

Abstract
Ichthyoses, including inherited disorders of lipid metabolism, display a permeability barrier abnormality in which the severity of the clinical phenotype parallels the prominence of the barrier defect. The pathogenesis of the cutaneous phenotype represents the consequences of the mutation for epidermal function, coupled with a “best attempt” by affected epidermis to generate a competent barrier in a terrestrial environment. A compromised barrier in normal epidermis triggers a vigorous set of metabolic responses that rapidly normalizes function, but ichthyotic epidermis, which is inherently compromised, only partially succeeds in this effort. Unraveling mechanisms that account for barrier dysfunction in the ichthyoses has identified multiple, subcellular, and biochemical processes that contribute to the clinical phenotype. Current treatment of the ichthyoses remains largely symptomatic: directed toward reducing scale or corrective gene therapy. Reducing scale is often minimally effective. Gene therapy is impeded by multiple pitfalls, including difficulties in transcutaneous drug delivery, high costs, and discomfort of injections. We have begun to use information about disease pathogenesis to identify novel, pathogenesis-based therapeutic strategies for the ichthyoses. The clinical phenotype often reflects not only a deficiency of pathway end product due to reduced-function mutations in key synthetic enzymes but often also accumulation of proximal, potentially toxic metabolites. As a result, depending upon the identified pathomechanism(s) for each disorder, the accompanying ichthyosis can be treated by topical provision of pathway product (eg, cholesterol), with or without a proximal enzyme inhibitor (eg, simvastatin), to block metabolite production. Among the disorders of distal cholesterol metabolism, the cutaneous phenotype in Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects (CHILD syndrome) and X-linked ichthyosis reflect metabolite accumulation and deficiency of pathway product (ie, cholesterol). We validated this therapeutic approach in two CHILD syndrome patients who failed to improve with topical cholesterol alone, but cleared with dual treatment with cholesterol plus lovastatin. In theory, the ichthyoses in other inherited lipid metabolic disorders could be treated analogously. This pathogenesis (pathway)-driven approach possesses several inherent advantages: (1) it is mechanism-specific for each disorder; (2) it is inherently safe, because natural lipids and/or approved drugs often are utilized; and (3) it should be inexpensive, and therefore it could be used widely in the developing world.
Introduction
Permeability barrier dysfunction as the “driver” of disease expression
Regardless of the underlying genetic abnormality, all of the ichthyoses studied to date have demonstrated a permeability barrier abnormality.1–5 Because permeability barrier requirements generally “drive” metabolic responses in the underlying epidermis, the clinical phenotypes in the ichthyoses almost certainly reflect a “best effort” attempt by the epidermis to normalize permeability barrier function.2 Notably, these metabolic responses to a compromised barrier, although only partially successful, nevertheless suffice to allow survival in a dry, terrestrial environment. Even in Harlequin ichthyosis, where few if any lipids are delivered to the stratum corneum (SC) interstices,6–8 the epidermis compensates with an intense, hyperplastic response noted by increased cell proliferation in response to a highly defective barrier that generates multiple layers of corneocytes (a “make more cells” imperative).2 In inherited disorders that affect the structural proteins of the corneocyte “bricks,” permeability barrier abnormalities result from downstream alterations in the extracellular matrix, albeit by divergent mechanisms; for example, transglutaminase 1 (TGM1)-negative lamellar ichthyosis and loricrin keratoderma represent disorders in which the key cross-linking enzyme and its principal substrate (loricrin), that form the corneocyte envelope, are affected (Figure 1). In both of these disorders, the corneocyte envelope is attenuated, which results in a defective corneocyte scaffold that leads in turn to fragmented and foreshortened lamellar membranes.9,10 These altered membranes result in an impaired barrier, with leakage of water via the extracellular pathway, as can be demonstrated with lanthanum, a water-soluble tracer that reflects the movement of water through the SC. The link between a defective corneocyte envelope and the paracellular route of increased transepidermal water loss (TEWL) in lamellar ichthyosis and loricrin keratoderma provides definitive proof that the corneocyte provides a scaffold necessary for the supramolecular organization of the lipid-enriched extracellular matrix.
Fig. 1.

Scaffold abnormalities account for barrier abnormality in lamellar ichthyosis and loricrin keratoderma. Milder clinical phenotype in loricrin keratoderma correlates with compensatory cross-linking of other cornified envelope (CE) precursors. In contrast, CE is absent or defective throughout stratum corneum (SC) in transglutaminase 1-negative lamellar ichthyosis, correlating with a more severe phenotype.
A different mechanism operates in epidermolytic ichthyosis (epidermolytic hyperkeratosis), where abnormal keratins (keratin 1 or 10) form dominant-negative keratin pairs that disrupt the cytoskeleton, thereby impeding lamellar body (LB) exocytosis.11 Once again, the barrier abnormality in epidermolytic hyperkeratosis is provoked by a defect in the extracellular matrix, but in epidermolytic hyperkeratosis, reduced secretion of lipids leads to impoverishment of lamellar bilayers.11 A similar cytoskeletal-based pathogenic mechanism appears to occur in filaggrin-deficient ichthyosis vulgaris,12 where mutations block the processing of profilaggrin into filaggrin, and unprocessed profilaggrin again appears to impede secretion of lamellar bodies. In inherited disorders of corneocyte proteins of diverse etiology, the protein abnormality ultimately provokes a defect in the extracellular lamellar membranes (“mortar”).4,9–11 This secondary defect in the extracellular matrix then allows accelerated, extracellular transcutaneous water movement (ie, the permeability barrier abnormality), which drives epidermal hyperplasia and results in a thickened (ichthyotic) SC.
Approach to assess pathogenesis
To elucidate the subcellular and biochemical mechanisms that account for the barrier abnormality, which in turn drives the cutaneous phenotype in many of the inherited disorders of ichthyoses, we have used a pathogenesis algorithm in patients and animal models (Figure 2), with sequential use of ultra-structural, cellular biologic, and biochemical assays to assess the basis for abnormal barrier function.4,13,14 The pathogenic algorithm that we apply to all of the ichthyoses begins with one key, functional end point: changes in permeability barrier homeostasis. To date, abnormal barrier function has been found in all of these disorders, regardless of mutation type, and we would not proceed with follow-up studies.
Fig. 2.

Assessment of disease pathogenesis in patients and animal models: algorithm and approach. ABCA12, aSMase, acid sphingomyelinase; ATP-binding cassette sub-family A member 12; β-GlcCer’ase, β-glucocerebrosidase; Cer, ceramide; EM, electron microscope; FA, fatty acid; GLC, gas liquid chromatography; shRNA, short hairpin RNA; TLC, thin layer chromatography.
Assuming that barrier function is abnormal, we next ascertain whether the barrier defect is due to corneocyte fragility or to defects in the SC extracellular matrix (always the case to date in both lipid metabolic disorders, and with mutations that alter corneocyte proteins). We then determine whether alterations in LB secretion, or in the quantities, organization, and/or maturation of the lamellar bilayers is/are abnormal. These results in turn dictate further biochemical and zymographic studies, as summarized in Figure 2.
Deployment of this algorithm can provide the cellular and biochemical basis for the cutaneous phenotype in each of the inherited ichthyoses, which in turn should dictate potential, pathogenesis-based therapeutic approaches for these disorders. To date, the permeability barrier abnormality (and phenotype) in all of the ichthyoses can be attributed to abnormalities in the supramolecular organization, synthesis, and/or secretion of the extracellular lamellar bi-layers.14 Notably, in all of the lipid-metabolic disorders that we have studied to date, metabolite accumulation or pathway product deficiency, or both, alter(s) lipid content and distribution, thereby disrupting the architecture of SC lamellar membranes13 (Table 1), which in turn provokes a barrier abnormality (and phenotype).
