in the brains of DFP-intoxicated rats.
Administration of NRG-1 at 1 h after DFP injection provided neuroprotection against delayed neuronal injury.
Acknowledgments
The research is supported by the CounterACT Program, National Institutes of Health Office of the Director, and the National Institute of Neurological Diseases and Stroke Grant Number U01 NS 057993, R21 NS072094, and National Center for Research Resources grants C06 RR-07571, G12-RR03034 and G20 RR14335. We thank Dr. Anthony Caggiano and Jennifer Iaci of Acorda Therapeutics, Inc. for generously providing NRG-GGF2 and support for the studies involving NRG-GGF2. The sponsors were not involved in the study design, collection, analysis, and interpretation of data, in the writing of the report or in the decision to submit the paper for publication.
Footnotes
Conflict of Interest Statement: Acorda Therapeutics, Inc. provided NRG-GGF2 and support for the studies involving NRG-GGF2. The sponsors were involved in the study design but not in the collection, analysis, and interpretation of data, in the writing of the report or in the decision to submit the paper for publication. Dr. Byron Ford has served as a paid consultant for Acorda Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carlsson T, Schindler FR, Hollerhage M, Depboylu C, Arias-Carrion O, Schnurrbusch S, Rosler TW, Wozny W, Schwall GP, Groebe K, Oertel WH, Brundin P, Schrattenholz A, Hoglinger GU. Systemic administration of neuregulin-1beta(1) protects dopaminergic neurons in a mouse model of Parkinson's disease. J Neurochem. 2011;117:1066–1074. doi: 10.1111/j.1471-4159.2011.07284.x. [DOI] [PubMed] [Google Scholar]
- Collombet JM, Masqueliez C, Four E, Burckhart MF, Bernabe D, Baubichon D, Lallement G. Early reduction of NeuN antigenicity induced by soman poisoning in mice can be used to predict delayed neuronal degeneration in the hippocampus. Neurosci Lett. 2006;398:337–342. doi: 10.1016/j.neulet.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Croslan DR, Schoell MC, Ford GD, Pulliam JV, Gates A, Clement CM, Harris AE, Ford BD. Neuroprotective effects of neuregulin-1 on B35 neuronal cells following ischemia. Brain Res. 2008;1210:39–47. doi: 10.1016/j.brainres.2008.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sarro G, Di Paola ED, De Sarro A, Vidal MJ. L-arginine potentiates excitatory amino acid-induced seizures elicited in the deep prepiriform cortex. Eur J Pharmacol. 1993;230:151–158. doi: 10.1016/0014-2999(93)90797-l. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol Sci. 2010;116:623–631. doi: 10.1093/toxsci/kfq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Rosen KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–458. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- Fujisawa Y, Kato K, Giulivi C. Nitration of tyrosine residues 368 and 345 in the beta-subunit elicits FoF1-ATPase activity loss. Biochem J. 2009;423:219–231. doi: 10.1042/BJ20090594. [DOI] [PubMed] [Google Scholar]
- Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, Li T, Liu X, Xu Y, Li X, Zhou M. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. 2010;55:1907–1914. doi: 10.1016/j.jacc.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Golomb BA. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc Natl Acad Sci U S A. 2008;105:4295–4300. doi: 10.1073/pnas.0711986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WP, Wang J, Li RX, Peng YW. Neuroprotective effects of neuregulin-1 in rat models of focal cerebral ischemia. Brain Res. 2006;1087:180–185. doi: 10.1016/j.brainres.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Milatovic D, Dettbarn WD. Nitric oxide modulates high-energy phosphates in brain regions of rats intoxicated with diisopropylphosphorofluoridate or carbofuran: prevention by N-tert-butyl-alpha-phenylnitrone or vitamin E. Arch Toxicol. 2001;75:346–356. doi: 10.1007/s002040100249. [DOI] [PubMed] [Google Scholar]
