

Abstract
Cancer stem cells (CSC), also termed “cancer initiating cells” or “cancer progenitor cells”, which have the ability to self-renew, proliferate, and maintain the neoplastic clone, have recently been discovered in a wide variety of pediatric tumors. These CSC are thought to be responsible for tumorigenesis, tumor maintenance, aggressiveness and recurrence due to inherent resistance to current treatment modalities such as chemotherapy and radiation. Oncolytic virotherapy offers a novel, targeted approach for eradicating pediatric CSC by utilizing mechanisms of cell killing that differ from conventional therapies. Moreover, oncolytic viruses have the ability to target specific features of CSC such as cell surface proteins, transcription factors, and the CSC microenvironment. Through genetic engineering, a wide variety of foreign genes may be expressed by oncolytic viruses to augment the oncolytic effect. We review the current data regarding the ability of several types of oncolytic viruses (herpes simplex virus-1 (HSV-1), adenovirus, reovirus, Seneca Valley virus, vaccinia virus, Newcastle disease virus, myxoma virus, vesicular stomatitis virus) to target and kill both CSC and tumor cells in pediatric tumors. We highlight advantages and limitations of each virus and potential ways next-generation engineered viruses may target resilient CSC.
Cancer affects nearly 15 out of every 100,000 children in the United States, and while survival rates have improved greatly over the past 30 years due to cooperative trials and advances in surgical techniques, chemotherapy and radiotherapy regimens, a significant subset of children, approximately 20%, succumb to their disease (1). Death can result from tumor progression or from ensuing toxicities caused by treatment. In the past, new treatment regimens have focused on increasing the dose of current therapies or combining multiple cytotoxic agents for patients with high-risk disease; however present regimens already approach the upper limits of tolerability. Therefore, simply increasing the dose of current therapies or expanding treatment regimens with more cytotoxic agents is likely to worsen toxicities with minimal improvement in survival rates. The latest research has focused on determining which cells are responsible for tumor recurrence and finding ways in which these cells may be targeted in order to decrease toxicity and enhance quality of life and survival rates for children with cancer.
Recently, the cells thought to be responsible for tumorigenesis, tumor maintenance, aggressiveness, and recurrence have been identified in a number of pediatric malignancies (2). Termed “cancer stem cells” (CSC), these malignant cells retain many of the capabilities of normal stem cells including the ability to differentiate into multiple cell types, to self-renew, to proliferate, and to maintain the neoplastic clone. Whether these are true stem cells is the subject of much debate; the cells may be in a further stage of differentiation than a true stem cell, and therefore have also been called “cancer progenitor cells”. The fact that these cells can initiate tumors has led some to describe them as “cancer-initiating cells”. The term “CSC” will be used throughout this review with the understanding that the actual nature of these cells is not entirely clear.
CSC are thought to create and reside in a specialized microenvironment or niche where tumor and CSC regulation occurs through oxygen tension, cell-to-cell interactions, the extracellular matrix, and the balance of signals received through embryonic signaling pathways (3-5). Importantly, CSC are characteristically resistant to traditional chemotherapy and radiotherapy, and consequently, are believed to be responsible for tumor recurrence (6, 7). Mechanisms that pediatric CSC use to resist current therapies include efficient DNA repair ability with preferential activation of DNA damage response, upregulation of anti-apoptotic genes, differential expression and phosphorylation of various kinases, increased expression of ATP-binding cassette (ABC) transporters, and relative quiescence (2).
One of the main challenges for researchers is developing methods to identify and distinguish these cells from other tumor cells and normal cells in order to gain a better understanding of CSC biology and to develop novel targeted therapies to attack and kill CSC. Current techniques to identify CSC rely on an assortment of markers including cell-surface, nuclear, or cytoplasmic proteins; transcription factors; enzymes and/or functional attributes (2). These unique features of CSC along with the niche in which they reside offer potential strategies for targeting CSC; novel approaches may direct an attack at CSC surface antigens, the niche, embryonic signaling and self-renewal pathways, angiogenesis, or mechanisms of resistance such as ABC transporters or DNA repair (2, 8).
One such innovative, targeted therapy with preclinical efficacy in a variety of pediatric malignancies that may be well-suited to eradicate resilient pediatric CSC is oncolytic virotherapy which kills tumor cells, releasing infectious virus to extend the therapy to neighboring tumor cells (9, 10). Oncolytic viruses can be deadly to cancer cells and CSC in three main ways: 1) viruses can directly target and attack tumor cells due to genetically engineered mutations that prevent viruses from either infecting or replicating in normal cells while permitting infection/replication in tumor cells; 2) some viruses can be engineered to express therapeutic foreign gene products that either directly or indirectly result in cell death; or 3) viruses which normally do not cause significant disease in humans may infect and kill tumor cells that contain altered signaling pathways or deficient interferon responses (Figure 1). Oncolytic viruses utilize methods of cell killing that differ from traditional therapies and thus are able to elude the typical mechanisms than CSC use to resist current chemotherapy and radiotherapy. Moreover, oncolytic viruses have the ability to target specific features of CSC such as cell surface proteins, transcription factors, and the CSC microenvironment. This review will focus on the current research regarding the ability of oncolytic viruses to target and kill CSC in pediatric tumors and highlight potential ways next-generation viruses may target resilient CSC. Viruses that have already entered clinical trials or have been used clinically in children include herpes simplex virus-1 (HSV), adenovirus, reovirus, Seneca Valley virus (SVV), vaccinia virus (VV), and Newcastle disease virus (NDV). Viruses that are being studied preclinically for possible use in children include myxoma virus (MYXV) and vesicular stomatitis virus (VSV). Table 1 provides a comparison of the benefits and limitations of the viruses to be discussed below.
Figure 1.
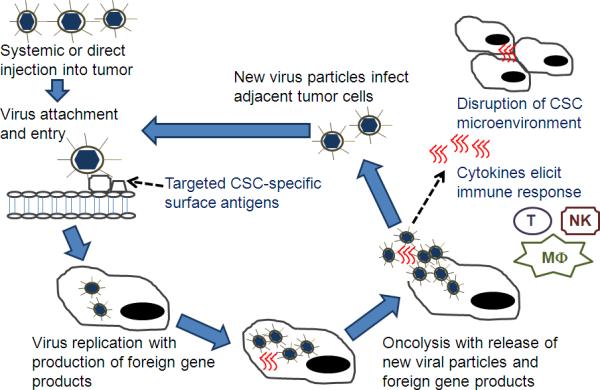
Oncolytic viral therapy +/- gene therapy may be used to target cancer stem cells (CSCs). Virus can be delivered systemically or via direction injection into the tumor bed. Viral mutations (e.g. deletion of virulence genes) or non-human host range prevents a productive infection in normal cells but permits infection in CSC. CSC-specific surface antigens may be targeted for viral entry. As viral replication ensues, foreign gene products are produced such as cytokines (e.g. interleukin-12), enzymes (e.g. chondroitinase), or other proteins (e.g. angiostatin). After host-cell lysis and release of foreign products, cytokines can result in an immune response (T cells (T), NK cells (NK), and macrophages (MΦ)) against CSC antigens in uninfected cells. Enzymes or inhibitory proteins can disrupt the CSC microenvironment. New viral particles can infect adjacent tumor cells.
Table 1.