Table 1.
Diagnostic ultrastructural features in the ichthyoses (modified from13)
| Feature | KHG/keratins | LB formation/contents | LB exocytosis | Lipid processing | Lamellar bilayers | Cornified envelopes | CD | CLE |
|---|---|---|---|---|---|---|---|---|
| Lipid metabolic | ||||||||
| ARCI (Ichthyin) | Normal/normal | Decreased | Decreased | N/A | N/A | N/A | Normal | N/A |
| ARCI (ABCA12) | Decreased/normal | ↓Contents | Normal | N/A | Largely absent | Normal | Persist | Normal |
| NLSDI | Normal/normal | Abnormal contents | Normal | Normal | L/non-L-PS | Normal | Normal | Abnormal |
| SLS | Normal/normal | Cytolysis; abnormal contents | Abnormal | Delayed | L/non-L-PS | Normal | Normal | Normal |
| Refsum Disease | Normal/normal | Abnormal shape & contents | Abnormal | Delayed | L/non-L-PS | Normal | Normal | Absent |
| CHH/CHILD | Normal/normal | Abnormal contents | Impaired | Delayed | L/non-L-PS | Normal | Normal | Normal |
| Gaucher Type II | Normal/normal | Normal | Normal | Impaired→ Absent | L/non-L-PS | Normal | Normal | Normal |
| RXLI | Normal/normal | Normal | Normal | Normal | L/non-L-PS | Normal | Persist | Normal |
| Lipid transporters | ||||||||
| HI | Abnormal/normal | Empty | N/A | N/A | Absent | Normal | Persist | |
| CEDNIK | ? | Empty | Impaired | NA | N/A | N/A | N/A | N/A |
| IPS | Normal/normal | Abnormal contents | Normal | Normal | L/non-L-PS | Normal | Normal | Normal |
| Structural proteins | ||||||||
| EI | Normal/abnormal | Normal | Impaired | Delayed | Decreased/fragmented | Persist | Persist | Normal |
| LI (TGM1) | Normal/normal | Normal | Normal | Normal | Fragmented | Absent/attenuated | Normal | Normal |
| LK | Normal/normal | Normal | Normal | Normal | Fragmented | Attenuated-lower SC | Normal | Normal |
| IV | Reduced/normal | Normal | Impaired | Impaired | Decreased, L/non-L-PS | Normal | Persist | ?Abnormal |
| Accelerated desquamation | ||||||||
| NS | Normal/abnormal | Normal | Accelerated | Impaired | Reduced/fragmented | Normal | Degraded | Normal |
| PSS, Type II | Normal | Normal | Normal | Normal | Normal | Normal | Degraded | Normal |
| Other | ||||||||
| En Confettis | Abnormal/abnormal | Normal | Abnormal | Impaired | Decreased | Absent | Absent | Normal |
Bolded features are particularly helpful in differential diagnosis.
ARCI, autosomal recessive congenital ichthyosis; CD, corneodesmosome; CEDNIK, cerebral dysgenesis–neuropathy–ichthyosis–keratoderma; CHH, Conradi-Hünermann-Happle syndrome; CHILD, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; CLE, corneocyte lipid envelope; EI, epidermolytic ichthyosis; HI, Harlequin ichthyosis; IPS, ichthyosis prematurity syndrome; IV, ichthyosis vulgaris; KHG keratohyaline granules; L/non-L-PS, lamellar/nonlamellar phase separation; LB, lamellar bodies; LI, lamellar ichthyosis; LK, loricrin keratoderma; NLSDI, neutral lipid storage disease with ichthyosis; N/A, not assessed or not applicable; NS, Netherton syndrome; PSS, Peeling skin syndrome; RD, Refsum disease; SC, stratum corneum; SLOS, Smith-Lemli-Opitz syndrome; SLS, Sjögren-Larsson syndrome; SP, serine protease; TGM1, transglutaminase 1; XLI, X-linked ichthyosis.
Pathogenesis of multisystem, cholesterol biosynthetic disorders
Postlanosterol synthesis of cholesterol requires multiple enzymatic reactions, including reduction of Δ7, Δ14, and Δ24 double bonds; removal of methyl groups at positions C4α, C4β, and C14 that generate cholesterol (C27) from lanosterol (a C30 sterol), as well as isomerization of the Δ8(9) double bond to Δ7(8); followed by desaturation at the Δ5 position to generate cholesterol. A subsequent 3-β-sulfation step generates cholesterol sulfate (CSO4) from cholesterol (Figure 3). An abnormal cutaneous phenotype has been described in seven of these eight syndromic disorders (lathosterolosis, congenital hemidysplasia with ichthyosiform erythroderma and limb defects [CHILD] syndrome, Conradi-Hünermann-Happle syndrome [CHH] or X-linked dominant chondrodysplasia punctata type 2 [CDPX2], sterol-C4-methyl oxidase like [SC4MOL] deficiency, and XLI) but less prominently in desmosterolosis and Smith-Lemli-Opitz syndrome (SLOS).15–23
Fig. 3.

Enzymatic stages in distal cholesterol metabolites and their associated clinical disorders. Syndromic disorders occur with mutations in 8 of the 10 post-lanosterol steps in cholesterol synthesis (bold), and ichthyosis occurs in 7 of these diseases (bold). *Neonatal lethal. CHILD, congenital hemidysplasia with ichthyosiform erythroderma and limb defects. (Adapted with permission from Elias PM, Crumrine D, Paller A, Rodriguez-Martin M, Williams ML. Pathogenesis of the cutaneous phenotype in inherited disorders of cholesterol metabolism: therapeutic implications for topical treatment of these disorders. Dermatoendocrinol 2011;3:100-6.)
In epidermis, cholesterol is one of the three key SC lipids, along with ceramides and free fatty acids (FFA), that form the extracellular lamellar bilayer system that mediates barrier function.24 The pathogenesis of the ichthyosis in the inborn errors of distal cholesterol metabolism to date can be attributed largely to deficiency of cholesterol in cell membranes (cholesterol is required for normal cell membrane function), coupled with accumulation of toxic sterol precursors, either or both of which can provoke structural alterations25–27 (Table 2). The formation of sterol metabolites that (1) downregulate cholesterol synthesis,28 (2) alter hedgehog pathway signaling (hedgehog normally is tethered to cell membranes by cholesterol),29,30 and/or (3) accelerated degradation of 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase by sterol metabolites,31 could also contribute to disease pathogenesis. Finally, in some cases, enzyme blockade could result in vitamin D deficiency if not compensated for by diet. Because 1,25(OH)2 vitamin D3 is a potent regulator of epidermal differentiation and proliferation, deficiency could have undesirable consequences.
Table 2.