- Haynes V, Traaseth NJ, Elfering S, Fujisawa Y, Giulivi C. Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. Am J Physiol Endocrinol Metab. 2010;298:E978–987. doi: 10.1152/ajpendo.00739.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A, Eisenkraft A, Finkelstein A, Schein O, Rotman E, Dushnitsky T. A decade after the Tokyo sarin attack: a review of neurological follow-up of the victims. Mil Med. 2007;172:607–610. doi: 10.7205/milmed.172.6.607. [DOI] [PubMed] [Google Scholar]
- Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD, et al. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. doi: 10.1126/science.256.5060.1205. [DOI] [PubMed] [Google Scholar]
- Iaci JF, Ganguly A, Finklestein SP, Parry TJ, Ren J, Saha S, Sietsma DK, Srinivas M, Vecchione AM, Caggiano AO. Glial growth factor 2 promotes functional recovery with treatment initiated up to 7 days after permanent focal ischemic stroke. Neuropharmacology. 2010;59:640–649. doi: 10.1016/j.neuropharm.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Jabbour A, Hayward CS, Keogh AM, Kotlyar E, McCrohon JA, England JF, Amor R, Liu X, Li XY, Zhou MD, Graham RM, Macdonald PS. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail. 2011;13:83–92. doi: 10.1093/eurjhf/hfq152. [DOI] [PubMed] [Google Scholar]
- Jett DA. Neurological aspects of chemical terrorism. Ann Neurol. 2007;61:9–13. doi: 10.1002/ana.21072. [DOI] [PubMed] [Google Scholar]
- Kadriu B, Guidotti A, Costa E, Auta J. Imidazenil, a non-sedating anticonvulsant benzodiazepine, is more potent than diazepam in protecting against DFP-induced seizures and neuronal damage. Toxicology. 2009;256:164–174. doi: 10.1016/j.tox.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, Pan W. Neuregulin-1-beta1 enters brain and spinal cord by receptor-mediated transport. J Neurochem. 2004;88:965–970. doi: 10.1046/j.1471-4159.2003.02224.x. [DOI] [PubMed] [Google Scholar]
- Kato T, Abe Y, Sotoyama H, Kakita A, Kominami R, Hirokawa S, Ozaki M, Takahashi H, Nawa H. Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: implication in neurodevelopmental hypothesis for schizophrenia. Mol Psychiatry. 2011;16:307–320. doi: 10.1038/mp.2010.10. [DOI] [PubMed] [Google Scholar]
- Kim YB, Hur G, Shin S, Sok D, Kang J, Lee Y. Organophosphate-induced brain injuries: delayed apoptosis mediated by nitric oxide. Environ Toxicol Pharm. 1999;7:147–152. doi: 10.1016/s1382-6689(99)00006-x. [DOI] [PubMed] [Google Scholar]
- Lemercier G, Carpentier P, Sentenac-Roumanou H, Morelis P. Histological and histochemical changes in the central nervous system of the rat poisoned by an irreversible anticholinesterase organophosphorus compound. Acta Neuropathol (Berl) 1983;61:123–129. doi: 10.1007/BF00697391. [DOI] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC. Cdk5 is involved in neuregulin-dependent activation of PI-3 kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278:35702–35709. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhang R, Guo YL, Mei YW. Effect of neuregulin on apoptosis and expressions of STAT3 and GFAP in rats following cerebral ischemic reperfusion. J Mol Neurosci. 2009;37:67–73. doi: 10.1007/s12031-008-9121-3. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu Z, Ford GD, Croslan DR, Cairobe T, Li Z, Ford BD. Neuroprotection by neuregulin-1 in a rat model of permanent focal cerebral ischemia. Brain Res. 2007;1184:277–283. doi: 10.1016/j.brainres.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Tewolde T, Ford G, Ford BD. Spatiotemporal pattern of neuronal injury induced by DFP in rats: A model for delayed neuronal cell death following acute OP intoxication. Toxicology and Applied Pharmacology. 2011;253:261–269. doi: 10.1016/j.taap.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough JH, Jr, McLeod CG, Jr, Nipwoda MT. Direct microinjection of soman or VX into the amygdala produces repetitive limbic convulsions and neuropathology. Brain Res. 1987;435:123–137. doi: 10.1016/0006-8993(87)91593-9. [DOI] [PubMed] [Google Scholar]
