

Abstract
Soluble guanylyl/guanylate cyclase (sGC) converts GTP to cGMP after binding nitric oxide, leading to smooth muscle relaxation and vasodilation. Impaired sGC activity is common in cardiovascular disease and sGC stimulatory compounds are greatly sought. sGC is a 150 kDa heterodimeric protein with two H-NOX domains (one with heme, one without), two PAS domains, a coiled-coil domain and two cyclase domains. Binding of NO to the sGC heme leads to proximal histidine release and stimulation of catalytic activity. To begin understanding how binding leads to activation, we examined truncated sGC proteins from Manduca sexta (tobacco hornworm) that bind NO, CO and stimulatory compound YC-1, but lack the cyclase domains. We determined the overall shape of truncated Ms sGC using analytical ultracentrifugation and small angle X-ray scattering (SAXS), revealing an elongated molecule 115 Å by 90 Å by 75 Å. Binding of NO, CO or YC-1 had little effect on shape. Using chemical cross-linking and tandem mass spectrometry, we identified 20 intermolecular contacts, allowing us to fit homology models of the individual domains into the SAXS-derived molecular envelope. The resulting model displays a central parallel coiled-coil platform upon which the H-NOX and PAS domains are assembled. The β1 H-NOX and α1 PAS domains are in contact and form the core signaling complex, while the α1 H-NOX domain can be removed without significant effect on ligand binding or overall shape. Removal of 21 residues from the C-terminus yields a protein with dramatically increased proximal histidine release rates upon NO binding.
Nitric oxide (NO) regulates numerous vital functions in animal physiology including blood pressure, memory formation, platelet aggregation, angiogenesis and tissue development.1 Dysregulation of NO signaling contributes to cardiovascular disease, cancer, poor wound healing, diabetes, asthma, and aging. NO is produced through the conversion of L-arginine to L-citrulline by nitric oxide synthase (NOS)2, 3 and may function in the cell where it is produced or in nearby cells (autocrine/paracrine signaling). The primary NO receptor is soluble guanylyl/guanylate cyclase (sGC), a heterodimeric protein of ~150 kDa that binds NO through a ferrous heme.4 NO binding stimulates cyclase activity, the production of cGMP from substrate GTP and the subsequent amplification of NO-dependent signaling cascades. In smooth muscle cells, this leads to a reduction in free cytosolic calcium concentration and smooth muscle relaxation, a mechanism closely tied to the regulation of blood pressure. While regulation of NOS is relatively well studied,5 the mechanisms underlying sGC regulation are poorly understood.6
Improving blood flow and lowering blood pressure in cardiovascular disease, the number one cause of death in the Western world, has long been a treatment goal. While some success has been achieved, only ~43% of those treated for hypertension have their condition under control.7 Overall, 36% of the adult population suffers from cardiovascular disease, a value that rises to 72% for those over age 60, and >80% for those over age 80. Since nitric oxide both lowers blood pressure and improves blood flow through its vasorelaxation and antiplatelet activities, NO signaling has long been a target for treating cardiovascular disease. For example, organic nitrates such as nitroglycerin, which is metabolized to release NO, have a >130 year history in treating angina pectoris.8 While current treatments are successful for some patients, some do not respond and many who do develop tolerance to the compounds, which then become ineffective.
Efforts to discover new treatments are increasingly focused on sGC, which is compromised in all forms of cardiovascular disease. One promising avenue for treatment involves compounds related to YC-1, a benzylindazole derivative, which stimulate sGC directly and act synergistically with NO binding.9 Several such compounds have entered pre-clinical or clinical trials, one of which (Riociguat) has reached phase III clinical trials.10, 11 While YC-1-family compounds provide a promising step forward, how they bind to sGC and how they stimulate catalytic activity is unknown. A second promising avenue involves compounds targeted to oxidized sGC, which loses heme during inflammation and is then degraded.12, 13 The new compounds replace missing heme, stimulate activity and stabilize the protein, preventing degradation.
The most common form of sGC is a heterodimeric enzyme with one α1 subunit of ~77 kDa and one heme-containing β1 subunit of ~70 kDa; α2 and β2 subunits also occur with an α2β1 complex of particular importance in the brain.4, 14 Each subunit consists of four domains: an N-terminal H-NOX (Heme-Nitric oxide / OXygen binding) domain,15 a central PAS (Per-ARNT-Sim) domain,16 a coiled-coil, and a C-terminal catalytic domain. Binding of NO to the ferrous b-type heme in the β1 H-NOX domain leads to dissociation of the proximal histidine and stimulation of cyclase activity. Binding of carbon monoxide (CO) can also stimulate sGC upon binding; however, stimulation is weak in the absence of YC-1 or related compounds and, importantly, does not require proximal histidine dissociation.17, 18
Understanding of NO, CO and YC-1 allosteric stimulation of sGC is impeded by the lack of sGC crystal structures. Insight is available through homology models for individual domains based on crystal structures of bacterial homologues. The H-NOX domain has been most heavily studied in this regard with crystal structures determined for the H-NOX from T. tengcongensis19, 20 and from Nosotoc sp.21 The crystal structure of the PAS domain from Nostoc punctiforme, which shows high homology with the sGC PAS domains, has also been determined.22 Additionally, the structure for a β1 coiled-coil homodimer from rat has been determined,23 and the structure of the α1/β1 heterodimeric cyclase domain is available (PDB entry 3UVJ, unpublished). However, the arrangement of these domains in sGC and the mechanism by which binding of stimulatory molecules leads to activation remains unknown.
We have developed truncated forms of guanylyl cyclase from the tobacco hornworm / hawkmoth (Manduca sexta) for biophysical study.24–26 Ms sGC is highly homologous to mammalian sGC proteins and responds to NO, CO and YC-1-compounds. Constructs lacking the α1/β1 catalytic domains can be bacterially expressed, leading to heterodimeric protein that is fully loaded with ferrous heme and which displays the expected heme spectra and ligand binding properties. Of particular interest is that C-terminally truncated Ms sGC retains allosteric response to YC-1-family compounds. Binding of YC-1 or BAY 41-2272 leads to an apparent closing of the heme pocket and enhanced CO and NO binding.25 Here, we have investigated domain-domain contacts in truncated Ms sGC using chemical cross-linking and high-resolution mass spectrometry, and determined a molecular envelope for the protein using small angle X-ray scattering (SAXS). These data have allowed us to assemble the first model for heterodimeric sGC and to probe the mechanism underlying allosteric regulation.
MATERIALS AND METHODS
Design and cloning of Ms sGC constructs
Several C-terminal truncations of Manduca sexta α1β1 sGC were utilized in the present study (Fig. 1). The N-terminal construct containing α1 49–450 and β1 1–380, referred to as Ms sGC NT13, was expressed in E. coli from a pET-Duet-1 vector as previously described.26 Construct Ms sGC NT19 contains the same α1 and β1 sequence as NT13, but has an additional C-terminal Strep purification tag (WSHPQFEK) on the α subunit. This was cloned by PCR amplification from the Ms sGC NT13 template using forward primer 5'-gatcggcgtggctagcttctgcaaagcgtttccatggc-3', and reverse primer 5'-gcgagcaaagcagtagacaaggaacgagagaagacctggagccacccacaattcgaaaaatgaaagcttagcctt-3'. The PCR product was cloned into the p-GEM T Easy vector, removed with restriction endonucleases NheI and HindIII, and subsequently ligated into the pETDuet-NT13 construct using the same restriction enzymes. The final construct, pETDuet-NT19, was transformed into pLysS cells. Construct Ms sGC NT21, which lacks the α1 H-NOX domain, was prepared by first excising the α1 subunit from the Ms sGC NT13 pET Duet1 vector with restriction endonucleases BamHI and NotI, purifying on a 1% agarose gel, excising the 6.6 kb gel band, and purifying with a GeneJET Gel Extraction Kit (Fermentas). The α1 subunit residues 272–699 from construct CT1 (unpublished data, footnote 2) was excised with BamHI and NotI, gel purified, and ligated into the pET-Duet1 vector containing NT13 β1 using a Rapid DNA Ligation kit (Thermo Scientific). A stop codon was inserted into the CT1 α1 sequence at position 451 using primer 5'-caaggaacgagagaagtaagtcagcctgctgcatttaatattcc-3'. Sequencing with pET-Duet1 primers UP1 (5'-atgcgtccggcgtaga-3') and UP2 (5'-ttgtacacggccgcataatc-3') confirmed appropriate sequence for expression of α1 272–450 and β1 1–380.
Figure 1.
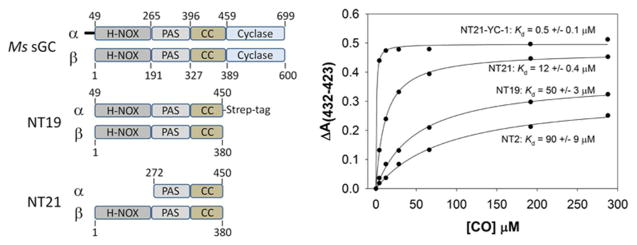
Ms sGC domain boundaries, expression constructs and equilibrium CO titration measurements. (Left) Shown are the predicted domain boundaries and boundaries for constructs NT19 and NT21. Not shown are constructs NT1 (α1 1–471, β1 1–401), NT2 (α1 49–471, β1 1–401) and NT13 (same as NT19 but without Strep-tag). (Right) Representative saturation binding curves. NT21 responds to YC-1 with a 6-fold tightening of the CO-dissociation constant. Also examined were NT2 + YC-1 (KdCO/YC-1 = 1.0 ± 0.1), NT13 (KdCO = 42 ± 5), NT13 + YC-1 (KdCO/YC-1 = 0.9 ± 0.1), and NT19 + YC-1 (KdCO/YC-1 = 0.8 ± 0.1).