Comparison of oncolytic viruses
| Virus | Virus type | Benefits | Limitations |
|---|---|---|---|
| HSV-1 | DNA | Neurotropic Ability to infect a wide variety of tumors Large amount of nonessential genes can be replaced with foreign DNA to enhance cytotoxicity Clinically available antiviral agents |
Systemic delivery may be limited by preexisting immunity and hepatic adsorption |
| Adenovirus | DNA | Ability to infect a wide variety of tumors with modifications of the fiber knob Large amount of nonessential genes can be replaced with foreign DNA to enhance cytotoxicity |
CAR variability in human cancers Systemic delivery may be limited by preexisting immunity, hepatic adsorption and toxicity |
| Reovirus | RNA | Wild-type virus causes mild to no disease Systemic delivery possible | Activated Ras or Ras effectors necessary Inability to enhance infection with foreign DNA |
| SVV | RNA | Virus does not cause human disease | Mechanism of infection unclear Inability to enhance infection with foreign DNA |
| VV | DNA | Ability to infect a wide variety of tumors Large amount of nonessential genes can be replaced with foreign DNA to enhance cytotoxicity |
Inefficient systemic delivery |
| NDV | RNA | Targets cancer cells with loss of interferon responsiveness Ability to express foreign DNA to enhance cytotoxicity Used safely in children with recurrent gliomas |
Immune-mediated clearance of virus |
| MYXV | DNA | Targets cancer cells with altered Akt signaling Does not cause human disease |
Limited preclinical data in pediatric cancers |
| VSV | RNA | Targets cancer cells with loss of interferon responsiveness | Limited preclinical data in pediatric cancers Uncertain tumor selective oncolytic effect |
CAR, coxsackie adenovirus receptor; MYXV, myxoma virus; NDV, Newcastle disease virus; SVV; Seneca Valley virus; VSV, vesicular stomatitis virus; VV, vaccinia virus
Herpes simplex virus
HSV is a double-stranded, enveloped DNA cytolytic virus that has shown promise in treating a variety of pediatric malignancies including brain tumors, neuroblastoma, and sarcomas (11). Deletions or mutations of essential HSV-1 genes (e.g. γ134.5 “neurovirulence gene”) required for effective viral replication in normal cells but not cancer cells enables the virus to target malignant cells (12). Furthermore, a large amount (up to 30 kilobase-pairs (kb)) of the HSV genome is nonessential for the virus to replicate in cancer cells, and consequently, can be substituted with foreign DNA that can be used in several ways to enhance viral efficacy such as by restoring the neurovirulence gene under control of a tumor specific promoter to improve replication in tumor cells, by producing enzymes that disrupt and inhibit the tumor microenvironment, or by generating cytokines that can stimulate an immune response against the tumor. Importantly, clinically available antiviral agents (e.g. acyclovir, ganciclovir) are effective against mutant HSV in the unlikely event that the virus causes toxicity to normal cells.
HSV is a neurotropic virus thus rendering neural malignancies like pediatric brain tumors and neuroblastomas, tumor types which have been reported to contain CSC, ideal treatment targets (13-17). Two mutant viruses, G207 and HSV1716, have been used safely without any dose limiting toxicities in adult patients with recurrent glioblastoma multiforme (GBM) (Table 2 summarizes viruses included in the text) (18, 19). G207 is a doubly-deleted genetically engineered HSV that was originally derived from the wild-type clinical isolate, HSV-1 (F). G207 HSV has had both copies of the γ134.5 gene deleted combined with an insertional deletion of the UL39 gene encoding ICP6 (the heavy chain for ribonucleotide reductase). Insertion of the lacZ gene encoding β-galactosidase effectively disables expression of ribonucleotide reductase while providing a useful marker. HSV1716 was produced by deleting both copies of γ134.5 from wild type HSV strain 17. Preclinical studies in neonatal mice and New World owl monkeys (Aotus nancymae) that are as sensitive to wild type HSV-1 as human neonates suggest that engineered HSV will be safe in pediatric patients as well (20, 21). Efficacy of mutant HSV has been demonstrated preclinically in pediatric gliomas, medulloblastomas, and neuroblastomas (22-25).
Table 2.
Summary of oncolytic viruses discussed in the text
| Virus | Deletions/Mutations | Foreign gene/promoter insertion | Pediatric tumors targeted in studies | References |
|---|---|---|---|---|
| HSV-1 | ||||
| G207 | Deletion in both copies of γ134.5 gene and disabling lacZ insertion in UL39 | None | Glioma, MDB, OST, RMS | 16, 21, 32 |
| HSV1716 | Deletion in both copies of γ134.5 gene | None | Non-CNS solid tumors | 22, 56 |
| M002 | Deletion in both copies of γ134.5 gene | Murine IL-12 gene insert | Glioma, RMS | 2, 23 |
| rQNestin34.5 | Deletion in γ134.5 gene and UL39 | ICP-34.5 under control of a synthetic nestin promoter | Glioma, NB | 17, 26 |
| rQT3 | Deletions in ICP6 and γ134.5 gene | Tissue inhibitor of MMP3, HSV-1 immediate early 4/5 promoter | NB, MPNST | 27 |
| VAE | Deletion in both copies of γ134.5 gene | Endostatin–angiostatin fusion gene insert | Glioma | 28 |
| Chase-ABC | Deletion of both copies of γ134.5 and in-frame gene-disrupting insertion of GFP within the ICP6 gene | Inserted Chase-ABC cDNA under the viral IE4/5 promoter within the ICP 6 locus | Glioma | 29 |
| Adenovirus | ||||
| OBP-301 | Deletion of native E1 promoter of Ad5 | Human hTERT promoter to drive E1A and E1B expression linked to an internal ribosome entry site | OST | 38 |
| AD5/3-Cox2L-D24 | Deletion of Rb binding region from the E1A gene | Inserted cyclooxygenase -2 (COX-2) promoter | NB | 41 |
| ICOVIR-5 | Deletion of Rb binding region from the E1A gene | Substitution of the E1A promoter for E2F-responsive elements, RGD-4C peptide motif insertion | NB | 42 |
| Delta-24-RGD | Deletion of Rb binding region from the E1A gene | Inserted RGD into the H1 loop of the fiber protein | Glioma | 44 |
| CRAd-Survivin-pk7 | Deletion of native E1 promoter of Ad5, polylysine modification in the fiber knob | Human survivin promoter to drive E1 expression | Glioma | 45 |
| Reovirus | ||||
| Reovirus type 3 Dearing | None | None | MDB, RMS, OST, EWS | 54, 55 |
| SVV | ||||
| SVV-001 | None | None | EWS, glioma, MDB, NB, rhabdoid tumor, RB, Wilms tumor | 57-61 |
| Vaccinia | ||||
| JX-594 | Deletion of both copies of TK gene | Human GM-CSF and lacZ insertion into the TK gene region | EWS, lymphoma, NB, RMS, Wilms tumor | 56, 66 |
| CIK-vvDD | Deletion of TK genes and vaccinia growth factor genes | None; cytokine-induced killer cells used as carrier vehicle | Lymphoma | 67 |
| NDV | ||||
| 73-T | None | None | NB, OST, | 70, 71 |
| MTH-68/H | Unknown | None | Glioma | 72 |
| MYXV | ||||
| MYXV | None | None | Rhabdoid tumors, NB, leukemia | 78-80 |
| VSV | ||||
| VSVΔm51 | Deletion inactivating the matrix protein | None | Glioma, rhabdoid tumors | 78, 82 |
Ad5, adenovirus serotype 5; CIK-vvDD, cytokine-induced killer double-deleted vaccinia virus; CNS, central nervous system; EWS, Ewing's sarcoma; GM-CSF, granulocyte colony-stimulating factor; hTERT, human telomerase reverse transcriptase; IL; interleukin; MDB, medulloblastoma; MMP3, metalloproteinases-3; MPNST; malignant peripheral nerve sheath tumor; MYXV, myxoma virus; NB, neuroblastoma; OST, osteosarcoma; RB, retinoblastoma; RGD, arginine-glycine-aspartic acid; RMS, rhabdomyosarcoma; SVV, Seneca valley virus; TK, thymidine kinase; VSV, vesicular stomatitis virus