Proposed pathogenesis-based therapy for inherited disorders of distal cholesterol metabolism
| Metabolic category | Incidence | Inheritance pattern | Affected protein (gene) | Normal function | Likely efficacy of pathway-based treatment | Proposed therapy |
|---|---|---|---|---|---|---|
| CHH (CDPX2) | Rare | X-linked dominant | Δ (8)-Δ (7) sterol isomerase emopamil-binding protein (EBP) | Distal cholesterol synthesis | Very likely (but often self-resolving) | Cholesterol ± HMG-CoA-R inhibitor |
| CHILD syndrome | Very rare | X-linked dominant | NAD(P)H steroid dehydrogenase-like protein (NSDHL) | Same | Yes (shown) | Same as CHH |
| SLOS | Fairly common | Recessive | 7-dehydroreductase (DHCR7) | Same | Very likely | Same as CHH |
| SC4MOL | Very rare | Recessive | Sterol-C4-methyl oxidase (SC4MOL) | Same | Very likely | Same as CHH |
| Lathosterolosis | Very rare | Recessive | Lathosterol-5-desaturase (Sc5d) | Same | Very likely | Same as CHH |
| Desmosterolosis | Very rare | Recessive | 24-dehydroreductase (DHCR24) | Same | Very likely | Same as CHH |
| XLI | 1:2,000–6,000 males | X-linked recessive | Steroid sulfatase (STS) | Same | Very likely | Sult2b inhibitor or cholesterol + HMG-CoAR inhibitor |
CDPX2, X-linked chondrodysplasia punctata type 2; CHH, Conradi-Hünermann-Happle syndrome; CHILD, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; HMGCoAr, 3-hydroxy-3-methylglutaryl coenzyme A reductase; SLOS, Smith-Lemli-Opitz syndrome; Sult2b, cholesterol sulfotransferase; XLI, X-linked ichthyosis.
CCH (CDPX2) and CHILD syndromes
CCH (CDPX2; OMIM #302960) is caused by reduced-function mutations in the gene expressing emopamil-binding protein (EBP), which catalyzes the conversion of 8(9)-cholestenol to lathosterol and causes ichthyosis in a mosaic pattern in affected females that can spontaneously resolve.32–35 In the X-linked dominant CHILD syndrome (OMIM #308050), reduced-function mutations in NSDHL, and occasionally EBP15,35 result in a failure to remove the C-4 methyl group from lanosterol (Figure 3). The skin in CHILD syndrome exhibits a unique cutaneous phenotype, consisting of circumscribed linear plaques surmounted by prominent wax like scales in a strictly unilateral distribution.30 Involved skin sites in both disorders conform to regions in which the mutant X-chromosome predominates.36,37
Although the density of LBs and LB secretion are normal in CHH, organelle contents display abnormal vesicular inclusions that fail to disburse normally at the stratum granulosum–corneum interface. As a result, maturation of lamellar bilayers is delayed, and normal lamellar bilayers are displaced by extensive areas of lamellar/nonlamellar phase separation.38 The ultrastructure of clinically affected skin in CHILD syndrome is much more abnormal than in CHH. The LBs form normally but display almost no internal lamellae, and most fuse into intracellular multivesicular bodies that are not secreted. As a result, the SC extracellular matrix is filled with interspersed lamellar and nonlamellar material.
SLOS, desmosterolosis, lathosterolosis, and SC4MOL deficiency
Although ichthyosis is not clinically apparent in SLOS (7-dehydrocholesterol reductase [DHCR7] deficiency; OMIM #270400), ultraviolet B-mediated photosensitivity39,40 and a propensity to develop eczema (Dr Rosalind Elias and Dr R. Steiner, personal communication) occur commonly. SLOS is a fairly common Mendelian trait (predicted incidence of ~1:6-10,000 conceptions), with more than 120 different reduced-function mutations in DHCR7 identified to date.40 DHCR7 deficiency impairs desmosterol (8-DHC) and 7-dehydrocholesterol (7-DHC) metabolism,24,28,41 resulting in elevated 7-DHC and 8-DHC blood levels, with proportionate reductions in serum cholesterol.15–19,34,40,42,43 These features are mimicked in Dhcr7−/− and Dhcr7+/− mice44 and in mice with a knock-in of the human T93M DHCR7 mutation.45
Although lathosterolosis (OMIM #607330) has not been described in the United States, several European patients have been reported, all with prominent ichthyosis.22,46–48 Two US patients have been described with desmosterolosis (OMIM #602398; reduced function mutations in 24-dehydrocholesterol reductase [DHCR24]), who display developmental and neurologic anomalies, but minimal ichthyosis.21,49 We are assessing the basis for the skin phenotype in 2 patients with SC4MOL deficiency, which presents with severe ichthyosis, developmental abnormalities, and psoriasiform features.23
X-linked ichthyosis
The pathogenesis of XLI (OMIM #308100) has been more fully delineated than for any of the other ichthyoses. As a result of steroid sulfatase deficiency in XLI, CSO4 accumulates in the outer epidermis,50–52 erythrocyte cell membranes,52,53 and lipoprotein fractions of plasma.53 Although CSO4 levels normally comprise approximately 1% of lipid mass in SC,54,55 CSO4 contents reach 10% to 12% in XLI.56
The prominence of epidermal vs other organ involvement in XLI reflects higher CSO4 levels in epidermis than in blood.52,53,56 Hydrolysis of CSO4 normally generates some of the cholesterol required for the barrier, but CSO4 also is a potent inhibitor of HMG-CoA reductase, further reducing cholesterol levels in XLI.56 Accumulation of CSO4 coupled with cholesterol deficiency, disrupts lamellar membrane architecture, together accounting for the barrier abnormality in XLI57,58 (Figure 4).
Fig. 4.

Pathogenesis of x-linked ichthyosis. HMG CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A. (Adapted with permission from Lai-Cheong JE, Elias PM, Paller AM. Pathogenesis-based therapies in ichthyosis. Dermatol Ther 2012; in press.)
Kinetic studies have demonstrated that the hyperkeratosis in XLI reflects delayed desquamation.59 The basis for this classic, retention-type of ichthyosis is persistence of corneodesmosomes at all levels of the SC (Figure 4). Two key serine proteases, kallikreins 7 (SC chymotryptic enzyme) and kallikrein 5 (SC tryptic enzyme), are primary mediators of corneodesmosomes degradation in vitro.60 CSO4 appears to increase SC retention through the known ability of this lipid to function as a serine protease inhibitor.57,61,62 Although the acidic pH of SC inhibits the activities of SC chymotryptic enzyme (kallikrein 7) and SC tryptic enzyme (kallikrein 5),63–65 the pH of the SC in XLI is even more acidic than normal.66 Hence, serine protease activity is reduced dramatically in XLI.57
The SC in XLI demonstrates abundant Ca++ in extracellular domains, which preferentially localizes along the external faces of opposing corneodesmosomes.57 The delayed degradation of corneodesmosomes in XLI could be partly due to leakage of Ca++ into the lower SC (due to the barrier defect), with formation of Ca++ bridges between adjacent corneodes-mosomes.57 Ca++, if present in sufficient quantities, can stabilize highly anionic SO4 groups (from persistent cholesterol sulfate) on lamellar bilayers.67 Indeed, CSO4-containing liposomes aggregate avidly in the presence of Ca++.68,69
Brief summary of pathogenesis and proposed therapy for other lipid metabolic disorders
Pathogenesis data are available for the following additional inherited disorders of lipid metabolism (Table 3):
Table 3.