Purification of recombinant M. sexta sGC from E. coli
Ms sGC NT13, NT19 and NT21 were expressed in E. coli and purified by a previously described procedure24, 26 with the following modifications. NT19 was expressed at 30 °C in BL21(DE3)pLysS cells, lysed by French press and the resulting supernatant applied to a pre-packed Ni2+-NTA affinity column (GE Healthcare, Piscataway, NJ). Protein was eluted from the Ni2+-NTA resin with 30 mM EDTA, loaded directly onto a StrepTactin Sepharose High Performance column (GE Healthcare, Piscataway, NJ), washed with equilibration buffer, eluted in approximately 1 mL of equilibration buffer containing 2.5 mM desthiobiotin, and loaded onto a sephacryl S-200 gel filtration column (GE Healthcare, Piscataway, NJ). NT21 expressed optimally from Rosetta(DE3)pLysS cells at 16 °C with shaking at 90 rpm for 15 h. Purification was as for NT13, using Ni2+-NTA and size exclusion chromatography. All gel filtration steps for NT13, NT19 and NT21 contained 1 mM tris(2-carboxyethyl)phosphine (TCEP) as a reductant to prevent aggregation.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman XL-1 analytical ultracentrifuge. Each two-sector sample cell contained 7.5 μM NT13 in one chamber (A280 ≈ 1.0) and sample buffer (50 mM KPO4 pH 7.4, 200 mM KCl, 5% glycerol) in the other as reference. For complexes containing CO and YC-1, NT13 was diluted into CO-saturated buffer containing 50 μM YC-1 (A330 ≈ 1.0). Spectra were measured pre- and post-ultracentrifugation to ensure that no protein had been lost to precipitation, and that CO had not been lost during the experiment. Centrifugation was performed at 40,000 rpm for 12 h at 4 °C and absorbance measured every 15 min at 280, 330 or 430 nm. Data were fitted using SEDFIT27 with the following parameters calculated by SEDNTERP:28 buffer density, 1.024 g/cm3; viscosity, 0.0189 g·cm−1·s−1 (Poise).
Chemical cross-linking and protein digestion
Cross-linking reagents BS2G (bis(sulfosuccinimidyl)glutarate-d0), BS3 (bis(sulfosuccinimidyl)suberate-d0), EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) and BMOE (bis(maleimido)ethane) were purchased from Pierce (Rockford, IL). Sequencing-grade trypsin was purchased from Promega (Madison, WI) and all other chemicals were purchased from Sigma-Aldrich unless otherwise stated. The homo-bifunctional cross-linking reagents BS2G and BS3 were prepared individually as stock solutions (50 mM) in DMSO shortly prior to addition, and added with a final concentration of 1 mM to samples containing 20 μM Ms sGC NT13 or NT19 (in 50 mM potassium phosphate buffer, pH 7.4, 300 mM KCl). All final reaction volumes were 50 μL. For the hetero-bifunctional cross-linker EDC, a stock solution was prepared containing 1 M EDC and 2.5 M NHS (N-hydroxyl succinimide) and was added to a 20 μM Ms sGC NT13 sample, leading to a final concentration of 20 mM EDC and 50 mM NHS, and a final volume of 50 μL. The reactions were carried out on ice for 1 h and were quenched by addition of NH4HCO3 (20 mM final concentration). Reactions with BMOE contained 70 μM NT19 and 6-fold excess cross-linker, were reacted for 1 h at room temperature, and quenched with the addition of 5 mM DTT. Cross-linked protein samples were visualized on a precast 10% polyacrylamide gel (Biorad), and stained with Coomassie brilliant blue. In-gel trypsin digestion was performed following the Mann protocol.29
Mass spectrometry for cross-linked peptide identification
An LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) fronted with a Proxeon nanoEasy HPLC (Thermo Fisher Scientific) coupled using a NanoMate nanospray source (Advion, Ithaca, NY) was used to analyze the tryptic digests from SDS-PAGE. The peptide sample was first loaded on a trap column (100 μm × 2 cm, C-18, Easy column, Thermo Fisher Scientific) with 100% solvent A (0.1% formic acid) at a flow rate determined by maximum 280 bar pressure loading, generally 20 μL/min. The sample was then separated on a nano column (75 μm × 10 cm, C-18, Easy column) using a 50 min gradient from 5% B (0.1% formic acid in acetonitrile) to 35% B, followed by a 5 min gradient to 85% B and then a 5 min gradient to 95% B at 300 nL/min. The peptides were directly introduced into the LTQ Orbitrap Velos using the NanoMate with the spray voltage set at 1.77 kV. To identify the cross-linked peptides, data-dependent MS/MS analysis (m/z 350–2000) was performed using MS acquisition software (Xcalibur 2.1, Thermo Fisher Scientific) in which a full high resolution MS scan at 30,000 resolution was followed by a maximum of ten MS/MS scans of the ten most intense precursor ions with charge states ≥ 4. In order to perform MS/MS of the less abundant ions, dynamic exclusion was set to select and fragment those ions once and then place the selected ion on an exclusion list for 45 s. Precursor ions were selected and excluded using the monoisotopic precursor selection (MIPS) feature of the LTQ Orbitrap Velos, which allowed a selected ion width of 10 ppm and required that the isotope pattern of the selected precursor fit a model for peptide ions calculated for similar mass. For MS/MS, precursor ions were selected with a mass width of 4 amu, and were fragmented in the ion trap at 50% relative energy with 30 ms activation time before transfer to the Orbitrap for mass measurement at 7,500 resolution. MS/MS spectra were converted to *.dta files using Thermo Proteome Discoverer 1.2 (Thermo Fisher Scientific) for de novo sequencing.
MS/MS data analysis
Tandem mass spectral data were converted into peak lists (.dta files), deconvoluted, and searched by an in-house program.30 Briefly, a list of potential peptides from trypsin digest was generated, and masses of linear peptide combinations that included cross-linker masses were searched against the corresponding peak list files. Calculated masses of b- and y-ion fragments were searched against the MS/MS peak list files, and lists of identified matches were scored with respect to the number of fragments identified. Independent verification of each match was made by manual comparison of the raw data from Xcalibur (Thermo Fisher) with MS/MS fragment lists from GPMAW (Lighthouse Data) and/or Protein Prospector.31 Cross-linked peptides were considered reliable if 10 or more fragments were identified, the calculated and experimental cross-linked peptide masses were agreeable, if fragments were observed that contain the cross-linker and portions of both peptides, and if the same cross-linked peptide was found in multiple scans.
Small-angle X-ray scattering and shape reconstruction
Small-angle X-ray scattering (SAXS) data were measured at Stanford Synchrotron Radiation Lightsource Beamline 4-2. Proteins were confirmed to be monodisperse at concentrations used for SAXS by dynamic light scattering (DLS), using a Wyatt DynaPro NanoStar. DLS size distribution histograms were calculated with DYNAMICS 6.12 (Wyatt Technology Corporation). Prior to SAXS measurement, Ms sGC NT proteins were concentrated using a Vivaspin 50 kDa cutoff filter (Sartorius Stedim Biotech, Goettingen, Germany). During the final concentration step, the proteins were washed with fresh gel filtration buffer three times and the filtrates collected for use in the blank SAXS measurements to ensure the buffer composition of the blank matched that of the sample. SAXS samples were prepared by diluting protein with the blank buffer supplemented with 1 mM fresh TCEP in series from 10 mg/mL to 1 mg/mL. CO complexes were prepared by dilution with a buffer saturated with CO gas. NO complexes were prepared by addition of a 100 μM solution of DEA/NO dissolved in 10 mM NaOH. YC-1 and BAY 41-2272 complexes were prepared by addition of a 4 mM solution of YC-1 or BAY 41-2272 in DMSO to the sample before addition of protein. Final DMSO concentrations in each sample never exceeded 2%. Buffer subtraction and data averaging were performed using SASTOOL.32 Molecular weights were calculated using an SSRL program based on the method of Orthaber, et.al.33 Rg and I(s) analyses were performed using PRIMUS34, and p(r) distribution function analysis was performed using GNOM.35 Quality scores based upon the Guinier plot were calculated using AutoRg.36 Ab initio shape reconstruction was performed, both with and without symmetry restraints, using DAMMIN or GASBOR.37 Averages of ten or more models were created using DAMAVER.38 UltraScan39 was used to calculate the sedimentation coefficient and stokes radius for the bead model shape reconstructions computed from the SAXS envelopes. CRYSOL40 was used for comparing model and experimental scattering curves. Molecular envelopes were displayed using Chimera.41
Homology modeling
Homology models of individual domains were generated using the bioinfobank meta server.42 After domain modeling, 20% of the protein remained unmodeled. To obtain models for this region, the sequence was submitted to the Robetta server: (http://robetta.bakerlab.org).43 For the individual domains, the models generated with Robetta were similar to those generated through homology modeling. In regions outside the domains, if the secondary structure was predicted to be helical or beta sheet, residues in those regions were constrained to adopt that conformation. Homology models were displayed with either Chimera41 or PyMol (Delano Scientific, San Carlos, CA, http://www.pymol.org/).