Recent research has examined the ability of mutant HSVs to kill CSC from pediatric neural tumors (Table 3 summarizes studies utilizing oncolytic viruses to target pediatric CSC). We showed that pediatric GBM xenograft D456MG contains CSC marked by expression of CD133 (prominin-1), a transmembrane protein with uncertain biological function expressed on the surface of neuronal and hematopoetic stem cells and CSC in a variety of malignancies (23). CSC isolated from this xenograft were as sensitive as non-CSC tumor cells (CD133- cells) to several different γ134.5-deleted viruses including M002, an engineered HSV that expresses murine interleukin (IL)-12. IL-12 is an example of a cytokine added to elicit an immune response via activation of natural killer (NK) and T cells. The pediatric GBM D456MG was more sensitive to killing than several adult GBM xenografts tested (2, 23). To increase tumor selectivity and enhance viral replication, Kambara et al. developed rQNestin34.5 which expresses ICP-34.5 under control of a synthetic nestin promoter (26). Nestin is an intermediate filament protein that is expressed in embryonic neuroglial cells and has been used as a CSC marker in a number of cancers including brain tumors and neuroblastoma. Mahller et al. used the rQNestin34.5 virus to infect and kill neuroblastoma CSC (17). The virus prevented the CSC from forming tumors in athymic nude mice suggesting that the CSC may be effectively targeted.
Table 3.
Summary of studies utilizing oncolytic viruses to target pediatric CSC by tumor type
| Tumor Type | Virus Type, Name | Outcome | References |
|---|---|---|---|
| Glioma | HSV-1, M002 | CD133+ CSC and CD133- tumor cells equally sensitive | 21 |
| MDB | SVV, wild-type | CD133+ CSC equally sensitive to CD133- cells | 58 |
| Not all xenografts sensitive | |||
| NB | HSV-1, rQNestin34.5 | Prevented CSC from forming tumors in athymic nude mice | 15 |
| MYXV, wild-type | CSC appear sensitive in preliminary studies | 78 | |
| VSV, VSV | Possible resistance of CSC in preliminary studies | 78 | |
| RMS | HSV-1, M002 | CD133+ CSC and CD133- tumor cells equally sensitive | U |
CSC, cancer stem cells; MDB, medulloblastoma; MYXV, myxoma virus; NB, neuroblastoma; RMS, rhabdomyosarcoma; U, unpublished
Another approach to targeting CSC with engineered HSV is to disrupt the microenvironment through specific proteins produced during viral replication to supplement the oncolytic effects of the virus. Engineered HSV rQT3 has deletions of ICP6 and γ134.5 and expresses human Tissue Inhibitor of Metallo-Proteinases 3 (TIMP3) (27). Matrix metalloproteinases are a group of endopeptidases that degrade the extracellular matrix and thus play a critical role in the tumor niche by permitting angiogenesis and invasion. RQT3-treated neuroblastoma and peripheral nerve sheath tumor xenografts not only showed delayed tumor growth but also had a reduced vascular density. Moreover, the treatment decreased circulating endothelial progenitors indicating a possible anti-angiogenesis effect of the virus. Zhu et al. developed an oncolytic HSV, VAE, which carries an exogenous Endo-Angio fusion gene. Endostatin and angiostatin are potent angiogenesis inhibitors. The virus not only infected and killed glioma CSC but also inhibited their vascular niche in vitro (28). Using a similar approach, Dmitrieva et al. examined the effect of Chase-ABC, an HSV that produces chondroitinase ABC which is a bacterial enzyme that removes the chondroitin sulfate from proteoglycans, a major component of the tumor extracellular matrix (29). Compared to a control virus, Chase-ABC spread throughout glioma spheroids more efficiently, and through degradation of the extracellular matrix, the virus showed enhanced replication and antitumor activity in vivo. It is important to note that the studies using VAE and Chase-ABC were conducted in glioma cells from adult tumors which in general are molecularly quite distinct from pediatric gliomas, and therefore the findings might not be extrapolatable to pediatric glioma CSC.
Even though HSV is a neurotropic virus, it is capable of killing cells from a wide variety of non-neural cancers including sarcomas, melanomas, colon, breast, lung, prostate and hepatic tumors and several adult human studies have demonstrated safety and antitumor effects (30-32). Preclinical efficacy of G207 in pediatric rhabdomyosarcoma and osteosarcoma cell lines was demonstrated by Bharatan et al (33). We have found that the CSC, marked by CD133, in both alveolar and embryonal rhabdomyosarcoma cell lines are equally sensitive compared to other tumor cells to killing with M002 (GK Friedman and GY Gillespie, unpublished results). The humanized IL-12 version of the M002 virus, M032 is being prepared for a phase I trial at the University of Alabama at Birmingham in adult patients with recurrent GBM. No other studies to date have examined the effect of HSV on non-neural pediatric solid tumor CSC. The first trial utilizing an engineered HSV, HSV1716, to be injected intratumorally in pediatric patients (13 and older) with recurrent or refractory extra-cranial solid tumors is on-going at Children's Hospital Medical Center in Cincinnati (ClinicalTrials.gov identifier NCT00931931).
Adenovirus
Adenovirus is a non-enveloped, non-integrated double-stranded DNA virus in the Adenoviridae family that has been studied extensively as a novel, oncolytic therapeutic. While wild-type adenoviruses can infect both dividing and non-dividing cells and cause respiratory, ophthalmic, or gastrointestinal illnesses in humans, attenuated conditionally replicative adenoviruses (CRAds) can target cancer cells with few side effects. Deletions in immediate-early (E1A) or early (E1B) adenovirus genes result in attenuated mutants that cannot bind normal cellular proteins that drive gene expression initiating and maintaining cellular proliferation needed for productive virus infection (34). These virus genome deletions do not effect its replication in cancer cells due to pathway defects such as p16/retinoblastoma (Rb) or p53. Most adenoviral gene therapy vectors including the most commonly used serotype 5 (Ad5) enter cells through the coxsackie adenovirus receptor (CAR), which is problematic because of highly variable (to absent) expression of CAR by tumor cells (35). For example, neuroblastoma and medulloblastomas tend to express a higher degree of CAR than gliomas, which tend to have lower and variable expression (36). Moreover, normal epithelial cells, neurons and astrocytes also express a high amount of CAR which could result in adverse treatment effects. Newer CRAds circumvent this limitation through modifications of the fiber knob of the viral capsid thereby altering the tropism of the virus and enabling infection of cancer cells through a CAR-independent mechanism (34). Similar to engineered HSV-1, foreign DNA can be inserted into CRAds to enhance viral efficacy by targeting cancer cells under control of a tumor-specific promoter, by inducing a tumor-specific immune response through various cytokines, and/or by directing attacks at the tumor microenvironment and angiogenesis (35).