Proposed pathogenesis-based therapy for other inherited disorders of lipid metabolism
| Metabolic category | Incidence | Inheritance pattern | Affected protein (gene) | Normal function | Likely efficacy of pathway-based treatment | Proposed therapy |
|---|---|---|---|---|---|---|
| Gaucher disease, type II | Uncommon | Recessive | Deglycosylates glucosylceramide | Generation of Cer, a key barrier lipid | Very likely | Cer + GC synthase inhibitor |
| Sjögren-Larsson syndrome | Rare | Recessive | Fatty aldehyde dehydrogenase (ALDH3A2) | Oxidation of aldehydes to FFA/isoprenoids | Very likely | ACC or FAS inhibitor + FFA or farnesoeate |
| Harlequin ichthyosis (HI) | Rare | Recessive | ATP-binding cassette (ABCA12, loss-of-function) | Transports GlcCer into LB | Possible | GlcCer alone, or in triple lipid mix |
| ARCI (Lamellar ichthyosis phenotype) | Rare | Recessive | ABCA12, missense | Same as HI | Very likely | GlcCer +/or PPAR-β/δ or LXR activator |
| Ichthyosis prematurity syndrome | Rare | Recessive | Fatty acid transport protein 4 (FATP4) | Imports and CoA esterifies FFA in keratinocytes | Possible | Appropriate FFA-CoA |
| Refsum disease | Rare | Recessive | Phytanoyl CoA hydroxylase (PAHX, PHYH) | Oxidates plant-derived branched FA | Likely | FFAs + ACC or FAS inhibitors (+ diet) |
| Neutral lipid storage disease | Rare | Recessive | CGI-58 acid lipase (ABHD5) | Generates DAG & FFA from TAG | Likely | Topical acylCer |
| ARCI (LOX mutations) | (rare) | Recessive | eLOX3 & 12RLOX | Oxygenates-ωC18 in acylCer | Very likely | Topical acylCer or ω-OH-Cer |
ACC, acetyl coenzyme A carboxylase; ARCI, autosomal recessive congenital ichthyosis; Cer, ceramide; CoA, coenzyme A; DAG, diacylglycerol; FAS, fatty acid synthase; FFA, free fatty acid; GC, glucocorticoid; GlcCer, glucosylceramide; LB, lamellar body, LXR, liver X receptor; PPAR, peroxisome proliferator-activated receptor; TAG, triacylglycerol.
Sjögren-Larsson syndrome [SLS], a disorder of FA metabolism, displays ichthyosis and severe neurologic features. Our ongoing studies suggest that accumulation of cytotoxic metabolites, coupled with FFA deficiency, produce ichthyosis in SLS.70 This predicts SLS could be treatable with topical FFAs and/or farnesoic acid, plus a topical acetyl-CoA carboxylase (ACC) or fatty acid synthase (FAS) inhibitor.
Neutral lipid storage disease with ichthyosis (NLSDI), a disorder of triacylglycerol and sphingolipid metabolism, in which ichthyosis and mild neurologic abnormalities both occur. Although triacylglycerols accumulate in NLSDI, we recently showed that acyl ceramide deficiency is responsible for pathogenesis of the cutaneous disease,71 suggesting that the ichthyosis should respond to replacement with topical acyl ceramide.
Refsum disease a disorder of FA metabolism, that displays ichthyosis and severe neurologil abnormalities. The corneocyte lipid envelope is absent in Refsum disease, suggesting that plant-derived, branched fatty acids cannot be utilized for acyl ceramide production72 or that the resultant acyl ceramide cannot be used as a substrate for the de-esterifying enzyme that generates ω-hydroxy-ceramide (ω-OH-ceramide) for corneocyte lipid envelope formation (Figure 5; see also arachidonate lipoxygenases [ALOX] mutations below). For these reasons, the cutaneous features of Refsum disease could be theoretically treatable by FFA or acyl ceramide with or without a topical inhibitor of diacylglycerol/triacylglycerol synthesis, although treatment of the neurologic and the systemic features would require dietary intervention.72,73
Some patients with autosomal recessive congenital ichthyosis (ARCI) have ATP-binding cassette subfamily A member 12 (ABCA12) missense mutations, as occur in Harlequin ichthyosis (see below). Glucosylceramides transport into nascent LB likely is reduced, but not absent.6,74 Topical, cell-permeant ceramide, along with a topical liver X receptor or peroxisome proliferator-activated receptor (PPAR) β/δ activator should thus benefit ARCI patients with residual ABCA12 expression because our recent studies have shown that both exogenous ceramide and liver X receptor/PPAR-β/δ activators upregulate ABCA12 expression.75 As noted above, ABCA12 is absent in Harlequin ichthyosis a nonsyndromic, but life-threatening recessive disorder of cornification,76 which might be treatable with topical glucosylceramide.
In neuronopathic (type II) Gaucher disease, which is due to loss-of-function mutations in β-glucocerebrosidase), we showed that the cutaneous phenotype reflects accumulation of glucosylceramide and decreased production of ceramide. The cutaneous phenotype, and perhaps the neurologic features of Gaucher disease type II patients, might be treatable with a glutamylcysteine synthase inhibitor and topical ceramide, although enzyme replacement is the current standard of therapy.
A defect in FFA transport due to loss of FATP4 function provokes ichthyosis prematurity syndrome.77 We are characterizing the pathogenesis of ichthyosis prematurity syndrome in a cohort of Norwegian patients, and in partially, rescued Fatp4 −/− mice that mirror the extent of reduced transporter function in ichthyosis prematurity syndrome. Specifically, we have shown that although FFAs are imported into keratinocytes, these FFAs fail to be acylated by CoA, a shared function of all FATPs.73 Strategies that upregulate CoA synthase, in theory, could be deployed as topical corrective therapy in ichthyosis prematurity syndrome.
In ARCI due to epidermal lipoxygenase-3 (eLOX3) or 12R-lipoxygenase (12RLOX) mutations, we recently found that a failure to oxygenate the ω-acyl linoleic acid in acyl ceramide results in a failure of ω-OH-ceramide to be de-esterified and covalently bound to the external face of the cornified envelope78 (Figure 5). It should be possible to treat these patients by provision of a topical ω-OH-ceramide.
Fig. 5.
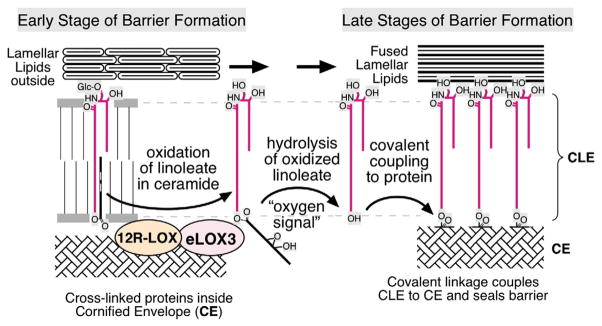
Metabolic steps leading to the formation of corneocyte lipid envelope (CLE). The two arachidonate lipoxygenases (ALOX) enzymes sequentially oxygenate the linoleate moiety in acyl ceramides, which then allows an as-yet-unidentified lipase to de-esterify acyl ceramide. The resultant pool of ω-OH-ceramide can then be covalently linked to the outer surface of the CE, forming the CLE.
Studies to date in relevant animal models
Although several relevant analogues of the diseases of interest are available, little is known about their cutaneous phenotypes.
Mouse models of SLOS
SLOS patients display a mild cutaneous phenotype, butDhcr7−/− mice display prominent ichthyosis, with neonatal lethality due to a putative permeability barrier abnormality, developmental retardation, poor feeding, and neurologic abnormalities.44 More pertinent to SLOS patients, who display residual enzyme function, our preliminary studies in Dhcr7+/− mice, with approximately 50% loss-of-function, display serum 7DHC and cholesterol levels comparable to patients with moderate SLOS.44 These mice demonstrate defective lamellar body contents and lamellar bilayer organization (Figure 6), predictive of a barrier abnormality in SLOS. The most common SLOS mutation (T93M) has been recapitulated in a transgenic knock-in model,45,79 but its skin phenotype has not yet been assessed. Studies in both of these mice could further delineate the basis for the clinical phenotype as well as the role of metabolite accumulation vs cholesterol depletion in SLOS.
Fig. 6.

(A) Abnormal lamellar bodies (arrows) and (B) lamellar bilayer architecture (arrows) in Dhcr7+/− mice (Mag bars = 0.2 μm).