Model assembly
The Ms sGC-NT19 model was built in an incremental fashion. First the conformation of the coiled-coil was determined, the β1 H-NOX, α1 PAS, α1 H-NOX, and β1 PAS were then incrementally added. In each step, candidate interfaces between the two components were generated using ZDOCK.44 The rotational sampling were set to be dense (-D parameter) and 50,000 conformations were generated for each dock run. The conformations that satisfied the cross-linking restraints were selected and one of those was taken forward for adding the remaining components. Finally, the model was refined using molecular dynamics in a structure-based force field45 with two biasing forces derived from the SAXS envelope and the cross-linking restraints. The former biasing force was employed using the flexible fitting program MDfit,46, 47 as used previously in a cryo-electron microscopy fitting study.48 MDfit is based on Gromacs49 and allowed us to simultaneously incorporate the later biasing of cross-linking restraints, which were modeled as distance restraints in Gromacs with the force constant 1000 kJ mol−1 nm−2.
The docking process started by identifying the coiled-coil conformation, since most cross-links included residues in this region. Among the 50,000 coiled-coil conformations generated using ZDOCK, many satisfied the cross-linking restraints. In order to select the best model based on the interactions energies, rescoring was performed using ZRANK50 and the top 1000 conformations were selected; only 86 of those conformations were found consistent with the cross-linking restraints. These conformations were then clustered into ten bins based on RMSD. The top-ranked model, which also belongs to the largest cluster, was then selected for docking with the β1 H-NOX domain. Docking of coiled-coil with β1 H-NOX resulted in a single cluster (~40 conformations among the 50,000 generated) that satisfied all four cross-linking restraints and the lowest-energy conformer among these was chosen. The coiled-coil/β1 H-NOX model was then docked with α1 PAS and α1 H-NOX domains, separately. Both dockings resulted in multiple candidate orientations that satisfied the cross-linking restraints, 4 and 50 candidates for α1 PAS and α1 H-NOX domains, respectively. These candidates were then combined in all possible combinations (200) and conformations with steric clashes between the α1 PAS and α1 H-NOX domains were removed, leading to 36 models containing the coiled-coil, α1 H-NOX, β1 H-NOX and α1 PAS domains. Further consideration of chain connectivity, cross-linking restraints and SAXS envelope fitting using the refinement procedure described above identified four potential models. The fittings of the models into the SAXS envelope were visually inspected and the most promising model was selected. To obtain the final Ms sGC-NT19 model, the β1 PAS domain was initially intuitively added to the most promising model, due to the lack of cross-linking restraints, and the new model refined using the protocol described above. The final model satisfies all of the experimental restraints and fits well in the SAXS envelope, but does not include residues α1 267–279 and β1 183–194, which link the H-NOX and PAS domains, or residues α1 391–406 and β1 317–336, which link the PAS and coiled-coil domains, since reliable models for these regions were unavailable. Also missing are the 6xHis and Strep purification tags.
CO binding affinity
CO dissociation constants for Ms sGC NT constructs were measured using a previously published procedure.24 The Ms sGC NT protein samples were prepared at 1 μM concentration in 50 mM KPO4 pH 7.4, 100 mM KCl, 5% glycerol, and placed in a septum-capped cuvette at room temperature (~22 °C). Buffer saturated with CO gas was assumed to contain 1 mM CO, based on CO solubility in water at room temperature.51 Aliquots from CO saturated buffer were added to the cuvette, mixed, and the spectrum measured. When present, compounds YC-1 (50 μM) or BAY 41-2272 (5–10 μM) were added before addition of CO. Binding of CO to heme was measured by the shift in unliganded Soret absorbance (430–434 nm) to that of the CO complex (423–424 nm) after accounting for dilution due to the addition of CO. Data were fitted to a single-site saturation ligand binding model using SigmaPlot (SPSS, Inc., Chicago, IL).
Proximal histidine release rates upon NO binding to heme
The rates for release of β1 His-105 upon NO binding to Ms sGC NT1, 2, 13, 19 and 21 were measured at 10 °C by mixing 1 μM protein and 10 μM NO in a RSM-1000 stopped-flow spectrophotometer (OLIS, Inc.), using a previously published procedure.24 Protein samples were prepared by deoxygenating buffer through bubbling of argon gas for at least 20 min, followed by addition of protein in a gas tight-syringe, and transfer to the stopped-flow device. NO solutions were prepared by addition of DEA/NO from a stock solution to argon-purged buffer in a gas-tight syringe and then connected to the stopped-flow device. DEA/NO decomposition was allowed to proceed for 20 min at room temperature before transfer to the instrument, where the solution was allowed to equilibrate to the desired temperature (5 min). Absorbance changes (A420) were fitted to single- or double-exponential equations using SigmaPlot; values reported are the average and standard deviation of 3 to 5 consecutive measurements.
RESULTS
Development of truncated sGC proteins for functional analyses
Functional studies of sGC are impeded by difficulty with obtaining robust material. We have developed sGC from Manduca sexta (Ms sGC) as a model system and have produced truncated recombinant heterodimeric proteins in sufficient quantity and purity for biophysical studies.24, 26 For the present studies, we employed sGC truncations lacking the C-terminal regions of both subunits, including the catalytic domains, and portions of the N-terminal region of the α1 subunit (Figure 1). The truncated proteins have ferrous heme, display CO and NO binding characteristics similar to that of the full-length enzyme, and retain binding of the allosteric stimulators YC-1 and BAY 41-2272.24 Construct NT224 contains the H-NOX, PAS, and coiled-coil domains of Ms sGC α1 and β1 (a 49–471, β 1–401), while truncation of ~20 residues from the C-terminus of both subunits leads to the more stable construct NT13 (α1 49–450, β1 1–380).26 For SAXS experiments, which require high purity and monodispersity at high concentration, we developed construct NT19, which contains an additional Streptavidin purification tag (WSHPQFEK). The additional purification step led to a protein with extremely high purity and which was monodisperse at concentrations up to 8 mg/mL as estimated by dynamic light scattering (see below). We also prepared protein NT21, which lacks the α1 H-NOX domain, to investigate the role of this domain and to help with identifying domain arrangement in the protein. This protein was also prepared in high purity and was monodisperse at high concentration.
As with NT2 and NT13,24, 26 NT19 and NT21 were heterodimeric and displayed typical Soret maxima for unliganded (433 nm), NO liganded (400 nm) and CO liganded (423 nm) complexes. The A432/A280 ratio was 1.6:1 and 1.9:1 for NT19 and NT21, respectively, indicating high – likely complete – heme incorporation. CO binding was slightly tighter to NT21 than to NT19 or NT2 (Figure 1). Interestingly, NT21 still responds to YC-1 (Figure 1), indicating YC-1does not bind to the α1 H-NOX domain.
Chemical cross-linking and mass spectrometry
We undertook chemical cross-linking coupled with high-resolution mass spectrometry to determine close contacts between sGC domains. Such methods can provide powerful restraints for model building.52 We chose the readily available amine-reactive cross-linkers BS2G and BS3, and the amine-carboxyl-reactive cross-linker EDC for these studies. The succinimidyl esters of BS2G and BS3 react with two nearby lysine residues, providing covalent attachments that can later be identified by mass spectrometry. The carbon spacer arms between the reactive groups differ in length (7.7 Å for BS2G, and 11.4 Å for BS3), allowing for the capture of lysines separated by differing distances. The EDC cross-linker is zero-length; one end reacts with free carboxyl groups of aspartate or glutamate, and the other with nearby lysine side chains, leading to a peptide bond between the two amino acids. It is therefore useful for detecting intermolecular salt bridges.
Both Ms sGC NT13 and Ms sGC NT19 proved amenable to cross-linking. These constructs differ by the presence of a C-terminal strep tag on the α-subunit of NT19. Covalently linked α1β1 subunits were readily identified on denaturing polyacrylamide gels (Figure 2A), allowing for the bands to be cut out and subjected to in-gel trypsin digestion, followed by mass spectrometry (Figure 2B). The peptides were analyzed on a hybrid linear ion trap-Orbitrap instrument and high charge-state peptides (≥4+) were selected for collision induced dissociation (CID) fragmentation. The observed precursor and fragment masses were matched to masses of predicted peptides. Tables 1 and 2 list, respectively, the 34 intermolecular and 26 intramolecular cross-linked peptides identified for NT13 and NT19; a representative MS/MS spectrum and fragment assignment is displayed in Figure 2B. A cysteine-reactive maleimide cross-linker (BMOE, 8Å) also successfully cross-linked α1 and β1 subunits; however, no cross-linked peptides were identified by our search methods.
Figure 2.
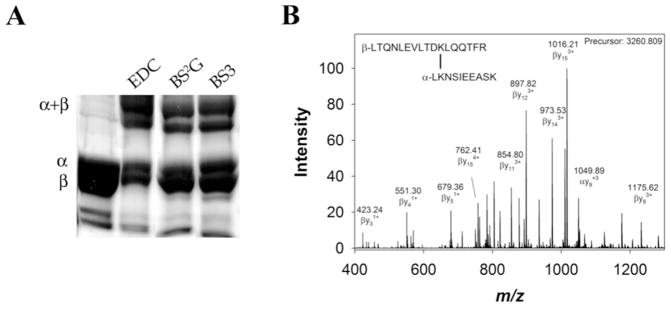
Representative cross-linking results. A) Cross-linked Ms sGC NT13, examined by SDS-PAGE and stained with Coomassie brilliant blue. Individual bands were cut from the gel, digested with trypsin and examined by tandem mass spectrometry. B) Representative cross- linked peptide MS/MS spectrum. BS2G cross-link α1 K434 to β1 K366 was found in the +4 charge state due to two amine termini and lysine/arginine at the trypsin cleavage sites, and displayed a 22 ppm error in observed vs. calculated precursor mass (Table 1). The cross-linked peptide was observed and selected for fragmentation in 7 scans in a single experiment. Between 7 and 36 fragments were identified in each scan, each with a mass accuracy of approximately 1 ppm. Representative fragments are labeled in the mass spectrum. Identified fragments in this spectrum included β1 fragments b4–10, y2–6, y8–15 (which contain the cross-linker and entire α1 peptide), α1 fragments y2–8, and α1 fragments b3–5 (which contain the cross-linker and entire β1 peptide).