In preclinical and clinical studies, CRAds have demonstrated safety and efficacy in pediatric extra-cranial solid tumors. Ewing's sarcoma cells expressed CAR and were highly sensitive to viral oncolysis by adenovirus (37). OBP-301 (Telomelysin), a CRAd with a human telomerase reverse transcriptase (hTERT) promoter driving expression of E1A and E1B genes linked to an internal ribosome entry site, was cytotoxic in osteosarcoma cell lines that expressed CAR and suppressed tumor growth in a murine osteosarcoma xenograft model (38). Telomerase plays an important role in tumorigenesis; telomerase activation results in cellular proliferation and can lead to mutagenesis and transformation, and telomerase appears to be overexpressed in CSC compared to other tumor cells (39). Thus therapeutics which target telomerase such as OBP-301 are promising agents to eradicate resistant CSC.
While there are no specific studies examining CRAds effect on neuroblastoma CSC, various adenoviruses have effectively targeted neuroblastoma cells, and two viruses have been used clinically in children with neuroblastoma (40-42). Pesonen et al. reported treatment with an oncolytic adenovirus AD5/3-Cox2L-D24 in a six year old boy with metastatic neuroblastoma resistant to several chemotherapy regimens including autologous transplant (41). The virus has a 24 base pair deletion in the Rb binding site of E1A and the native E1A promoter is replaced with the cyclooxygenase-2 (COX-2) promoter. Cyclooxygenase-2 is believed to play an important role in tumorigenesis and cell survival by stimulating cell growth, invasiveness, and neovascularization, which are similar functions attributed to CSC (43). Injection of 1011 viral particles into the primary tumor bed resulted in a 71% regression of the primary tumor and clearance of metastatic bone marrow disease. Side effects included mild fever, diarrhea, stomach pains and elevated liver enzymes that resolved in two weeks. In a separate study, four children (2 to 5 years of age) with refractory metastatic neuroblastoma received several doses of Ad-DM-E2F-K-Delta24RGD (ICOVIR-5), a CRAd that contains a deletion in E1A, a substitution of the E1A promoter for E2F-responsive elements, and an RGD-4C peptide motif inserted into the adenoviral fiber to enhance adenoviral tropism (42). The virus was delivered intravenously by autologous mesenchymal stem cells, which may engraft in tumor stroma, and was well tolerated with side effect of fever in three patients and an elevated liver alanine aminotransferase in one patient that resolved in 96 hours. While three of the patients had no response, one patient had a very good partial response suggesting that further investigation would be worthwhile. Currently, there are no known on-going studies of CRAds in children.
Although not specific to pediatric brain CSC, adenovirus vectors have shown promise in killing brain CSC from adult glioma cell lines. Using Delta-24-RGD, a CRAd with the Rb binding region deleted from the E1A gene and an inserted RGD (arginine-glycine-aspartic acid) into the H1 loop of the fiber protein allowing the virus to enter cells via αvβ3 and αvβ5 integrins independent of CAR, Jiang et al. demonstrated that xenografts derived from glioma CSC were sensitive to killing by the virus, and treatment resulted in prolonged survival in glioma bearing mice (44). The glioma CSC expressed high levels of CAR and integrins and were targeted due to a defective Rb pathway not present in normal brain cells. Delta-24-RGD is currently in a phase I trial in adults with recurrent malignant gliomas (NCT00805376). To improve the selectivity of adenovirus for malignant gliomas, Nandi et al. developed CRAd-Survivin-pk7, an Ad5 virus with a human survivin promoter to drive E1 expression and a polylysine modification in the fiber knob to selectively bind heparan sulfate proteoglycans overexpressed in gliomas (45). Survivin is a member of the inhibitor of apoptosis family of proteins which is overexpressed on adult gliomas but downregulated in normal tissue (46). CRAd-Survivin-pk7 effectively targeted CD133+ glioma CSC (45). Additionally, the survivin promoter was radio-inducible with low dose radiation increasing the cytotoxicity of CRAad-Survivin-pk7 in glioma cells. This effect was more pronounced in the CD133+ glioma cells suggesting that the CSC may have increased proliferative capacity following low dose radiation. Of note, survivin expression in pediatric neural tumors is quite variable. Zhang et al. found only 1 of 26 pediatric brain tumors (5 GBM, 4 low-grade astrocytomas, 10 juvenile pilocytic astrocytomas, and 7 ependymomas) showed moderate levels of survivin; however in other studies, ependymomas, medulloblastomas, and neuroblastomas demonstrated elevated survivin expression which correlated with poor outcomes (47-50). Thus the potential benefit of CRAd-Survivin-pk7 to target pediatric CSC is unclear.
Skog et al. suggest that Ad5 may not be the best vector for targeting glioma CSC, and other vectors with an adenovirus serotype 16 (Ad16) and chimpanzee serotype 23 (CV23) backbone should be evaluated as alternatives (51). Ad5 was the least efficient serotype whereas Ad16 and CV23 were the most effective at killing both CD133+ and CD133- cells. With significant biological differences in pediatric versus adult brain tumors, further studies are needed to determine if pediatric brain CSC can be effectively targeted by CRAds.
Reovirus
Reovirus (respiratory enteric orphan virus) is a non-enveloped, double-stranded, segmented RNA virus that has shown potential as an oncolytic, targeted agent. The virus only causes mild respiratory or gastrointestinal symptoms, if any, in humans. It is limited by cellular activation of protein kinase R (PKR) which subsequently phosphorylates eukaryotic initiation factor 2α resulting in inhibition of viral gene translation and an ineffective infection in normal cells (52). Activated Ras or Ras pathway effector proteins, which are commonly found in human tumors, prevents PKR activation thereby permitting viral gene translation and resulting in an effective lytic infection. Importantly, Ras activation has been shown to be an important mediator of tumorigenesis in various tumor types and may initiate tumor formation by expanding the stem cell population (53). Thus the benign nature of the virus, its ability to target cancer cells with upregulated Ras signaling pathway, and the capability to deliver the virus systemically make it an appealing oncolytic virus.
In preclinical studies reovirus has been effective against pediatric malignancies. Yang et al. found most medulloblastoma (MDB) cell lines and MDB primary cultures from surgical specimens were sensitive to killing by human reovirus type 3 Dearing (54). Not only was survival significantly increased in an in vivo mouse model, but spinal and leptomeningeal metastases, which are relatively common in MDB patients, were decreased with intrathecal injections of the virus. Using the same strain of virus, Hingorani et al. demonstrated efficacy of reovirus delivered systemically to treat rhabdomyosarcoma, osteosarcoma and Ewing's sarcoma cell lines in the flank of athymic nude mice (55). Combining the virus with radiation or cisplatin enhanced the therapeutic effect.