Mouse models of desmosterolosis and lathosterolosis
Although Dhcr24−/− mice are neonatal lethal due to a failure of epidermal development in utero and a lethal postnatal barrier defect.80,81 Dhcr24+/− mice (a reasonable model of desmosterolosis) survive and show ultrastructural evidence of a barrier abnormality, similar to Dhcr7 +/− mice (Elias, et al., unpublished observations). Finally, we are assessing the cutaneous features in transgenic Sc5d +/− mice that display residual enzyme expression, comparable to humans with lathosterolosis.
Related work in insulin-induced gene 1 (Insig-1) and -2 (Epi-insig) double knockout mice
The Brown and Goldstein group recently described an animal model with aberrant cholesterol synthesis (epidermal-localized deficiency in insulin-induced gene 1 [Insig-1] with germ-line deletion of Insig-2 [Insig 1/2] double knockout mice),82 a model that mimics ichthyosis follicularis with alopecia and photophobia (IFAP) syndrome. These intracellular proteins normally restrict sterol regulatory element-binding proteins (SREBPs) to the endoplasmic reticulum. In their absence, SREBPs migrate to the Golgi apparatus, where proteases cleave the 125-kD SREBPs to mature 65-kD proteins that migrate to the nucleus in an unrestricted fashion, resulting in the continuous stimulation of several genes involved in cholesterol synthesis. Although this model differs fundamentally from the inherited disorders of distal cholesterol metabolism, where cholesterol synthesis instead is impeded, their cutaneous phenotype responded to topical simvastatin, which lowers sterol metabolites and epidermal cholesterol levels.82
Animal models of other lipid metabolic disorders
We also are assessing the basis of the cutaneous phenotype in several other lipid metabolic disorders. Most thoroughly characterized are the transgenic β-glucocerebrosidase-deficient (Gaucher) mice, where the severe, cutaneous phenotype in −/− mice is neonatal lethal, but mice that survive display a prominent ichthyosis when enzyme levels are reduced (<10% of normal) but not absent due to both accumulation of glucosylceramide and ceramide deficiency.83–85 We also are assessing a transgenic mouse model of ichthyosis prematurity syndrome (IPS), which is due to reduced-function mutations in FATP4. Although −/− mice are neonatal lethal, they can be partially rescued by epidermal targeting of Fatp4, which restores epidermal transporter activity in a mosaic pattern.86 As noted, we have identified that disease pathogenesis reflects failure of acyl CoA synthase activity, leading to FFA accumulation and detergentlike toxicity.73
Patients with neutral lipid storage disease (NLSDI)87 and cgi-58−/− transgenic mice88 both display a severe barrier abnormality. The CGI-58 mutation leads to a defect in the lipolysis of a pool of triglycerides that provides the linoleic acid required for acyl ceramide synthesis. Because the cutaneous phenotype results from a selective deficiency of N-acyl, ω-esterified ceramides (ie, acyl ceramide), these mice (and patients) could be rescued by provision of linoleic acid or acyl ceramide.89
We also are studying fatty aldehyde dehydrogenase-deficient (faldh−/−, faldh+/−, and wild type) mice with William Rizzo, who first identified the responsible enzyme abnormality for SLS.90 We recently found cytotoxic abnormalities and evidence of FA deficiency in SLS epidermis, suggesting that the pathogenesis of SLS reflects both metabolite accumulation and pathway-product depletion.70 Finally, a subgroup of ARCI patients displays mutations in two epidermis-localized lipoxygenases (ie, eLOX3 or 12RLOX). We are assessing the basis for the cutaneous phenotype in 12RLox−/− mice. A characteristic (diagnostic) ultrastructural feature of this disorder is absence of the corneocyte lipid envelope due to lack of covalently bound ω-OH-ceramide.91 The biochemical basis for the phenotype can now be attributed to the substrate specificity of the ALOX enzymes for the ω-esterified linoleate moiety in acyl ceramide, which normally are de-esterified, generating ω-OH-ceramide, allowing their covalent attachment to the cornified envelope (Figure 5).
Therapeutic interventions to date
In all of the lipid metabolic disorders, blockade of metabolite production alone, even if temporarily useful,82 cannot be used as monotherapy for the cutaneous phenotype because the end products of these pathways (ie, cholesterol, ceramide, and FFA) are all required to prevent development of a permeability barrier abnormality (Figure 7), which in turn inevitably leads to an ichthyotic phenotype.92,93 Topical cholesterol alone was ineffective in two patients with CHILD syndrome (Drs Amy Paller [Northwestern University] and Marina Rodriquez-Martin [Canary Islands University Hospital]—point mutation in G83Dp Gly83 Asp in NSHDL, and nonsense mutation [c.317C>A; p.S106x] in NSHDL, respectively), but both patients responded to cotherapy with cholesterol plus lovastatin.94 Both excessive scale and epidermal hyperplasia diminished greatly by 6 to 8 weeks of treatment94 (Figure 8). Although the primary rationale for provision of the pathway product (cholesterol) is to avoid epidermal dysfunction due to lipid deficiency (Figure 7), coprovision of cholesterol also could be beneficial by further downregulating HMG-CoA reductase activity by negative feedback regulation. Despite knowledge of disease pathogenesis, mechanism-targeted therapies may not always be effective if multiple downstream pathways contribute to disease pathogenesis.
Fig. 7.

Pathogenesis-based therapy for inherited disorders of distal cholesterol metabolism (modified from Paller et al94). HMG CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A. (Adapted with permission from Elias PM, Crumrine D, Paller A, Rodriguez-Martin M, Williams ML. Pathogenesis of the cutaneous phenotype in inherited disorders of cholesterol metabolism: therapeutic implications for topical treatment of these disorders. Dermatoendocrinol 2011;3:100-6.)
Fig. 8.
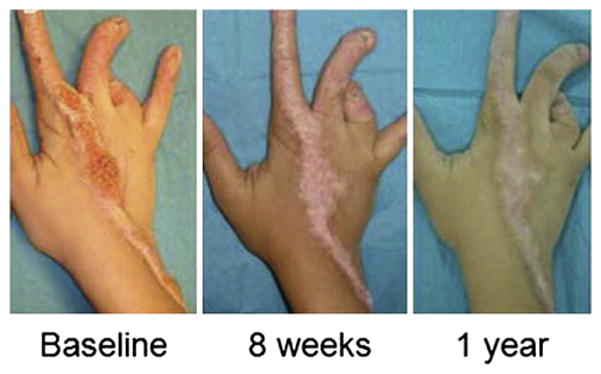
Congenital hemidysplasia with ichthyosiform erythro-derma and limb defects (CHILD) syndrome: Response to topical cholesterol and lovastatin (modified from Paller et al94).
Biomedical significance
Current therapy of the ichthyoses is purely symptomatic, and often irrational (eg, when removal of excess scale interferes with a potentially homeostatic response that allows patients to survive in a harsh, terrestrial environment). At the other extreme, corrective gene therapy, although seductive in concept, remains a distant dream, with high costs and many potential pitfalls. We are identifying cellular and biochemical mechanisms that lead to the cutaneous phenotype in inherited disorders of lipid metabolism. In several disorders of distal cholesterol metabolism, the cutaneous phenotype can range from severe (as in CHILD syndrome, SC4MOL deficiency, and lathosterolosis), to moderately severe (as in CHH/CDPX2 and XLI), or mild to inapparent (as in SLOS and desmosterolosis). Although most of the distal cholesterol disorders are rare, others are quite common (eg, SLOS occurs in about 1 in 6–10,000 conceptions20; XLI in about 1:2000–6000 males).13
New pathogenic insights could be followed by rapid translation into readily deployable, topical therapies, which could be tested initially in disease-appropriate animal models. Identification of effective therapies in the animal models could then provide the approach that would be most likely to help affected patients. Specifically, this approach leverages pathogenesis data into topical therapies that override biochemical abnormalities, potentially initiating an entirely new departure for the therapy of the ichthyoses in the inherited lipid metabolic disorders. The proposed therapies fully exploit the unique accessibility of the skin, allowing rapid validation of this topical approach. In addition, topical lipids and lipid-soluble drugs are often inexpensive and readily delivered across the SC. Because we already have encouraging preliminary evidence in two CHILD patients, we focus our initial proof-of-concept on disorders of distal cholesterol metabolism, where we have deployed dual-therapy with the pathway product, cholesterol (already used topically in personal care products and does not raise serum cholesterol levels), plus a topical, generic statin (simvastatin), which is deployed at much higher dosages than for the treatment of hyperlipidemia. The cutaneous manifestations of most, if not all of the disorders of distal cholesterol metabolism should be treatable safely and effectively by this approach. If successful, the pathogenesis-based approach could initiate a paradigm shift in how inherited ichthyoses will be treated in the future.