Table 1.
α1/β1 Intermolecular Cross-Links
| Cross-linker | α1 Peptide | Residue | β1 Peptide | Residue | Precursor Mass (MH+) | Error (ppm) |
|---|---|---|---|---|---|---|
| BS2G (7.7 Å) | 92–101 | K98 | 356–372 | K366 | 3343.808 | 16 |
| 254–273 | K255 | 28–40 | K28 | 3585.800 | 23 | |
| 254–273 | K255 | 356–372 | K366 | 4211.143 | 7 | |
| 430–434 | K432 | 1–26 | K15 | 3899.969 | 12 | |
| 429–434 | K432 | 356–372 | K366 | 2932.624 | 11 | |
| 433–442 | K434 | 342–366 | K366 | 4156.138 | 12 | |
| 433–442 | K434 | 356–372 | K366 | 3260.809 | 22 | |
| 435–448 | K442 | 356–372 | K366 | 3717.969 | 11 | |
| 443–448 | K446 | 356–372 | K366 | 2859.543 | 8 | |
|
| ||||||
| BS3 (11.4 Å) | 92–101 | K98 | 373–380 | K378 | 2413.274 | 4 |
| 340–344 | K343 | 169–188 | K170 | 3090.564 | 3 | |
| 430–434 | K432 | 356–372 | K366 | 2818.544 | 2 | |
| 429–434 | K432 | 356–372 | K366 | 2974.649 | 0 | |
| 429–442 | K432 | 356–372 | K366 | 3833.035 | 6 | |
| 430–434 | K432 | 373–380 | K378 | 1787.979 | 7 | |
| 429–434 | K432 | 373–380 | K378 | 1944.078 | 8 | |
| 429–442 | K432 | 373–380 | K366 | 2802.469 | 12 | |
| 433–442 | K434 | 356–372 | K366 | 3302.785 | 3 | |
| 433–442 | K434 | 373–380 | K380 | 2272.224 | 6 | |
| 435–446 | K442 | 356–372 | K366 | 3474.920 | 25 | |
| 435–448 | K442 | 373–380 | K378 | 2729.413 | 6 | |
| 443–448 | K446 | 356–372 | K366 | 2901.578 | 0 | |
| 435–448 | K446 | 356–372 | K366 | 3759.980 | 2 | |
| 443–448 | K446 | 373–380 | K380 | 1871.003 | 10 | |
| 449-Strep9 | K450 | 373–380 | K378 | 2570.316 | 3 | |
| 449-Strep9 | Strep-K9 | 356–372 | K366 | 3600.876 | 1 | |
| Strep1–9 | Strep-K9 | 356–372 | K366 | 3343.730 | 4 | |
| Strep1–9 | Strep-K9 | 373–380 | K378 | 2313.175 | 5 | |
|
| ||||||
| EDC (0 Å) | 254–273 | E256 | 28–40 | K28 | 3529.734 | 12 |
| 277–293 | K286 | 189–207 | E196 | 4087.173 | 15 | |
| 340–344 | E340 | 169–188 | K170 | 2934.515 | 5 | |
| 355–368 | E366 | 373–380 | K378 | 2414.309 | 10 | |
| 412–421 | E418 | 73–95 | K85 | 3714.971 | 27 | |
| 443–448 | K446 | 16–27 | E20 | 3083.537 | 6 | |
Table 2.
Intramolecular Cross-links
| α1 Intramolecular Cross-Links
| ||||||
|---|---|---|---|---|---|---|
| Cross-linker | α1 Peptide | Residue | α1 Peptide | Residue | Precursor Mass (MH+) | Error (ppm) |
| BS2G (7.7 Å) | 115–127 | K122 | 196–220 | K216 | 4217.293 | 12 |
| 217–221 | K220 | 241–255 | K253 | 2483.419 | 12 | |
| 254–273 | K255 | 287–297 | K296 | 3580.778 | 18 | |
|
| ||||||
| BS3 (11.4 Å) | 115–127 | K122 | 196–220 | K216 | 4259.298 | 5 |
| 123–142 | K127 | 217–221 | K220 | 3097.607 | 9 | |
| 217–221 | K220 | 254–273 | K253 | 2222.257 | 8 | |
| 217–221 | K220 | 241–255 | K253 | 2525.432 | 5 | |
| 217–221 | K220 | 254–273 | K255 | 2821.421 | 3 | |
| 254–273 | K255 | 287–297 | K296 | 3680.792 | 4 | |
| 254–273 | K255 | 428–434 | K432 | 3225.611 | 6 | |
| 287–297 | K296 | 340–344 | K343 | 2213.222 | 5 | |
| 430–434 | K432 | 435–448 | K442 | 2347.212 | 7 | |
| 429–434 | K432 | 443–448 | K446 | 1644.908 | 8 | |
| 430–434 | K432 | 449-Strep9 | K450 | 2188.110 | 5 | |
| 430–434 | K432 | Strep1–9 | Strep-K9 | 1930.970 | 7 | |
| 429–434 | K432 | Strep1–9 | Strep-K9 | 2087.076 | 4 | |
| 433–442 | K434 | 443–448 | K446 | 1973.044 | 11 | |
| 433–442 | K434 | 449-Strep9 | K450 | 2672.359 | 3 | |
| 435–446 | K442 | 449-Strep9 | K450 | 2844.400 | 5 | |
| 435–448 | K442 | 449-Strep9 | K450 | 3129.544 | 5 | |
| 443–448 | K446 | 449-Strep9 | K450 | 2271.136 | 6 | |
|
| ||||||
| EDC (0 Å) | 243–253 | K253 | 254–273 | E256 | 3418.716 | 18 |
| 243–253 | K253 | 254–273 | E264 | 3418.744 | 26 | |
| β1 Intramolecular Cross-Links
| ||||||
|---|---|---|---|---|---|---|
| Cross-linker | β1 Peptide | Residue | β1 Peptide | Residue | Precursor Mass (MH+) | Error (ppm) |
| BS2G (7.7 Å) | 1–26 | K15 | 152–163 | K156 | 4592.254 | 10 |
| 373–380 | K378 | 356–372 | K366 | 3158.749 | 17 | |
|
| ||||||
| BS3 (11.4 Å) | 356–372 | K366 | 373–380 | K380 | 3200.742 | 3 |
The high mass accuracy of precursor and fragment peptides provided confidence in the assignments; mass errors for cross-linked peptides were generally less than 0.1 AMU (Tables 1 and 2), and mass errors for fragment peptides were ~1 ppm. Cross-linked peptides were considered reliable if 10 or more fragments were identified, the calculated and experimental peptide masses were agreeable, and fragments were observed that contained the cross-linker and portions of both peptides. Additionally, many cross-links were identified in both BS2G and BS3 cross-linking experiments, and in both the NT13 and NT19 experiments, highlighting the reproducibility of our methods.
A summary of the intermolecular cross-links and restraints used in model building can be found in Table 3 and are shown graphically in Figures 3 and 4. It is clear from the extensive cross-links between residues in the predicted coiled-coil region that the coiled-coil likely does form, and, furthermore, has parallel α1β1 strands (Figure 3). Sixteen unique cross-linked peptides were found within the α1β1 coiled-coil, which spans residues α1 419–450 and β1 355–380; an additional cross-link was found in NT19 between β1 Lys 380 and Lys 9 of the C-terminal α1 Strep-tag. Most of these cross-links are only consistent with a parallel coiled-coil model (Figure 3). In particular, cross links formed between α1 442–β1 378, α1 446–β1 380, α1 450–β1 378 and α1 Strep-tag 9–β1380 are 42–49 Å apart in the antiparallel model, far longer than the 23 Å maximum Cα-Cα distance that can be spanned by the BS3 linker. In contrast, the maximum Cα-Cα distance spanned by linkers is 16 Å for the parallel model. This agrees with the conclusions of Ma and co-workers, who preferred a parallel coiled-coil arrangement based on electrostatic arguments despite the crystal structure of the rat sGC β-homodimer containing an antiparallel coiled-coil.23
Table 3.
Summary of Chemical Cross-Links and Modeling Distance Restraints
| α1 residue | Domain | β1 residue | Domain | Distance (Å) |
|---|---|---|---|---|
| K98 | H-NOX | K366 | coiled-coil | 21 |
| K98 | H-NOX | K378 | coiled-coil | 25 |
| K255 | H-NOX | K28 | H-NOX | 21 |
| K255 | H-NOX | K366 | coiled-coil | 21 |
| E256 | H-NOX | K28 | H-NOX | 13 |
| K286 | PAS | E196 | PAS | 13 |
| E340 | PAS | K170 | H-NOX | 13 |
| K343 | PAS | K170 | H-NOX | 25 |
| E366 | PAS | K378 | coiled-coil | 13 |
| E418 | coiled-coil | K85 | H-NOX | 13 |
| K432 | coiled-coil | K15 | H-NOX | 21 |
| K432 | coiled-coil | K366 | coiled-coil | 21 |
| K432 | coiled-coil | K378 | coiled-coil | 25 |
| K434 | coiled-coil | K366 | coiled-coil | 21 |
| K434 | coiled-coil | K380 | coiled-coil | 25 |
| K442 | coiled-coil | K366 | coiled-coil | 21 |
| K442 | coiled-coil | K378 | coiled-coil | 25 |
| K446 | coiled-coil | E20 | H-NOX | 13 |
| K446 | coiled-coil | K366 | coiled-coil | 21 |
| K450 | coiled-coil | K378 | coiled-coil | 25 |
Figure 3.