The only study examining the effects of reovirus on CSC was in breast cancer. Breast CSC, marked by CD24-CD44+ expression and over-expression of aldehyde dehydrogenase, were as sensitive to killing by reovirus as the non-CSC (56). Notably, there were similar levels of Ras in the CSC and the other tumor cells suggesting that reovirus may effectively target chemotherapy and radiotherapy resistant CSC. Further studies are needed to determine if reovirus can effectively target CSC from pediatric tumors.
Multiple phase I and phase II studies of reovirus injected into the tumor bed or systemically have been or are currently being conducted in adult patients with CNS and extra-cranial solid tumors (9). The virus is being tested as monotherapy or in combination with chemotherapy or radiation. Objective responses and disease stabilization have been reported with few side effects ranging from flu-like symptoms to mild gastrointestinal symptoms to neutropenia. No severe dose limiting toxicities have been reported to date. The first trial utilizing reovirus in pediatric patients (3-21 years of age) with relapsed or refractory extracranial solid tumors is currently accruing patients (NCT01240538; Children's Oncology Group (COG) ADVL1014).
Seneca Valley virus
SVV is a recently discovered single-stranded, non-enveloped, non-integrating RNA virus in the family Picornaviridae. The conditionally replication-competent virus does not cause disease in humans but has potent cytolytic activity in some cancer cell types. The mechanism by which SVV produces a productive infection in cancer cells has not been fully elucidated; however cell surface receptor interactions with the virus appear to be an important component, and viral replication is at least partially mediated through autophagy (57, 58). SVV has been used safely without dose limiting toxicities in a phase I clinical trial in adults with advanced solid tumors (59).
In preclinical studies SVV has effectively killed cancer cells in a variety of pediatric solid tumors. Reddy et al. demonstrated sensitivity of Ewing's sarcoma, medulloblastoma, neuroblastoma, and retinoblastoma cell lines to SVV when injected systemically (57). Very high doses up to 1 × 1014 were tolerated in immunocompetent mice. Wadhwa et al. found that a single tail vein injection of SVV was able to treat invasive retinoblastoma and prevent CNS metastatic disease in a murine model (60). Testing of SVV by the Pediatric Preclinical Testing Program (PPTP) confirmed a marked cytotoxic effect of the virus in some neuroblastoma cell lines (61). Additionally, rhabdomyosarcoma cell lines were highly sensitive to the virus, and there was an objective response seen in at least one rhabdoid tumor, Wilms tumor, and GBM cell line. While several Ewing's sarcoma cell lines were sensitive to SVV in vitro, this effect was lost in vivo. Osteosarcoma and medulloblastoma cell lines were resistant to killing by the virus. Based on the promising results by Reddy et al. and the PPTP, the first trial utilizing SVV (NCT01048892; COG ADVL0911) in children 3 to 21 years old with relapsed or refractory neuroblastoma, rhabdomyosarcoma, or rare tumors with neuroendocrine features is currently accruing patients.
Recently, Yu et al. conducted the first study of the ability of SVV to kill pediatric CSC. In a panel of 10 primary human medulloblastoma xenografts, half of the tumors were sensitive to killing with SVV (58). The CD133+ medulloblastoma CSC and CD133- tumor cells were equally sensitive in permissive xenografts and similarly resistant in prohibitive xenografts in all cell lines tested suggesting that the CSC were no more resistant to the virus than other tumor cells. The variable sensitivity of cells of the same tumor type seen by Yu et al. and the PPTP should provide excellent models for determining barriers to tumor-selective replication.
Vaccinia virus
Vaccinia virus (VV) is a double-stranded, enveloped DNA virus in the poxvirus family that was first utilized as a vaccination against smallpox and more recently has been attenuated for use as a cancer therapeutic. Mutated viruses have a deletion in both copies of the thymidine kinase (TK) gene (62). The TK-deleted virus requires thymidine triphosphate for DNA synthesis which is provided by dividing cells thus leading to preferential replication in dividing cells and tumor cell specificity. Another promising approach to prevent infection in normal tissue involves deleting the B18R gene, which counteracts type I-interferons. The B18R-deleted mutant results in interferon-mediated enhanced virus inactivation in normal cells (63). Like HSV and adenovirus, a large portion of the genome may be replaced with foreign DNA to augment oncolysis. Several viruses which target cancer specific antigens and/or induce an immune response through expression of various cytokines have been used in human adult trials (reviewed in 64). VV that express anti-angiogenesis proteins have been used successfully in preclinical studies to treat human adult solid tumors (65). The first pediatric trial (NCT01169584) is currently testing the safety of JX-594, a VV that expresses human granulocyte macrophage colony-stimulating factor (GM-CSF) to induce a tumor-specific cytotoxic immune response, in children 2 years to 21 years old with refractory or recurrent solid tumors including neuroblastoma, rhabdomyosarcoma, lymphoma, Wilms tumor and Ewing's sarcoma.
Two recent studies highlight the potential of VV to target and kill CSC. Lun et al. tested JX-594 against CSC in a panel of high-grade glioma cell lines (66). Most cells from five separate cell lines grown in serum-free medium as neurospheres, free floating clumps of cells thought to be enriched for CSC, were killed by the virus. The self-renewal ability of the cells grown in neurospheres was inhibited by JX-594 infection. This study is limited by the lack of pediatric glioma cell lines and the lack of a specific CSC marker used to identify the CSC. Not all cells within a neurosphere are undifferentiated CSC, and in fact, most may be differentiated tumor cells. Nevertheless, the decrease in the number of neurospheres formed after infection with JX-594 suggests that the virus can target and kill some glioma CSC. Using a Western Reserve strain of VV with mutations in TK and viral growth genes that is delivered to tumor cells by an ex vivo expanded NK-T cell population (CIK-vvDD), Contag et al. demonstrated that CIK-vvDD targeted and killed residual murine lymphoma cells with stem-like features including the ability to initiate tumors and resistance to chemotherapy and radiation (67). No specific CSC marker exists for murine lymphoma and no human lymphomas were used in the experiments. Thus further studies are necessary to determine if this dual biotherapy can indeed target and kill human CSC.
Newcastle disease virus
Newcastle disease virus (NDV) is a negative-sense, single-stranded RNA paramyxovirus that is highly infectious in poultry but causes only mild flu-like symptoms in humans. Tumor cell tropism of the virus is thought to be dependent on defective interferon responsiveness or cell resistance to apoptosis (68, 69). In preclinical studies, NDV strain 73-T selectively targeted and killed pediatric cancers including neuroblastoma, osteosarcoma, and Wilms tumor (70, 71). With reverse genetic technology, recombinant viruses that express foreign genes like GM-CSF, interferon-γ, IL-2, or tumor necrosis factor α are being tested (69).
While there are no specific studies examining the effect of NDV on pediatric CSC, attenuated NDV has been used safely with demonstrated efficacy in children with high-grade gliomas (72-74). Csatary et al. reported on three children (1.5 to 12 years old) with high-grade gliomas who received NDV MTH-68/H after failure with conventional therapies. All three patients exhibited significant tumor regression and improvement in neurological function while receiving the virus repeatedly over several years (72). These exciting results strongly suggest that further study of NDV to target pediatric malignancies would be worthwhile.