Acknowledgments
Joan Wakefield provided superb editorial assistance.
Footnotes
This work was administered by the Northern California Institute for Research and Education, with resources of the Veterans Affairs Medical Center, San Francisco, California.
References
- 1.Williams ML, Coleman RA, Placezk D, Grunfeld C. Neutral lipid storage disease: a possible functional defect in phospholipid-linked triacylglycerol metabolism. Biochim Biophys Acta. 1991;1096:162–9. doi: 10.1016/0925-4439(91)90055-e. [DOI] [PubMed] [Google Scholar]
- 2.Williams ML, Elias PM. From basket weave to barrier. Unifying concepts for the pathogenesis of the disorders of cornification. Arch Dermatol. 1993;129:626–9. doi: 10.1001/archderm.129.5.626. [DOI] [PubMed] [Google Scholar]
- 3.Bouwstra JA, Ponec M. The skin barrier in healthy and diseased state. Biochim Biophys Acta. 2006;1758:2080–95. doi: 10.1016/j.bbamem.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Schmuth M, Gruber R, PME, Williams M. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol. 2007;23:231–56. doi: 10.1016/j.yadr.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams ML, Elias PM. Genetically transmitted, generalized disorders of cornification. The ichthyoses Dermatol Clin. 1987;5:155–78. [PubMed] [Google Scholar]
- 6.Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914–8. doi: 10.1001/archderm.142.7.914. [DOI] [PubMed] [Google Scholar]
- 7.Elias PM, Fartasch M, Crumrine D, Behne M, Uchida Y, Holleran WM. Origin of the corneocyte lipid envelope (CLE): observations in harlequin ichthyosis and cultured human keratinocytes. J Invest Dermatol. 2000;115:765–9. doi: 10.1046/j.1523-1747.2000.00124-5.x. [DOI] [PubMed] [Google Scholar]
- 8.Dale BA, Holbrook KA, Fleckman P, Kimball JR, Brumbaugh S, Sybert VP. Heterogeneity in harlequin ichthyosis, an inborn error of epidermal keratinization: variable morphology and structural protein expression and a defect in lamellar granules. J Invest Dermatol. 1990;94:6–18. doi: 10.1111/1523-1747.ep12873301. [DOI] [PubMed] [Google Scholar]
- 9.Schmuth M, Fluhr JW, Crumrine DC, et al. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis) J Invest Dermatol. 2004;122:909–22. doi: 10.1111/j.0022-202X.2004.22431.x. [DOI] [PubMed] [Google Scholar]
- 10.Elias PM, Schmuth M, Uchida Y, et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11:248–56. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- 11.Schmuth M, Yosipovitch G, Williams ML, et al. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol. 2001;117:837–47. doi: 10.1046/j.0022-202x.2001.01471.x. [DOI] [PubMed] [Google Scholar]
- 12.Gruber R, Elias PM, Crumrine D, et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol. 2011;178:2252–63. doi: 10.1016/j.ajpath.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elias P, Williams M, Crumrine D, Schmuth M. Ichthyoses—clinical, biochemical, pathogenic, and diagnostic assessment. Basel: S. Kargar AG; 2010. [Google Scholar]
- 14.Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 16.Haas D, Kelley RI, Hoffmann GF. Inherited disorders of cholesterol biosynthesis. Neuropediatrics. 2001;32:113–22. doi: 10.1055/s-2001-16618. [DOI] [PubMed] [Google Scholar]
- 17.Moebius FF, Fitzky BU, Glossmann H. Genetic defects in postsqualene cholesterol biosynthesis. Trends Endocrinol Metab. 2000;11:106–14. doi: 10.1016/s1043-2760(00)00235-6. [DOI] [PubMed] [Google Scholar]
- 18.Herman GE. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12(Spec1):R75–88. doi: 10.1093/hmg/ddg072. [DOI] [PubMed] [Google Scholar]
- 19.Porter FD. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr Opin Pediatr. 2003;15:607–13. doi: 10.1097/00008480-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.FitzPatrick DR, Keeling JW, Evans MJ, et al. Clinical phenotype of desmosterolosis. Am J Med Genet. 1998;75:145–52. [PubMed] [Google Scholar]
- 22.Krakowiak PA, Wassif CA, Kratz L, et al. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum Mol Genet. 2003;12:1631–41. doi: 10.1093/hmg/ddg172. [DOI] [PubMed] [Google Scholar]
- 23.He M, Kratz L, Michel LJ, et al. Mutations in the SC4MOL gene encoding a novel methyl sterol oxidase cause psoriasiform dermatitis, microcephaly and developmental delay. Nat Precedings. 2008 doi: 10.1172/JCI42650. Available at: http://hdl.handle.net/10101/npre.2008.2163.1. [DOI] [PMC free article] [PubMed]
- 24.Feingold KR. The regulation and role of epidermal lipid synthesis. Adv Lipid Res. 1991;24:57–82. doi: 10.1016/b978-0-12-024924-4.50007-9. [DOI] [PubMed] [Google Scholar]
- 25.Singh P, Saxena R, Paila YD, Jafurulla M, Chattopadhyay A. Differential effects of cholesterol and desmosterol on the ligand binding function of the hippocampal serotonin(1A) receptor: implications in desmosterolosis. Biochim Biophys Acta. 2009;1788:2169–73. doi: 10.1016/j.bbamem.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez-Acebes S, de la Cueva P, Fernandez-Hernando C, et al. Desmosterol can replace cholesterol in sustaining cell proliferation and regulating the SREBP pathway in a sterol-Delta24-reductase-deficient cell line. Biochem J. 2009;420:305–15. doi: 10.1042/BJ20081909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabatini K, Mattila JP, Kinnunen PK. Interfacial behavior of cholesterol, ergosterol, and lanosterol in mixtures with DPPC and DMPC. Biophys J. 2008;95:2340–55. doi: 10.1529/biophysj.108.132076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjorkhem I. Are side-chain oxidized oxysterols regulators also in vivo? J Lipid Res. 2009;50(suppl):S213–8. doi: 10.1194/jlr.R800025-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper MK, Wassif CA, Krakowiak PA, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–13. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 30.Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl. 2006;95:16–23. doi: 10.1111/j.1651-2227.2006.tb02384.x. [DOI] [PubMed] [Google Scholar]
- 31.DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609–21. doi: 10.1038/cr.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derry JM, Gormally E, Means GD, et al. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–90. doi: 10.1038/10350. [DOI] [PubMed] [Google Scholar]
- 33.Braverman N, Lin P, Moebius FF, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999;22:291–4. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 34.Ikegawa S, Ohashi H, Ogata T, et al. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet. 2000;94:300–5. doi: 10.1002/1096-8628(20001002)94:4<300::aid-ajmg7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–35. doi: 10.1002/(sici)1096-8628(20000214)90:4<328::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 36.Happle R. Lyonization and the lines of Blaschko. Hum Genet. 1985;70:200–6. doi: 10.1007/BF00273442. [DOI] [PubMed] [Google Scholar]