- McLeod CG, Jr, Singer AW, Harrington DG. Acute neuropathology in soman poisoned rats. Neurotoxicology. 1984;5:53–57. [PubMed] [Google Scholar]
- Newmark J. The birth of nerve agent warfare: lessons from Syed Abbas Foroutan. Neurology. 2004;62:1590–1596. doi: 10.1212/01.wnl.0000124519.85516.50. [DOI] [PubMed] [Google Scholar]
- Okudera H. Clinical features on nerve gas terrorism in Matsumoto. J Clin Neurosci. 2002;9:17–21. doi: 10.1054/jocn.2001.1020. [DOI] [PubMed] [Google Scholar]
- Okumura T, Hisaoka T, Yamada A, Naito T, Isonuma H, Okumura S, Miura K, Sakurada M, Maekawa H, Ishimatsu S, Takasu N, Suzuki K. The Tokyo subway sarin attack--lessons learned. Toxicol Appl Pharmacol. 2005;207:471–476. doi: 10.1016/j.taap.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Petras JM. Neurology and neuropathology of Soman-induced brain injury: an overview. J Exp Anal Behav. 1994;61:319–329. doi: 10.1901/jeab.1994.61-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosler TW, Depboylu C, Arias-Carrion O, Wozny W, Carlsson T, Hollerhage M, Oertel WH, Schrattenholz A, Hoglinger GU. Biodistribution and brain permeability of the extracellular domain of neuregulin-1-beta1. Neuropharmacology. 2011;61:1413–1418. doi: 10.1016/j.neuropharm.2011.08.033. [DOI] [PubMed] [Google Scholar]
- Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, Giulivi C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429:545–552. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Chang E, Igarashi T, Noble LJ. Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 2001;917:45–54. doi: 10.1016/s0006-8993(01)02905-5. [DOI] [PubMed] [Google Scholar]
- Shih T, Whalley CE, Valdes JJ. A comparison of cholinergic effects of HI-6 and pralidoxime-2-chloride (2-PAM) in soman poisoning. Toxicol Lett. 1991;55:131–147. doi: 10.1016/0378-4274(91)90128-s. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Chiang MF, Yang HI, Thajeb P, Li H. Neuregulin-1 reduces ischemia-induced brain damage in rats. Neurobiol Aging. 2004;25:935–944. doi: 10.1016/j.neurobiolaging.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Soltaninejad K, Abdollahi M. Current opinion on the science of organophosphate pesticides and toxic stress: a systematic review. Med Sci Monit. 2009;15:RA75–90. [PubMed] [Google Scholar]
- Talmage DA, Role LW. Multiple personalities of neuregulin gene family members. J Comp Neurol. 2004;472:134–139. doi: 10.1002/cne.20091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal-Cevik I, Kilinc M, Gursoy-Ozdemir Y, Gurer G, Dalkara T. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 2004;1015:169–174. doi: 10.1016/j.brainres.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jiang J, Ford G, Ford BD. Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem Biophys Res Commun. 2004;322:440–446. doi: 10.1016/j.bbrc.2004.07.149. [DOI] [PubMed] [Google Scholar]
- Xu Z, Croslan DR, Harris AE, Ford GD, Ford BD. Extended therapeutic window and functional recovery after intraarterial administration of neuregulin-1 after focal ischemic stroke. J Cereb Blood Flow Metab. 2006;26:527–535. doi: 10.1038/sj.jcbfm.9600212. [DOI] [PubMed] [Google Scholar]
- Xu Z, Ford GD, Croslan DR, Jiang J, Gates A, Allen R, Ford BD. Neuroprotection by neuregulin-1 following focal stroke is associated with the attenuation of ischemia-induced pro-inflammatory and stress gene expression. Neurobiol Dis. 2005;19:461–470. doi: 10.1016/j.nbd.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Yanagisawa N, Morita H, Nakajima T. Sarin experiences in Japan: acute toxicity and long-term effects. J Neurol Sci. 2006;249:76–85. doi: 10.1016/j.jns.2006.06.007. [DOI] [PubMed] [Google Scholar]