Myxoma virus
Like VV, MYXV is a large, double-stranded DNA poxvirus that can accommodate therapeutic foreign genes by replacing up to 25 kb. The natural host range of MYXV is rabbits, and the virus does not cause disease in human but is cytotoxic to cancer cells through altered Akt signaling (75). Akt signaling plays a critical role in cell survival, growth and proliferation, and recently the Akt pathway has been implicated in regulating the survival of CSC following radiation (76). Furthermore, Akt inhibition has been shown to preferentially kill brain CSC relative to other brain tumor cells and reduce tumor invasiveness (77). These data suggest that MYXV may be an excellent candidate to eradicate CSC.
To date, there are very few studies examining the sensitivity of pediatric tumors or CSC to MYXV. A high percentage of rhabdoid tumors, an aggressive pediatric malignancy, responded completely to a single intratumoral injection of MYXV in mice (78). Preliminary studies in neuroblastoma suggest that CSC may be sensitive to infection by MYXV (79). Lastly, adult human acute myeloid leukemic stem and progenitor cells were sensitive to killing by MYXV while normal hematopoetic stem and progenitor cells were not affected by the virus (80). Based on these promising studies, further evaluation of MYXV in pediatric cancers and CSC is warranted.
Vesicular stomatitis virus
VSV is an enveloped, negative-sense, single-stranded RNA rhabdovirus that mainly infects livestock and only rarely causes a flu-like syndrome in humans. Similar to NDV, normal human cells are believed to be protected by the exquisite sensitivity of VSV to the host cell interferon response, whereas cancer cells may be targeted by the virus due to a loss of interferon responsiveness (81). A mutant attenuated form of the virus, VSVΔm51, which has a single amino acid deletion of methionine-51 of the matrix protein to provide additional protection for normal cells by restoring interferon mediated responses, has shown efficacy in treating human rhabdoid tumors and gliomas (78, 82). Human osteosarcoma and Ewing's sarcoma were sensitive to infection with a different mutant VSV whereas a human synovial sarcoma line was very resistant (83). There are no reported studies on the ability of VSV to target and kill pediatric CSC although preliminary results suggest that neuroblastoma CSC may be resistant (79). Further studies are needed to confirm this finding, elucidate the mechanism of resistance, and determine if other pediatric CSC display resistance to VSV. Additionally, a recent report by Yasmeen et al. suggests that VSV may replicate well in some normal human cells, therefore a greater understanding of VSV replication is likely needed before the virus is advanced to human clinical studies (84).
Future Directions
Over the past decade, great progress has been made in the field of oncolytic virotherapy. Several viruses have been translated from the laboratory to the clinics to hopefully benefit children with chemo- and radioresistant malignancies, and several more viruses are likely to be used in clinical trials in the future. Each virus has unique benefits and limitations as an oncolytic agent which may help to determine how it is utilized and what tumors are targeted clinically (Table 1). Besides the viruses covered in this review, several viruses, which have not been tested for efficacy against CSC nor been used clinically in pediatric patients, including neuroattenuated poliovirus, modified measles virus, and parvovirus have shown promise preclincally in targeting pediatric malignancies including neuroblastoma and medulloblastoma (85-87). With the discovery of CSC, future research must focus on ways in which oncolytic viruses can be harnessed to target and eradicate these cells. As CSC biology is revealed, next generation viruses can be developed which target specific CSC antigens, the signaling pathways which regulate CSC, and the CSC microenvironment. Table 4 summarizes potential mechanisms oncolytic viruses may use to infect and kill CSC. Importantly, since CSC may share many of the characteristics of non-transformed stem cells that play a vital role in the developing child as well as in tissue repair and maintenance, the specificity of targeted viruses towards transformed CSC (but not normal stem cells) must be considered as next generation viruses are developed and oncolytic virotherapy is moved to clinical trials in children.
Table 4.
Mechanisms by which oncolytic viruses may target CSC
| Oncolytic Virus | Potential mechanisms to target CSC |
|---|---|
| Herpes Simplex Virus-1 | Deletion of gene(s) necessary for viral replication in normal cells but not CSC Increasing virulence by restoring ICP-34.5 under a CSC protein promoter Disrupting the CSC microenvironment Inhibiting angiogenesis Enlisting an immune response at CSC antigens Combining virus with radiation +/- chemotherapy |
| Adenovirus | Deletion of gene(s) necessary for viral replication in normal cells but not CSC Increasing virulence and tumor selectivity through a CSC protein promoter Disrupting the CSC microenvironment Inhibiting angiogenesis Enlisting an immune response at CSC antigens Combining virus with radiation +/- chemotherapy Utilizing carrier vehicle to deliver virus to CSC |
| Reovirus | Targeting Activated Ras or Ras effectors in CSC Combining virus with radiation +/- chemotherapy |
| Seneca valley virus | Unknown; possibly mediated through induction of autophagy |
| Vaccinia virus | Deletion of gene(s) necessary for viral replication in normal cells but not CSC Enlisting an immune response at CSC antigens Inhibiting angiogenesis Utilizing carrier vehicle to deliver virus to CSC Combining virus with radiation +/- chemotherapy |
| Newcastle disease virus | Targeting CSC with defective interferon responsiveness or resistance to apoptosis Enlisting an immune response at CSC antigens |
| Myxoma virus | Targeting CSC with altered Akt signaling |
| Vesicular stomatitis virus | Targeting CSC with defective interferon responsiveness |
Strategies to enhance viral efficacy include improving virus delivery, tumor specificity and virus replication, reducing virus clearance, and increasing the tumor-directed immune response. Combination therapy with chemotherapeutics, radiation, monoclonal antibodies, small molecule inhibitors, and/or other oncolytic viruses will likely be necessary to eliminate CSC and achieve superior outcomes. Low dose chemotherapy with agents like cyclophosphamide can reduce the anti-viral immune response and thus enhance oncolysis (88). Virotherapy may complement high dose chemotherapy regimens by providing a unique cell-cycle independent mechanism of cell killing. Oncolytic viruses and radiation may act synergistically; viruses can sensitize cells to radiation, and radiation can enhance viral infection, replication, and gene expression resulting in greater tumor cell death (89). Monoclonal antibodies and small molecule inhibitors can complement oncolytic virotherapy by altering regulatory pathways, increasing viral replication, and enhancing the induction of apoptosis (90). Lastly, combination therapy with viruses that have different mechanisms of attack will likely provide a synergistic effect. Through these various combination therapies, oncolytic virotherapy offers great promise in targeting resistant CSC and thereby decreasing toxicity and enhancing the quality of life and survival rates for children with cancer.
Acknowledgments
Funding: G.K.F., K.A.C., and E.A.B. are supported by the Kaul Pediatric Research Institute, and the National Institutes of Health (CA071933, CA097247).