- 37.Happle R. X-linked dominant chondrodysplasia punctata. Review of literature and report of a case. Hum Genet. 1979;53:65–73. doi: 10.1007/BF00289453. [DOI] [PubMed] [Google Scholar]
- 38.Emami S, Hanley KP, Esterly NB, Daniallinia N, Williams ML. X-linked dominant ichthyosis with peroxisomal deficiency. An ultrastructural and ultracytochemical study of the Conradi-Hunermann syndrome and its murine homologue, the bare patches mouse. Arch Dermatol. 1994;130:325–36. doi: 10.1001/archderm.130.3.325. [DOI] [PubMed] [Google Scholar]
- 39.Anstey AV, Ryan A, Rhodes LE, et al. Characterization of photosensitivity in the Smith-Lemli-Opitz syndrome: a new congenital photosensitivity syndrome. Br J Dermatol. 1999;141:406–14. doi: 10.1046/j.1365-2133.1999.03032.x. [DOI] [PubMed] [Google Scholar]
- 40.Yu H, Patel SB. Recent insights into the Smith-Lemli-Opitz syndrome. Clin Genet. 2005;68:383–91. doi: 10.1111/j.1399-0004.2005.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bommannan D, Menon GK, Okuyama H, Elias PM, Guy RH. Sonophoresis. II. Examination of the mechanism(s) of ultrasound-enhanced transdermal drug delivery. Pharm Res. 1992;9:1043–7. doi: 10.1023/a:1015806528336. [DOI] [PubMed] [Google Scholar]
- 42.Clayton PT, Kalter DC, Atherton DJ, Besley GT, Broadhead DM. Peroxisomal enzyme deficiency in X-linked dominant Conradi-Hunermann syndrome. J Inherit Metab Dis. 1989;12(suppl 2):358–60. doi: 10.1007/BF03335422. [DOI] [PubMed] [Google Scholar]
- 43.Tsai JC, Guy RH, Thornfeldt CR, Gao WN, Feingold KR, Elias PM. Metabolic approaches to enhance transdermal drug delivery. 1. Effect of lipid synthesis inhibitors. J Pharm Sci. 1996;85:643–8. doi: 10.1021/js950219p. [DOI] [PubMed] [Google Scholar]
- 44.Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res. 2006;47:1535–41. doi: 10.1194/jlr.M600141-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcos J, Shackleton CH, Buddhikot MM, Porter FD, Watson GL. Cholesterol biosynthesis from birth to adulthood in a mouse model for 7-dehydrosterol reductase deficiency (Smith-Lemli-Opitz syndrome) Steroids. 2007;72:802–8. doi: 10.1016/j.steroids.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunetti-Pierri N, Corso G, et al. Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3beta-hydroxysteroid-delta5-desaturase. Am J Hum Genet. 2002;71:952–8. doi: 10.1086/342668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi M, D’Armiento M, Parisi I, et al. Clinical phenotype of lathosterolosis. Am J Med Genet A. 2007;143A:2371–81. doi: 10.1002/ajmg.a.31929. [DOI] [PubMed] [Google Scholar]
- 48.Jiang XS, Backlund PS, Wassif CA, Yergey AL, Porter FD. Quantitative proteomics analysis of inborn errors of cholesterol synthesis: identification of altered metabolic pathways in DHCR7 and SC5D deficiency. Mol Cell Proteomics. 2010;9:1461–75. doi: 10.1074/mcp.M900548-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet. 2002;113:315–9. doi: 10.1002/ajmg.b.10873. [DOI] [PubMed] [Google Scholar]
- 50.Elias PM, Williams ML, Maloney ME, et al. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive X-linked ichthyosis. J Clin Invest. 1984;74:1414–21. doi: 10.1172/JCI111552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epstein EH, Jr, Williams ML, Elias PM. Steroid sulfatase, X-linked ichthyosis, and stratum corneum cell cohesion. Arch Dermatol. 1981;117:761–3. [PubMed] [Google Scholar]
- 52.Bergner EA, Shapiro LJ. Metabolism of 3H-dehydroepiandrosterone sulphate by subjects with steroid sulphatase deficiency. J Inherit Metab Dis. 1988;11:403–15. doi: 10.1007/BF01800429. [DOI] [PubMed] [Google Scholar]
- 53.Epstein EH, Jr, Krauss RM, Shackleton CH. X-linked ichthyosis: increased blood cholesterol sulfate and electrophoretic mobility of low-density lipoprotein. Science. 1981;214:659–60. doi: 10.1126/science.6945674. [DOI] [PubMed] [Google Scholar]
- 54.Long SA, Wertz PW, Strauss JS, Downing DT. Human stratum corneum polar lipids and desquamation. Arch Dermatol Res. 1985;277:284–7. doi: 10.1007/BF00509081. [DOI] [PubMed] [Google Scholar]
- 55.Ranasinghe AW, Wertz PW, Downing DT, Mackenzie IC. Lipid composition of cohesive and desquamated corneocytes from mouse ear skin. J Invest Dermatol. 1986;86:187–90. doi: 10.1111/1523-1747.ep12284246. [DOI] [PubMed] [Google Scholar]
- 56.Williams ML, Elias PM. Stratum corneum lipids in disorders of cornification: increased cholesterol sulfate content of stratum corneum in recessive x-linked ichthyosis. J Clin Invest. 1981;68:1404–10. doi: 10.1172/JCI110391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elias PM, Crumrine D, Rassner U, et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol. 2004;122:314–9. doi: 10.1046/j.1523-1747.2003.22258.x. [DOI] [PubMed] [Google Scholar]
- 58.Zettersten E, Man MQ, Sato J, et al. Recessive x-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol. 1998;111:784–90. doi: 10.1046/j.1523-1747.1998.00386.x. [DOI] [PubMed] [Google Scholar]
- 59.Frost P, Weinstein GD, Van Scott EJ. The ichthyosiform dermatoses. II. Autoradiographic studies of epidermal proliferation. J Invest Dermatol. 1966;47:561–7. doi: 10.1038/jid.1966.185. [DOI] [PubMed] [Google Scholar]
- 60.Ekholm IE, Brattsand M, Egelrud T. Stratum corneum tryptic enzyme in normal epidermis: a missing link in the desquamation process? J Invest Dermatol. 2000;114:56–63. doi: 10.1046/j.1523-1747.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 61.Sato J, Denda M, Nakanishi J, Nomura J, Koyama J. Cholesterol sulfate inhibits proteases that are involved in desquamation of stratum corneum. J Invest Dermatol. 1998;111:189–93. doi: 10.1046/j.1523-1747.1998.00244.x. [DOI] [PubMed] [Google Scholar]
- 62.Williams ML. Lipids in normal and pathological desquamation. Adv Lipid Res. 1991;24:211–62. doi: 10.1016/b978-0-12-024924-4.50012-2. [DOI] [PubMed] [Google Scholar]
- 63.Hachem JP, Crumrine D, Fluhr J, Brown BE, Feingold KR, Elias PM. pH directly regulates epidermal permeability barrier homeostasis, and stratum corneum integrity/cohesion. J Invest Dermatol. 2003;121:345–53. doi: 10.1046/j.1523-1747.2003.12365.x. [DOI] [PubMed] [Google Scholar]
- 64.Hachem JP, Fowler A, Behne M, Fluhr J, Feingold K, Elias P. Increased stratum corneum pH promotes activation and release of primary cytokines from the stratum corneum attributable to activation of serine proteases. J Invest Dermatol. 2002;119:258. [Google Scholar]