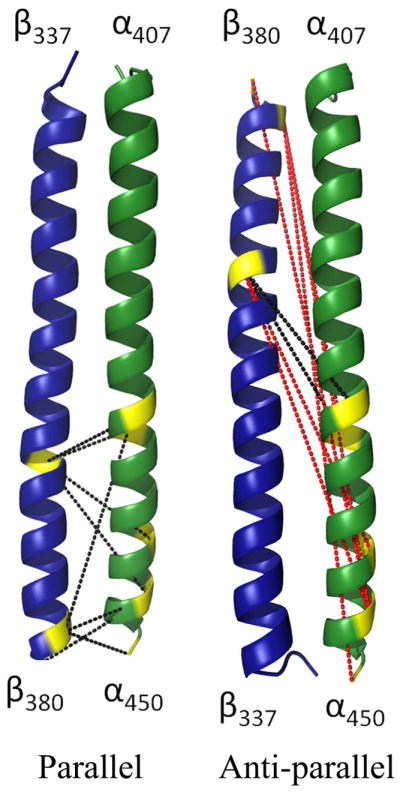
Cross-links within the Ms sGC coiled-coil. Displayed are homology models built for the Ms sGC α1 / β1 coiled-coil in both parallel and anti-parallel orientations. Highlighted in yellow are lysine residues found cross-linked with BS2G or BS3 (7.7 Å and 11.4 Å spacer arms, respectively). Black dashes indicate Ca distances in the model between 6–19 Å, appropriate for cross-linking; red dashes indicate distances between 30–48 Å, unlikely to be found in cross-links. These data confirm a parallel coiled-coil arrangement in sGC.
Figure 4.

Expanded diagram of intermolecular cross-links between Ms sGC α1 and β1 subunits. Displayed are homology models of each Ms sGC domain, with residues found in BS2G, BS3 or EDC cross-links highlighted in yellow and connected by black dashes. Cross-links within the coiled-coil have been removed for clarity.
Nine cross-links were found between the coiled-coil and other domains in Ms sGC NT proteins; eight of these were between the α1 and β1 strands. Five additional cross-links were found between the other domains, four of which were between α1 and β1 subunits. Taken together, these data suggest major inter-domain contacts between subunits on opposite chains. In particular, the α1 PAS domain appears closely associated with the β1 H-NOX domain, which, in turn, is closely associated with the α1 coiled-coil.
Analytical ultracentrifugation indicates sGC is elongated
Little is known about the general shape of sGC or how it changes in response to allosteric effectors. We employed sedimentation velocity analytical ultracentrifugation, which is sensitive to molecular shape, to begin investigating these properties in Ms sGC NT13 and NT21. Each protein sedimented as a single species during the long (~12 h) runs, and yielded a calculated molecular weight generally within 5% of the actual value (Table 4).
Table 4.
Biophysical Parameters Determined by Analytical Ultracentrifugationa
| Complex | S | S (20,w) | MWT (kDa) | Frictional Ratio (f/f0) | Stokes Radius |
|---|---|---|---|---|---|
| NT13 | 2.6 ± 0.10 | 5.5 | 87 | 1.3 | 38 |
| + YC-1 | 2.4 ± 0.12 | 5.0 | 91 | 1.5 | 43 |
| + CO/YC-1 | 2.4 ± 0.08 | 5.0 | 86 | 1.4 | 40 |
|
| |||||
| NT13 SAXS Bead Model | 2.4 | - | - | 1.3 | 38 |
|
| |||||
| NT21 | 2.2 ± 0.02 | 4.5 | 64 | 1.3 | 34 |
| + CO/YC-1 | 2.0 ± 0.03 | 4.0 | 66 | 1.5 | 39 |
Displayed are sedimentation coefficient (S) values at experimental conditions and extrapolated to 20 °C in H2O (S(20,w)), and the molecular weight, frictional ratio, and Stokes radius all computed from the sedimentation coefficient distribution. Each value reported is the average of a single sample measured at multiple wavelengths (400–432 nm for heme Soret; 280 nm for protein backbone; 330 nm for YC-1, when present). The error in S is reported as standard deviation from c(S) distribution peak integration. Error between the raw data and the Lamm equation fit was small in all cases (between 0.009 and 0.015) and is not reported in the table. Each sedimentation velocity experiment was reproduced 2–3 times and the results correlate well (not shown). Hydrodynamic parameters computed from UltraScan for a molecular enveloped for NT13 are consistent with the size measured from ultracentrifugation (not shown). The calculated molecular weights correlate well for both NT13 and NT21 (90.3 kDa and 65.0 kDa, respectively). Data measured in the presence of NO behaved badly and are not included (see text).
Sedimentation was followed by absorption of the heme Soret band (400–432 nm, depending on the complex), the protein (280 nm) or compound YC-1, when present (330 nm). Analysis of the data acquired for each wavelength yielded the same sedimentation coefficients and frictional ratios. Unliganded Ms sGC NT13 displayed a sedimentation coefficient (S) of 2.6, and frictional ratio (f/f0) of 1.3 (Table 4), indicative of an elongated molecule (a spherical molecule would have f/f0 = 1). A similar value (f/f0 = 1.35) was obtained from models derived from SAXS experiments (described below). Sedimentation of Ms sGC NT21, which has the entire α1 H-NOX domain removed (~25 kDa), followed the same trend as NT13 (Table 4). It too behaved as an elongated molecule and has the same frictional ratio of as NT13. Binding of both CO and YC-1 led to an increase in frictional ratio. Thus, the α1 H-NOX domain appears not to have a functional role in YC-1 binding.
The sedimentation velocity data for the NO complexes fit well only when the molecular weight of the protein is significantly underestimated, hence we consider these results unreliable and they have not been included in Table 4. It may be that some denaturing or dissociation of the protein occurs during the several hours of the sedimentation experiment. Similar problems do not occur in the small-angle scattering experiments (see below), which are conducted on a much shorter time scale (minutes versus hours between sample preparation and the end of data collection).
Small-angle X-ray scattering
After confirming that the protein constructs were monomeric in solution by gel filtration, analytical ultracentrifugation, and dynamic light scattering (Figure 5A, B), we performed small-angle X-ray scattering experiments to determine molecular envelopes for Ms sGC-NT constructs and to uncover any large structural changes that might occur upon ligand binding. In SAXS experiments, two parameters can be determined from the scattering curves. First, the radius of gyration (Rg) of the molecule is extracted from the linear relationship between scattering intensity I(s) and angle (s2, Å−2) at low scattering angles.53, 54 Second, Dmax, the maximum radial distance, is extracted from the distribution function, p(r), which is determined by Fourier transform of the scattering intensity and is a representation of the distribution of radial distances throughout the macromolecule.53, 54 We performed SAXS measurements for Ms sGC constructs NT2, NT13, NT19 and NT21 (Table 4). Of these, NT19 and NT21, had the highest purity and least tendency to aggregate at high concentration and yielded the best scattering curves. Both NT19 and NT21 had a slight increase in Rg at concentrations above 5 mg/ml, indicating a slight attractive interaction. For both NT19 and NT21, the molecular mass estimated from I(0) is within 5% of the actual value.
Figure 5.

Representative DLS and SAXS data. DLS size distribution histograms are shown for Ms sGC NT19 (A) and Ms sGC NT21 (B) (2 mg/ml each). The histograms were derived from a regularization analysis of the autocorrelation curve (insets). The intensity autocorrelation (IA) was evaluated as a function of time delay between 1 μs and 0.1 s. Ms sGC NT19 displays 8.0% polydispersity, while NT21 displays 9.6% polydispersity. C) Guinier plots for all NT19 and NT21 complexes, computed from merged data sets (1.3 mg/ml and 5.0 mg/ml, respectively, for the low angle and high angle data). D) Overlay of the NT19 and NT21 pair distance distribution functions p(r), computed using GNOM. E) Overlay of scattering curves for NT19, NT19 + NO, and NT19 + CO and BAY 41-2272. The curves show very little difference in scattering intensity distribution, indicating little change in shape upon complex formation. F) A similar overlay of for NT21 complexes (uncorrected), which also indicate little change in scattering intensity upon complex formation.
Unliganded NT19 has an Rg value of 34 Å and a Dmax value of 115 Å. The molecule is elongated and the calculated frictional ratio (f/f0 = 1.35) agrees with that obtained from ultracentrifugation experiments. Surprisingly, unliganded NT21, which lacks the α1 H-NOX domain, displayed nearly the same Rg and Dmax values as NT19, despite loss of the 25 kDa domain. Addition of NO or CO and BAY 41-2272, or CO and YC-1, had little effect on the scattering profiles or derived quantities for either NT19 or NT21 (Figure 5), indicating that overall shape changes upon YC-1 binding, or NO binding and proximal histidine release, are small. Examination of SAXS intensity decay as a function of scattering angle (Kratky plot), which is sensitive to protein disorder,55 indicates that NT19 and NT21 are well-folded but display some flexibility. Ligand addition had little effect on scattering decay.