References
- 1.Ries LAG, Melbert D, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2005. National Cancer Institute; Bethesda, MD: 2008. http://seer.cancer.gov/csr/1975_2005/ [Google Scholar]
- 2.Friedman GK, Gillespie GY. Cancer stem cells and pediatric solid tumors. Cancers (Basel) 2011;3:298–318. doi: 10.3390/cancers3010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sneddon JB, Werb Z. Location, location, location: The cancer stem cell niche. Cell Stem Cell. 2007;1:607–11. doi: 10.1016/j.stem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26:1818–30. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 5.Moore SW. Developmental genes and cancer in children. Pediatr Blood Cancer. 2009;52:755–60. doi: 10.1002/pbc.21831. [DOI] [PubMed] [Google Scholar]
- 6.Vlashi E, McBride WH, Pajonk F. Radiation responses of cancer stem cells. J Cell Biochem. 2009;108:339–42. doi: 10.1002/jcb.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rich JN, Bao S. Chemotherapy and cancer stem cells. Cell Stem Cell. 2007;1:353–5. doi: 10.1016/j.stem.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Yang ZJ, Wechsler-Reya RJ. Hit 'em where they live: Targeting the cancer stem cell niche. Cancer Cell. 2007;11:3–5. doi: 10.1016/j.ccr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Hammill AM, Conner J, Cripe TP. Oncolytic virotherapy reaches adolescence. Pediatr Blood Cancer. 2010;55:1253–63. doi: 10.1002/pbc.22724. [DOI] [PubMed] [Google Scholar]
- 10.Lacroix J, Witt O, Sclehofer JR, Rommelaere J. Therapeutic exploitation of oncolytic viruses in pediatric oncology. Drugs Fut. 2010;35:1015. [Google Scholar]
- 11.Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther. 2009;17:1125–35. doi: 10.1038/mt.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–6. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 13.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 14.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor MD, Poppleton H, Fuller C, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–35. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Chiou SH, Kao CL, Chen YW, et al. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS One. 2008;3:e2090. doi: 10.1371/journal.pone.0002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahller YY, Williams JP, Baird WH, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One. 2009;4:e4235. doi: 10.1371/journal.pone.0004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 19.Rampling R, Cruickshank G, Papanastassiou, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 20.Hunter WD, Martuza RL, Feigenbaum F, et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J Virol. 1999;73:6319–26. doi: 10.1128/jvi.73.8.6319-6326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radbill AE, Reddy AT, Markert JM, et al. Effects of G207, a conditionally replication-competent oncolytic herpes simplex virus, on the developing mammalian brain. J Neurovirol. 2007;13:118–29. doi: 10.1080/13550280601187177. [DOI] [PubMed] [Google Scholar]
- 22.Markert JM, Coen DM, Malick A, Mineta T, Martuza RL. Expanded spectrum of viral therapy in the treatment of nervous system tumors. J Neurosurg. 1992;77:590–4. doi: 10.3171/jns.1992.77.4.0590. [DOI] [PubMed] [Google Scholar]
- 23.Friedman GK, Langford C, Coleman J, et al. Engineered herpes simplex virus efficiently infects and kills CD133+ glioma cells that express CD111. J Neurooncol. 2009;95:199–209. doi: 10.1007/s11060-009-9926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasner TM, Kesari S, Brown SM, Lee VM, Fraser NW, Trojanowski JQ. Therapy of a murine model of pediatric brain tumors using a herpes simplex virus type-1 ICP34.5 mutant and demonstration of viral replication within the CNS. J Neuropathol Exp Neurol. 1996;55:1259–69. doi: 10.1097/00005072-199612000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Parikh NS, Currier MA, Mahller YY, et al. Oncolytic herpes simplex virus mutants are more efficacious than wild-type adenovirus Type 5 for the treatment of high-risk neuroblastomas in preclinical models. Pediatr Blood Cancer. 2005;44:469–78. doi: 10.1002/pbc.20268. [DOI] [PubMed] [Google Scholar]
- 26.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–9. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 27.Mahller YY, Vaikunth SS, Ripberger MC, et al. Tissue inhibitor of metalloproteinase-3 via oncolytic herpesvirus inhibits tumor growth and vascular progenitors. Cancer Res. 2008;68:1170–9. doi: 10.1158/0008-5472.CAN-07-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu G, Su W, Jin G, et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59–69. doi: 10.1016/j.brainres.2011.03.050. [DOI] [PubMed] [Google Scholar]
- 29.Dmitrieva N, Yu L, Viapiano M, et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17:1362–72. doi: 10.1158/1078-0432.CCR-10-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737–47. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 31.Fong Y, Kim T, Bhargava A, et al. A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectal cancer. Mol Ther. 2009;17:389–94. doi: 10.1038/mt.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16:4005–15. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- 33.Bharatan NS, Currier MA, Cripe TP. Differential susceptibility of pediatric sarcoma cells to oncolysis by conditionally replication-competent herpes simplex viruses. J Pediatr Hematol Oncol. 2002;24:447–53. doi: 10.1097/00043426-200208000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Ribacka C, Hemminki A. Virotherapy as an approach against cancer stem cells. Curr Gene Ther. 2008;8:88–96. doi: 10.2174/156652308784049372. [DOI] [PubMed] [Google Scholar]
- 35.Short JJ, Curiel DT. Oncolytic adenoviruses targeted to cancer stem cells. Mol Cancer Ther. 2009;8:2096–2102. doi: 10.1158/1535-7163.MCT-09-0367. [DOI] [PubMed] [Google Scholar]
- 36.Persson A, Fan X, Salford LG, Widegren B, Englund E. Neuroblastomas and medulloblastomas exhibit more Coxsackie adenovirus receptor expression than gliomas and other brain tumors. Neuropathology. 2007;27:233–6. doi: 10.1111/j.1440-1789.2007.00767.x. [DOI] [PubMed] [Google Scholar]
- 37.Rice AM, Currier MA, Adams LC, et al. Ewing sarcoma family of tumors express adenovirus receptors and are susceptible to adenovirus-mediated oncolysis. J Pediatr Hematol Oncol. 2002;24:527–33. doi: 10.1097/00043426-200210000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki T, Tazawa H, Hasei J, et al. Preclinical evaluation of telomerase-specific oncolytic virotherapy for human bone and soft tissue sarcomas. Clin Cancer Res. 2011;17:1828–38. doi: 10.1158/1078-0432.CCR-10-2066. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, He K, Goldkorn A. Telomerase targeted therapy in cancer and cancer stem cells. Clin Adv Hematol Oncol. 2011;9:442–55. [PubMed] [Google Scholar]
- 40.Geoerger B, van Beusechem VW, Opolon P, et al. Expression of p53, or targeting towards EGFR, enhances the oncolytic potency of conditionally replicative adenovirus against neuroblastoma. J Gene Med. 2005;7:584–94. doi: 10.1002/jgm.703. [DOI] [PubMed] [Google Scholar]
- 41.Pesonen S, Helin H, Nokisalmi P, et al. Oncolytic adenovirus treatment of a patient with refractory neuroblastoma. Acta Oncol. 2010;49:117–9. doi: 10.3109/02841860903071369. [DOI] [PubMed] [Google Scholar]
- 42.García-Castro J, Alemany R, Cascalló M, et al. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010;17:476–83. doi: 10.1038/cgt.2010.4. [DOI] [PubMed] [Google Scholar]
- 43.Rizzo MT. Cyclooxygenase-2 in oncogenesis. Clin Chim Acta. 2011;412:671–87. doi: 10.1016/j.cca.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 44.Jiang H, Gomez-Manzano C, Aoki H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–4. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 45.Nandi S, Ulasov IV, Tyler MA, Sugihara, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–84. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Houdt WJ, Haviv YS, Lu B, et al. The human survivin promoter: a novel transcriptional targeting strategy for treatment of glioma. J Neurosurg. 2006;104:583–92. doi: 10.3171/jns.2006.104.4.583. [DOI] [PubMed] [Google Scholar]