- 65.Hachem JP, Roelandt T, Schurer N, et al. Acute acidification of stratum corneum membrane domains using polyhydroxyl acids improves lipid processing and inhibits degradation of corneodesmosomes. J Invest Dermatol. 2010;130:500–10. doi: 10.1038/jid.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohman H, Vahlquist A. The pH gradient over the stratum corneum differs in X-linked recessive and autosomal dominant ichthyosis: a clue to the molecular origin of the “acid skin mantle”? J Invest Dermatol. 1998;111:674–7. doi: 10.1046/j.1523-1747.1998.00356.x. [DOI] [PubMed] [Google Scholar]
- 67.Epstein EH, Williams ML, Elias PM. The epidermal cholesterol sulfate cycle. J Am Acad Dermatol. 1984;10:866–8. doi: 10.1016/s0190-9622(84)80144-9. [DOI] [PubMed] [Google Scholar]
- 68.Abraham W, Wertz PW, Landmann L, Downing DT. Stratum corneum lipid liposomes: calcium-induced transformation into lamellar sheets. J Invest Dermatol. 1987;88:212–4. doi: 10.1111/1523-1747.ep12525375. [DOI] [PubMed] [Google Scholar]
- 69.Hatfield RM, Fung LW. A new model system for lipid interactions in stratum corneum vesicles: effects of lipid composition, calcium, and pH. Biochemistry. 1999;38:784–91. doi: 10.1021/bi981421k. [DOI] [PubMed] [Google Scholar]
- 70.Rizzo WB, S’Aulis D, Jennings MA, Crumrine DA, Williams ML, Elias PM. Ichthyosis in Sjogren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443–51. doi: 10.1007/s00403-009-1022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uchida Y, Cho Y, Moradian S, et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J Invest Dermatol. 2010;130:2497–9. doi: 10.1038/jid.2010.145. [DOI] [PubMed] [Google Scholar]
- 72.Menon E, Orso E, Schmitz G, Crumrine D, Elias P. Abnormalities in ultrastructure of epidermal lamellar bodies and corneocyte lipid envelope in Refsum disease. Society for Investigative Dermatology Annual Meeting; 2011; Phoenix, AZ. 2011. p. S52. [Google Scholar]
- 73.Khnykin D, Crumrine D, Uchida Y, et al. Ichthyosis prematurity syndrome: lipid metabolic abnormalities lead to an epidermal barrier defect and predispose to atopy. SID Annual Meeting 2011; 2011; Phoenix: JID. 2011. p. S59. [Google Scholar]
- 74.Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319–21. doi: 10.1038/jid.2009.57. [DOI] [PubMed] [Google Scholar]
- 75.Jiang YJ, Lu B, Kim P, et al. PPAR and LXR activators regulate ABCA12 expression in human keratinocytes. J Invest Dermatol. 2008;128:104–9. doi: 10.1038/sj.jid.5700944. [DOI] [PubMed] [Google Scholar]
- 76.Man MQ, Feingold KR, Thornfeldt CR, Elias PM. Optimization of physiological lipid mixtures for barrier repair. J Invest Dermatol. 1996;106:1096–101. doi: 10.1111/1523-1747.ep12340135. [DOI] [PubMed] [Google Scholar]
- 77.Klar J, Schweiger M, Zimmerman R, et al. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet. 2009;85:248–53. doi: 10.1016/j.ajhg.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng Y, Beier D, Brash A, Elias P, Crumrine D. Formation and function of the corneocyte lipid envelope. 2011 SID Annual Meeting; 2011; Phoenix, AZ. 2011. [Google Scholar]
- 79.Correa-Cerro LS, Wassif CA, Kratz L, et al. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet. 2006;15:839–51. doi: 10.1093/hmg/ddl003. [DOI] [PubMed] [Google Scholar]
- 80.Mirza R, Hayasaka S, Takagishi Y, et al. DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis. J Invest Dermatol. 2006;126:638–47. doi: 10.1038/sj.jid.5700111. [DOI] [PubMed] [Google Scholar]
- 81.Mirza R, Qiao S, Murata Y, Seo H. Requirement of DHCR24 for postnatal development of epidermis and hair follicles in mice. Am J Dermatopathol. 2009;31:446–52. doi: 10.1097/DAD.0b013e318196f10c. [DOI] [PubMed] [Google Scholar]
- 82.Evers BM, Farooqi MS, Shelton JM, et al. Hair growth defects in Insig-deficient mice caused by cholesterol precursor accumulation and reversed by simvastatin. J Invest Dermatol. 2010;130:1237–48. doi: 10.1038/jid.2009.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holleran WM, Ziegler SG, Goker-Alpan O, et al. Skin abnormalities as an early predictor of neurologic outcome in Gaucher disease. Clin Genet. 2006;69:355–7. doi: 10.1111/j.1399-0004.2006.00589.x. [DOI] [PubMed] [Google Scholar]
- 84.Sidransky E, Fartasch M, Lee RE, et al. Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease. Pediatr Res. 1996;39:134–41. doi: 10.1203/00006450-199601000-00020. [DOI] [PubMed] [Google Scholar]
- 85.Holleran WM, Ginns EI, Menon GK, et al. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–64. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moulson CL, Lin MH, White JM, Newberry EP, Davidson NO, Miner JH. Keratinocyte-specific expression of fatty acid transport protein 4 rescues the wrinkle-free phenotype in Slc27a4/Fatp4 mutant mice. J Biol Chem. 2007;282:15912–20. doi: 10.1074/jbc.M701779200. [DOI] [PubMed] [Google Scholar]
- 87.Demerjian M, Crumrine DA, Milstone LM, Williams ML, Elias PM. Barrier dysfunction and pathogenesis of neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome) J Invest Dermatol. 2006;126:2032–8. doi: 10.1038/sj.jid.5700332. [DOI] [PubMed] [Google Scholar]
- 88.Radner FP, Streith IE, Schoiswohl G, et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin bar rier defect in mice lacking comparative gene identification-58 (CGI-58) J Biol Chem. 2010;285:7300–11. doi: 10.1074/jbc.M109.081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ujihara M, Nakajima K, Yamamoto M, et al. Epidermal triglyceride levels are correlated with severity of ichthyosis in Dorfman-Chanarin syndrome. J Dermatol Sci. 2010;57:102–7. doi: 10.1016/j.jdermsci.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 90.Rizzo WB. Sjogren-Larsson syndrome: Molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1–9. doi: 10.1016/j.ymgme.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zheng Y, Yin H, Boeglin WE, et al. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: A proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem. 2011;286:24046–56. doi: 10.1074/jbc.M111.251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feingold KR, Man MQ, Proksch E, Menon GK, Brown BE, Elias PM. The lovastatin-treated rodent: a new model of barrier disruption and epidermal hyperplasia. J Invest Dermatol. 1991;96:201–9. doi: 10.1111/1523-1747.ep12461153. [DOI] [PubMed] [Google Scholar]
- 93.Menon GK, Feingold KR, Mao-Qiang M, Schaude M, Elias PM. Structural basis for the barrier abnormality following inhibition of HMG CoA reductase in murine epidermis. J Invest Dermatol. 1992;98:209–19. doi: 10.1111/1523-1747.ep12555880. [DOI] [PubMed] [Google Scholar]
- 94.Paller AS, van Steensel MA, Rodriguez-Martin M, Sorrell J, Heath C, Crumrine D, et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol. 2011;131:2242–8. doi: 10.1038/jid.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]