Ab initio methods have been developed for reconstruction of low-resolution molecular envelopes for protein molecules using SAXS data. We determined molecular envelopes for NT19 and NT21 using DAMMIN,37 which calculates an arrangement of dummy atoms whose computed scattering profile is minimized against that of the experimental SAXS data.37 Final envelopes were the average of at least 10 starting envelopes, calculated using DAMAVER.38 For NT19, the computed molecular envelope has a volume of 178 × 103 Å3, an appropriate size for two H-NOX and two PAS domains. NT21 has a similar envelope that is narrower at one end and has a calculated volume of 135 × 103 Å3, a difference of 43 × 103 Å3, an appropriate volume difference for a molecule lacking one H-NOX domain. The differing envelope shapes in conjunction with the chemical cross-linking results described above provide guidance for model building (Figure 6).
Figure 6.
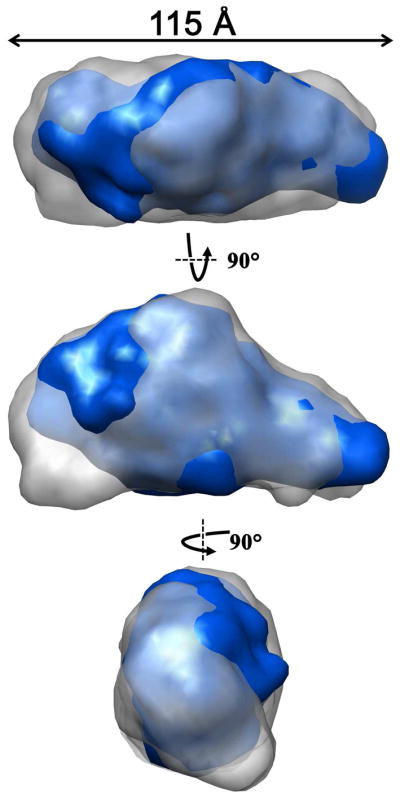
Overlay of DAMMIN reconstructions for NT19 (gray) and NT21 (blue). NT21, which lacks the α H-NOX domain, has a significant volume (~25%) missing from the envelope, although it retains the same Dmax. The straight arrow indicates the maximum length measurement derived from the SAXS analysis. Displayed with Chimera.
The α1 and β1 subunits of sGC arise from gene duplication, suggesting the overall molecular structure may display two-fold pseudosymmetry. However, imposing P2 symmetry in DAMMIN resulted in a significantly poorer chi-square for the fit to the scattering curve, indicating that the Ms sGC-NT constructs are not highly symmetric. As a further check on the reliability of the reconstruction, we calculated the predicted hydrodynamic properties of the NT13 bead model, using UltraScan,39 for comparison with the sedimentation experiments. The predicted values were in good agreement with those measured by ultracentrifugation (Table 4).
Molecular modeling
Homology models of the Ms sGC α1 and β1 H-NOX, PAS and coiled-coil domains were built using Modeller,56 as previously described.24 These models are based on structures of related bacterial H-NOX proteins (2O09),21 PAS proteins (2P04, 2P08),22 and the recent rat β coiled-coil homodimer (3HLS).23 Several intramolecular cross-links were found within domains that were consistent with the homology models, including β1 K15 - β1K156 (β1 H-NOX domain), α1 K122 - α1 K216 and α1 K127 - α1 K220 (a H-NOX domain). No intramolecular cross-links were found that were inconsistent with the homology models. Intervening sequences connecting these domains were built using the Robetta server.43 We considered the Robetta-built domain linker regions to be unreliable, except where helices are predicted to exist.
The domain homology models are expected to have the correct overall folds; however, their arrangement in space is completely unknown. We used a combination of surface matching, cross-link restraints and fitting to the SAXS molecular envelopes to obtain three-dimensional models of Ms sGC-NT19.
Since most cross-links included residues in the predicted coiled-coil region, we began by modeling coiled-coil arrangements. In the crystal structure of the rat β1 coil, a homodimeric antiparallel coiled-coil was found. Nonetheless, the authors concluded that the heterodimeric α1β1 protein is likely parallel, based on sequence matching.23 We examined surface complementarity using program ZDOCK44 and found both parallel and antiparallel arrangements were possible. However, as noted above, only a parallel coiled-coil arrangement is consistent with our cross-linking data (Figure 3).
To add other domains to the model, starting models were generated using ZDOCK,44 culling those that were inconsistent with chain connectivity or obviously in conflict with the cross-linking data, and then using a course-grained flexible-fitting molecular dynamics approach for minimizing deviations from the cross-linking results (Figure 6) while also maximizing agreement with the molecular envelope generated by SAXS. Docking of the β1 H-NOX domain was the most robust in this approach, followed by the α1 PAS domain. 200 starting models were initially generated for the coiled-coil, H-NOX and α1 PAS domains, reduced to 36 models based on geometry considerations and further reduced to 4 models based on the flexible fitting refinement. These 4 were all similar in domain placement, differing only in the rotational positioning of the domains. One of these was taken forward for inclusion of the β1 PAS domain, which was the least constrained (one cross-link plus chain connectivity).
The final model fits well with the SAXS molecular envelope (Figure 7). Comparison of the experimental and calculated scattering curves yields χ = 5.6, a reasonable value considering that domain linkers (~10% of the model) and solvent are missing, the individual domains are based on homology modeling, and the model is fit to the SAXS envelope, not directly to the scattering curve. The most reliable region of the model is the placement of the β1 H-NOX near the center of the parallel coiled-coil region, mostly in contact with the α1 coiled-coil strand. A slight curve was introduced into the coiled-coil during the flexible fitting to the SAXS envelope, around the β1 H-NOX domain. The α1 PAS domain lies close to both the β1 H-NOX domain and the C-termini of the coiled-coil region. This arrangement of the β1 H-NOX near the α1 PAS, organized onto the parallel coiled-coil near where the cyclase domains are connected, is likely to provide the heart of the signaling system, as discussed below.
Figure 7.

Ms sGC model. A) Restrained model for Ms sGC NT19 fitted to the SAXS molecular envelope. B) Stereo view of the restrained model. The α1 subunit is shown in green, β1 in blue. The N- and C- termini and all domains are labeled. Linker regions between individual domains are not included in the model (α1 267–279, α1 391–406, β1 183–194 and β1 317–336).
Placement of the remaining two domains is less reliable but allows for two general observations. First, the α1 H-NOX and β1 PAS positions do not appear to mirror those for β1 H-NOX and α1 PAS, and do not appear to be organized on the coiled-coil in the same way. Second, α1 H-NOX is positioned such that its removal would not overly change the shape of Ms sGC, as expected from the SAXS and analytical ultracentrifugation experiments (Figure 5), nor greatly alter β1 H-NOX activity, as expected from the CO binding experiments (Figure 1).
Stopped-flow kinetic measurements of β1 His 105 release upon NO binding
sGC initially forms a transient six-coordinate intermediate upon binding NO followed by release of the proximal histidine (β1 His 105), yielding a five-coordinate nitrosyl heme. Each of these is readily detected by absorption spectroscopically due to their distinct Soret absorption maxima, which occur at 433, 420 and 400 nm, respectively, for the unliganded, six-coordinate and five-coordinate species. We previously showed that the six-coordinate intermediate persists sufficiently long in Ms sGC NT1 for characterization by stopped-flow spectroscopy, yielding a histidine dissociation rate of 12.8 s−1.24 We examined NO-dependent proximal histidine release for the other NT constructs used in the present study and, surprisingly, found that constructs NT13, NT19 and NT21, all of which are shortened with respect to NT1 and NT2 by 21 residues on α1 and 20 residues on β1, have greatly increased proximal histidine release rates. In all three cases, release was faster than could be measured in our stopped-flow device (Table 6). Additionally, the truncations in NT13/NT19 lead to an ~2-fold lower CO dissociation constant in the absence of YC-1 (Figure 1), and truncation combined with removal of the α1 H-NOX domain in NT21 led to a decrease in CO affinity of nearly 8-fold. The C-terminal truncations are near the end of the predicted coiled-coil region, and close to where the β1 H-NOX domain lies in our model, suggesting these residues are in direct contact with the β1 H-NOX domain and have a packing arrangement that holds the heme domain in a more open conformation (discussed below).
Table 6.
Histidine Release Rates for Six-Coordinate Nitrosyl Complexa
| Ms sGC protein | k6-5 (s ) |
|---|---|
| NT1(ref. 24) | 12.8 ± 0.4 |
| NT1 (this work) | 11.1 ± 0.5 |
| NT2 | 14.5 ± 0.6 |
| NT13 | >100b |
| NT19 | >100b |
| NT21 | >100b |
Rate constants for proximal histidine release from the transient six-coordinate nitrosyl complex to the more stable five-coordinate nitrosyl complex. Measured at 10 °C in a stopped-flow spectrophotometer.
Unobserved.
DISCUSSION
We have produced a molecular model of truncated sGC from Manduca sexta, the first such model for any sGC. We obtained the overall shape of the molecule from SAXS and sedimentation velocity measurements and determined points of domain contact through chemical cross-linking and tandem mass spectrometry. We arranged homology-modeled H-NOX, PAS and coiled-coil domains into the molecular envelope using flexible fitting and cross-linking constraints. The model that emerges from our studies is one in which sGC is an elongated molecule with functional domains organized around a central parallel coiled-coil segment. A key region near the C-terminus of the coiled-coil brings together the β1 H-NOX, α1 PAS and the cyclase domains. This arrangement suggests a model for allostery in which domains inhibit one another through direct contact in a manner that can be overcome by ligand binding to any one of the domains. We discuss the rationale for this model and functional implications in what follows.