- 47.Zhang JG, Kruse CA, Driggers L, et al. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol. 2008;88:65–76. doi: 10.1007/s11060-008-9534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Preusser M, Wolfsberger S, Czech T, Slavc I, Budka H, Hainfellner JA. Survivin expression in intracranial ependymomas and its correlation with tumor cell proliferation and patient outcome. Am J Clin Pathol. 2005;124:543–9. doi: 10.1309/PP2G5GAAFKV82DTG. [DOI] [PubMed] [Google Scholar]
- 49.Aidida C, Berrebi D, Peucdhmaur M, Reyes-Mugica M, Altieri DC. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet. 1998;351:882–3. doi: 10.1016/S0140-6736(05)70294-4. [DOI] [PubMed] [Google Scholar]
- 50.Fangusaro JR, Jian Y, Holloway MP, et al. Survivin, Survivin-2B and Survivin-deltaEx32 expression in medulloblastoma: biologic markers of tumour morphology and clinical outcome. Br J Cancer. 2005;92:359–65. doi: 10.1038/sj.bjc.6602317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skog J, Edlund K, Bergenheim AT, Wadell G. Adenoviruses 16 and CV23 efficiently transduce human low-passage brain tumor and cancer stem cells. Mol Ther. 2007;15:2140–5. doi: 10.1038/sj.mt.6300315. [DOI] [PubMed] [Google Scholar]
- 52.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–62. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan MP, Quatela SE, Philips MR, Settleman J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol. 2008;28:2659–74. doi: 10.1128/MCB.01661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang WQ, Senger D, Muzik H, et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63:3162–72. [PubMed] [Google Scholar]
- 55.Hingorani P, Zhang W, Lin J, Liu L, Guha C, Kolb EA. Systemic administration of reovirus (Reolysin) inhibits growth of human sarcoma xenografts. Cancer. 2011;117:1764–74. doi: 10.1002/cncr.25741. [DOI] [PubMed] [Google Scholar]
- 56.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther. 2009;17:972–9. doi: 10.1038/mt.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy PS, Burroughs KD, Hales LM, et al. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J Natl Cancer Inst. 2007;99:1623–33. doi: 10.1093/jnci/djm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu L, Baxter PA, Zhao X, et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro Oncol. 2011;13:14–27. doi: 10.1093/neuonc/noq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rudin CM, Poirier JT, Senzer NN, et al. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res. 2011;17:888–95. doi: 10.1158/1078-0432.CCR-10-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wadhwa L, Hurwitz MY, Chévez-Barrios P, Hurwitz RL. Treatment of invasive retinoblastoma in a murine model using an oncolytic picornavirus. Cancer Res. 2007;67:10653–6. doi: 10.1158/0008-5472.CAN-07-2352. [DOI] [PubMed] [Google Scholar]
- 61.Morton CL, Houghton PJ, Kolb EA, et al. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr Blood Cancer. 2010;55:295–303. doi: 10.1002/pbc.22535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–7. [PubMed] [Google Scholar]
- 63.Kirn DH, Wang Y, Le Boeuf F, et al. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther. 2011;11:595–608. doi: 10.1517/14712598.2011.558838. [DOI] [PubMed] [Google Scholar]
- 65.Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010;21:127–34. doi: 10.1016/j.cytogfr.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lun X, Chan J, Zhou H, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18:1927–36. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Contag CH, Sikorski R, Negrin RS, et al. Definition of an enhanced immune cell therapy in mice that can target stem-like lymphoma cells. Cancer Res. 2010;70:9837–45. doi: 10.1158/0008-5472.CAN-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol. 2011;85:6015–23. doi: 10.1128/JVI.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravindra PV, Tiwari AK, Sharma B, Chauhan RS. Newcastle disease virus as an oncolytic agent. Indian J Med Res. 2009;130:507–13. [PubMed] [Google Scholar]
- 70.Reichard KW, Lorence RM, Cascino CJ, et al. Newcastle disease virus selectively kills human tumor cells. J Surg Res. 1992;52:448–53. doi: 10.1016/0022-4804(92)90310-v. [DOI] [PubMed] [Google Scholar]
- 71.Lorence RM, Reichard KW, Katubig BB, et al. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J Natl Cancer Inst. 1994;86:1228–33. doi: 10.1093/jnci/86.16.1228. [DOI] [PubMed] [Google Scholar]
- 72.Csatary LK, Gosztonyi G, Szeberenyi J, et al. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- 73.Freeman AI, Zakay-Rones Z, Gomori JM, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221–8. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 74.Wagner S, Csatary CM, Gosztonyi G, et al. Combined treatment of pediatric high-grade glioma with the oncolytic viral strain MTH-68/H and oral valproic acid. APMIS. 2006;114:731–43. doi: 10.1111/j.1600-0463.2006.apm_516.x. [DOI] [PubMed] [Google Scholar]
- 75.Wang G, Barrett JW, Stanford M, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci USA. 2006;103:4640–5. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–48. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027–36. doi: 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Y, Lun X, Zhou H, et al. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin Cancer Res. 2008;14:1218–27. doi: 10.1158/1078-0432.CCR-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cripe TP, Wang PY, Marcato P, Mahller YY, Lee PW. Targeting cancer-initiating cells with oncolytic viruses. Mol Ther. 2009;17:1677–82. doi: 10.1038/mt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim M, Madlambayan GJ, Rahman MM, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23:2313–7. doi: 10.1038/leu.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–5. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 82.Lun X, Senger DL, Alain T, et al. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546–57. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- 83.Paglino JC, van den Pol AN. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J Virol. 2011;85:9346–58. doi: 10.1128/JVI.00723-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yasmeen A, Zhang L, Al Moustafa AE. Does the vesicular stomatitis virus really have a selective oncolytic effect in human cancer? Int J Cancer. 2010;126:2509–10. doi: 10.1002/ijc.24922. [DOI] [PubMed] [Google Scholar]
- 85.Toyoda H, Wimmer E, Cello J. Oncolytic poliovirus therapy and immunization with poliovirus-infected cell lysate induces potent antitumor immunity against neuroblastoma in vivo. Int J Oncol. 2011;38:81–7. [PubMed] [Google Scholar]
- 86.Studebaker AW, Kreofsky CR, Pierson CR, Russell SJ, Galanis E, Raffel C. Treatment of medulloblastoma with a modified measles virus. Neuro Oncol. 2010;12:1034–42. doi: 10.1093/neuonc/noq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lacroix J, Leuchs B, Li J, et al. Parvovirus H1 selectively induces cytotoxic effects on human neuroblastoma cells. Int J Cancer. 2010;127:1230–9. doi: 10.1002/ijc.25168. [DOI] [PubMed] [Google Scholar]
- 88.Kumar S, Gao L, Yeagy B, Reid T. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther. 2008;10:371–9. [PubMed] [Google Scholar]
- 89.Touchefeu Y, Vassaux G, Harrington KJ. Oncolytic viruses in radiation oncology. Radiother Oncol. 2011;99:262–70. doi: 10.1016/j.radonc.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 90.Nguyen TL, Tumilasci VF, Singhroy D, Arguello M, Hiscott J. The emergence of combinatorial strategies in the development of RNA oncolytic virus therapies. Cell Microbiol. 2009;11:889–97. doi: 10.1111/j.1462-5822.2009.01317.x. [DOI] [PubMed] [Google Scholar]