Overall shape and ligand-induced conformational changes in sGC
Prior to the present studies, no information was available concerning the overall shape for sGC or how this shape changes during activation. We have shown, using SAXS and SV-AUC, that a truncated heterodimeric sGC containing approximately two-thirds of the protein but lacking the cyclase domains is an elongated molecule of approximately 115 Å by 90 Å by 75 Å. Removal of the α1 H-NOX domain creates a molecule of roughly the same length but somewhat thinner at one end (Tables 4 and 5, Figure 5).
Table 5.
Molecular Size Determined by SAXS4
| Protein | Complex | Guinier plot RG (Å)a (quality)b | Dmax (Å) | Vol. (Å3)c |
|---|---|---|---|---|
| NT19 | Unliganded | 33.7 ± 0.03 (0.93) | 115 | 178 × 103 |
| NO | 33.6 ± 0.02 (0.94) | 115 | - | |
| CO/YC-1 | 33.6 ± 0.03 (0.82) | 115 | - | |
| NT21 | Unliganded | 33.0 ± 0.02 (0.86) | 115 | 135 × 103 |
| NO | 32.0 ± 0.02 (0.82) | 115 | - | |
| CO/YC-1 | 30.7 ± 0.03 (0.80) | 115 | - |
RG: Radius of gyration.
Quality score from program AutoRg based upon the Guinier plot.
Volume of the bead model calculated by DAMAVER.
sGC is an allosteric protein in which the binding of NO or YC-1-family compounds leads to cyclase activation,18, 57 while modifications such as phosphorylation lead to cyclase inhibition.58–60 Likewise, binding of nucleotide to the cyclase domains can alter NO binding to the sGC heme.61–63 The truncated proteins used in the present study retain allosteric response to YC-1, which, on binding, leads to higher affinity and reduced off rates for CO and NO (references24, 25, Figure 1). To probe the nature of the conformational changes associated with allosteric response, we undertook SAXS and SV-AUC measurements both in the absence and presence of allosteric compounds. For the SAXS measurements, the Ms sGC NT proteins were monodisperse under all conditions examined, despite the higher concentrations required (up to 150 μM). These measurements indicated only minor changes in shape take place upon ligand binding (Table 5).
In contrast, the SV-AUC measurements indicate a small decrease in sedimentation coefficient on binding CO and YC-1 (Table 4). Such deviations are often interpreted as changes in protein conformation, but can also be due to changes in hydrodynamic parameters or oligomerization state.27, 64 There is no indication of dimer dissociation in these data, as evidenced by the relatively narrow sedimentation coefficient distribution obtained, nor were there indications of dissociation in the SAXS data, suggesting monomer/dimer transitions are not responsible for the observed changes. No large changes in shape were detected by SAXS upon ligand binding, suggesting that a substantial conformational change is not responsible for the observed changes in sedimentation coefficient. The most likely explanation is that CO/YC-1 binding to Ms sGC NT leads to a change in hydrodynamic properties, for example a change in surface hydrophobicity, yielding a protein complex with altered sedimentation but with roughly the same overall conformation. Taken together, the ultracentrifugation and SAXS results indicate that the allosteric changes occurring in Ms sGC NT proteins upon ligand binding are small in magnitude but lead to a change in surface properties. Consistent with this are the ~4 Å shifts observed in Nostoc H-NOX on binding NO.21 More complete explanations await higher resolution models of sGC.
Chemical cross-linking reveals parallel coiled-coil and domain contacts
We undertook chemical cross-linking coupled with tandem mass spectrometry to uncover domain and subunit contacts in Ms sGC NT. Sixteen unique cross-linked peptides were found within the α1β1 coiled-coil. These data make clear that the predicted coiled-coil likely does form, and, furthermore, has parallel α1β1 strands (Figure 5). This arrangement is in agreement with Ma and co-workers, who predicted a parallel arrangement based on electrostatic arguments despite their structure showing an antiparallel β1β1 homodimeric coiled-coil for the rat sGC β1 sequence.23
Nine cross-links were found between the coiled-coil and other domains in Ms sGC NT proteins (Table 3), consistent with the coiled-coil domain serving as an organizing center. Eight of these were between the α1 and β1 subunits. Five additional cross-links were found between the other domains, four of which were between α1 and β1 subunits. Taken together, these data suggest the major inter-domain contacts are between domains on opposite chains. In particular, the α1 PAS domain appears closely associated with the β1 H-NOX domain, which, in turn, is closely associated with the α1 coiled-coil.
Assembling a model for sGC NT
Our homology models for the H-NOX and PAS domains are based upon bacterial homologues, and for the coiled-coil domain based on the numerous examples in the literature. The 62 residues linking these domains were also modeled but are of uncertain reliability since there are no homologous structures available for these regions. Additionally, we generated a homology model for the catalytic domain based on the crystal structure of the closely related adenylyl cyclase. Twenty-three intra-domain cross-links were found by mass spectrometry (Table 2), all of which were consistent with the homology models, providing confidence in the reliability of these models. In particular, five unique cross-links were found in the α1 H-NOX domain, all consistent with our homology model, supporting the prediction that it retains the H-NOX fold despite having lost the ability to bind heme. One cross-link each was found for the α1 PAS and the β1 H-NOX domains. Only the β1 PAS domain fold was unconfirmed by cross-linking.
With reliable domain models in place, we assembled and energy minimized a domain arrangement that was consistent with our cross-linking data and our overall molecular envelopes. In this model, the parallel coiled-coil segment provides an organizing center for the other domains, particularly for the α1 PAS and β1 H-NOX domains, which are in contact. While there are insufficient data available for mapping residues in direct contact, two conclusions of functional importance are possible. First, although the α1 and β1 subunits are clearly gene duplications, sGC appears to be asymmetric. The α1 H-NOX domain, in particular, can be removed without major effect on overall shape while the β1 H-NOX is in the center of the molecule (Figure 7). These data are consistent with those of Koglin and Behrends who showed that a heterodimeric human sGC retains sensitivity to NO and YC-1 after deletion of the first 259 residues of the α1 subunit.65 Second, one face of the coiled-coil is exposed in the model and may represent the cyclase domain binding surface. Such a model places the cyclase domains in contact with the β1 H-NOX and α1 PAS domains, in perfect position for direct regulation through ligand binding. This arrangement is consistent with two previous studies, one showing that addition of purified β1 H-NOX domain could inhibit sGC cyclase domains that were expressed independently, indicating direct binding,66 and a second study making use of fluorescent fusion proteins and Förster resonance energy transfer (FRET), which concluded that the heme and catalytic domains are in close proximity.67 The model is also consistent with the α1 and β1 PAS domains forming part of the dimer interface, as has been suggested based on homodimer formation by a bacterial homologue.22 A cross-link between the N-termini of α1 PAS (Lys 286) and β1 PAS (Glu 196) constrain the domains in the model into an arrangement reminiscent of the bacterial homodimer.
Coiled-coil truncation affects proximal histidine release
NO binding to sGC yields a transient six-coordinate intermediate followed by proximal histidine release and a five-coordinate nitrosyl complex. The six-coordinate intermediate is readily observed by stopped-flow spectroscopy for both full-length sGC68, 69 and C-terminally truncated Ms sGC-NT1 (reference24, Table 6). Unexpectedly, truncation of an additional 20 amino acids from each chain in Ms sGC-NT, as occurs in Ms sGC NT13, NT19 and NT21, yields a protein with very rapid proximal histidine release, which cannot be observed by stopped-flow spectroscopy (Table 6). Additionally, Ms sGC NT13, NT19 and NT21 all show tighter CO binding (Fig. 1). The truncated amino acids (α1 451–471, β1 381–400) are thought to complete the end of the coiled-coil followed by a proline-induced turn (α1 460, β1 390) and the start of a new helix, based on the β1/β1 coiled-coil structure.23 The increased proximal histidine release rate in the shorter proteins suggests that the missing residues interfere with an NO-dependent change in sGC conformation. Interestingly, our cross-linking data place the α1 PAS domain in direct contact with the C-terminal end of the coiled-coil (cross-link α1 E366–β1 K378), and also in direct contact with the β1 H-NOX domain, near the heme pocket (α1 E340–β1 K170 and α1 K343–β1 K170, Figure 4). This arrangement suggests the α1 PAS domain opposes the NO-dependent conformational change in sGC. The α1 PAS domain F helix provides the contact residues. In PAS kinase, flexibility of the PAS domain F helix was proposed to provide inhibitory interactions with adjacent domains that are relaxed upon ligand binding.70
A model for allostery in sGC function
NO, CO and YC-1 allosterically stimulate sGC, but how binding leads to activation remains unclear. Our data are consistent with a model in which the α1 PAS domain inhibits NO and CO binding to heme through enhancing their release rates. Binding of YC-1 relieves this inhibition, leading to higher CO and NO affinity (reference24, 25, Figure 1). Removal of the α1 PAS domain also relieves inhibition, yielding a heterodimeric protein with high CO affinity and loss of YC-1 response (unpublished data, footnote 2). Taken together, these data suggest YC-1 family compounds serve to enhance NO and CO binding by removal of an α1 PAS barrier to conformational change.
Inhibition of cyclase activity is also thought to involve a direct interaction between the β1 H-NOX domain and the cyclase domains. Winger et al. demonstrated that the isolated β1 H-NOX domain can directly bind to the cyclase domains and inhibit cyclase activity when added in trans; they went on to suggest that NO binding to heme in the β1 H-NOX domain would relieve this autoinhibitory interaction.66 Although the cyclase domains are not included in Ms sGC-NT, our model suggests these domains may assemble onto the coiled-coil domain, opposite to where the β1 H-NOX sits, allowing for direct cyclase–β1 H-NOX contact. In this arrangement, α1 PAS is positioned to inhibit β1 H-NOX, which, in turn, is positioned to inhibit cyclase. We suggest binding of YC-1 family compounds relieves the α1 PAS–β1 H-NOX inhibition, which in turn reduces the β1 H-NOX–cyclase inhibition. Binding of NO, and to a lesser extent CO, also relieves the β1 H-NOX–cyclase inhibition. Binding of both would doubly oppose autoinhibition, leading to the observed synergy between binding of YC-1 and CO/NO for stimulating sGC catalysis. How YC-1 binds to sGC is not yet known, but may involve binding to the α1 PAS domain (unpublished data, footnote 2), to the β1 H-NOX domain,71 or to both, perhaps at the α1 PAS–β1 H-NOX domain interface.
Acknowledgments
Funding sources: This work was supported by National Institutes of Health grants HL062969 and GM077390 (WRM), T32 GM008804 (BGF), American Heart Association grant 10PRE2630177 (BGF) and National Science Foundation Grant 0744732 (TF). Orbitrap mass spectrometry data were acquired by the Arizona Proteomics Consortium at the University of Arizona, supported by NIH/NCRR grant S10 RR028868-01. SAXS measurements were carried out at the Stanford Synchrotron Radiation Lightsource, supported by the Department of Energy and by the National Institutes of Health (NCRR and NIGMS).
We are deeply grateful to Dr. Thomas Weiss and Dr. Tsutomu Matsui (SSRL) for assistance with collection and interpretation of SAXS data, and to Dr. Katrina Miranda for providing DEA/NO. We thank Dr. Ah-Lim Tsai and Dr. Vladimir Berka (University of Houston) for preliminary kinetic measurements on NT13 indicating rapid loss of the proximal histidine upon NO binding. We thank Dr. Chad Park for assistance with ultracentrifugation measurements. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
The abbreviations used are
- YC-1
3-(5'-hydroxymethyl-2'furyl)-1-benzylindazole
- BS2G
bis(sulfosuccinimidyl)glutarate-d0
- BS3
bis(sulfosuccinimidyl)suberate-d0
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- BMOE
bis(maleimido)ethane
- SAXS
small-angle X-ray scattering
- DLS
dynamic light scattering
- SV-AUC
sedimentation velocity analytical ultracentrifugation
References
- 1.Ignarro LJ, editor. Nitric Oxide Biology and Pathobiology. 2. Academic Press; San Diego: 2010. [Google Scholar]
- 2.Li H, Poulos TL. Structure-function studies on nitric oxide synthases. J Inorg Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Stuehr DJ, Tejero J, Haque MM. Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- 5.Schulz R, Rassaf T, Massion PB, Kelm M, Balligand JL. Recent advances in the understanding of the role of nitric oxide in cardiovascular homeostasis. Pharmacol Ther. 2005;108:225–256. doi: 10.1016/j.pharmthera.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Pyriochou A, Papapetropoulos A. Soluble guanylyl cyclase: more secrets revealed. Cell Signal. 2005;17:407–413. doi: 10.1016/j.cellsig.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murrell W. Nitro-glycerine as a remedy for angina pectoris. Lancet. 1879;113:113–115. [Google Scholar]
- 9.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belik J. Riociguat, an oral soluble guanylate cyclase stimulator for the treatment of pulmonary hypertension. Curr Opin Investig Drugs. 2009;10:971–979. [PubMed] [Google Scholar]
- 11.Mittendorf J, Weigand S, Alonso-Alija C, Bischoff E, Feurer A, Gerisch M, Kern A, Knorr A, Lang D, Muenter K, Radtke M, Schirok H, Schlemmer KH, Stahl E, Straub A, Wunder F, Stasch JP. Discovery of riociguat (BAY 63–2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. ChemMedChem. 2009;4:853–865. doi: 10.1002/cmdc.200900014. [DOI] [PubMed] [Google Scholar]
- 12.Martin F, Baskaran P, Ma X, Dunten PW, Schaefer M, Stasch JP, Beuve A, van den Akker F. Structure of cinaciguat (BAY 58-2667) bound to Nostoc H-NOX domain reveals insights into heme-mimetic activation of the soluble guanylyl cyclase. J Biol Chem. 2010;285:22651–22657. doi: 10.1074/jbc.M110.111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meurer S, Pioch S, Pabst T, Opitz N, Schmidt PM, Beckhaus T, Wagner K, Matt S, Gegenbauer K, Geschka S, Karas M, Stasch JP, Schmidt HH, Muller-Esterl W. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res. 2009;105:33–41. doi: 10.1161/CIRCRESAHA.109.198234. [DOI] [PubMed] [Google Scholar]
- 14.Taqatqeh F, Mergia E, Neitz A, Eysel UT, Koesling D, Mittmann T. More than a retrograde messenger: nitric oxide needs two cGMP pathways to induce hippocampal long-term potentiation. J Neurosci. 2009;29:9344–9350. doi: 10.1523/JNEUROSCI.1902-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Moglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 18.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: Implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 19.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma X, Sayed N, Baskaran P, Beuve A, van den Akker F. PAS-mediated Dimerization of Soluble Guanylyl Cyclase Revealed by Signal Transduction Histidine Kinase Domain Crystal Structure. J Biol Chem. 2008;283:1167–1178. doi: 10.1074/jbc.M706218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma X, Beuve A, van den Akker F. Crystal structure of the signaling helix coiled-coil domain of the beta1 subunit of the soluble guanylyl cyclase. BMC structural biology. 2010;10:2. doi: 10.1186/1472-6807-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu X, Murata LB, Weichsel A, Brailey JL, Roberts SA, Nighorn A, Montfort WR. Allostery in recombinant soluble guanylyl cyclase from Manduca sexta. J Biol Chem. 2008;283:20968–20977. doi: 10.1074/jbc.M801501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu X, Feng C, Hazzard JT, Tollin G, Montfort WR. Binding of YC-1 or BAY 41-2272 to Soluble Guanylyl Cyclase Induces a Geminate Phase in CO Photolysis. J Am Chem Soc. 2008;130:15748–15749. doi: 10.1021/ja804103y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritz BG, Hu X, Brailey JL, Berry RE, Walker FA, Montfort WR. Oxidation and loss of heme in soluble guanylyl cyclase from Manduca sexta. Biochemistry. 2011;50:5813–5815. doi: 10.1021/bi200794c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lebowitz J, Lewis MS, Schuck P. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 2002;11:2067–2079. doi: 10.1110/ps.0207702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 30.Li W, O'Neill HA, Wysocki VH. SQID-XLink: implementation of an intensity-incorporated algorithm for cross-linked peptide identification. Bioinformatics (Oxford, England) 2012;28:2548–2550. doi: 10.1093/bioinformatics/bts442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame AL. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol Cell Proteomics. 2008;7:2386–2398. doi: 10.1074/mcp.M800021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. Biological small-angle x-ray scattering facility at the Stanford synchrotron radiation laboratory. J Appl Crystallogr. 2007;40:S453–S458. [Google Scholar]
- 33.Orthaber D, Bergmann A, Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J Appl Crystallogr. 2000;33:218–225. [Google Scholar]
- 34.Svergun DI, Konarev PV, Volkov VV, Sokolova AV, Koch MHJ. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 35.Svergun DI. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 36.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr. 2007;40:S223–S228. [Google Scholar]
- 37.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demeler B. UltraScan: A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. In: Scott DJ, Harding SE, Rowe AJ, editors. Analytical Ultracentrifugation: Techniques and Methods. The Royal Society of Chemistry; Cambridge: 2005. pp. 210–229. [Google Scholar]
- 40.Svergun D, Barberato C, Koch MHJ. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 41.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics (Oxford, England) 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 43.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 45.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 2010;38:W657–661. doi: 10.1093/nar/gkq498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitford PC, Noel JK, Gosavi S, Schug A, Sanbonmatsu KY, Onuchic JN. An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields. Proteins. 2009;75:430–441. doi: 10.1002/prot.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grubisic I, Shokhirev MN, Orzechowski M, Miyashita O, Tama F. Biased coarse-grained molecular dynamics simulation approach for flexible fitting of X-ray structure into cryo electron microscopy maps. J Struct Biol. 2010;169:95–105. doi: 10.1016/j.jsb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed A, Whitford PC, Sanbonmatsu KY, Tama F. Consensus among flexible fitting approaches improves the interpretation of cryo-EM data. J Struct Biol. 2012;177:561–570. doi: 10.1016/j.jsb.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 50.Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 51.Weast RC, editor. CRC Handbook of Chemistry and Physics. 70. CRC Press, Inc; Boca Raton: 1989. [Google Scholar]
- 52.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 53.Mertens HD, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J Struct Biol. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 54.Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter. 2006;5(Unit 5.6) doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friebe A, Mullershausen F, Smolenski A, Walter U, Schultz G, Koesling D. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- 58.Ramanathan S, Mazzalupo S, Boitano S, Montfort WR. Thrombospondin-1 and angiotensin II inhibit soluble guanylyl cyclase through an increase in intracellular calcium concentration. Biochemistry. 2011;50:7787–7799. doi: 10.1021/bi201060c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Z, Sayed N, Pyriochou A, Roussos C, Fulton D, Beuve A, Papapetropoulos A. Protein kinase G phosphorylates soluble guanylyl cyclase on serine 64 and inhibits its activity. Arterioscler Thromb Vasc Biol. 2008;28:1803–1810. doi: 10.1161/ATVBAHA.108.165043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meurer S, Pioch S, Gross S, Muller-Esterl W. Reactive oxygen species induce tyrosine phosphorylation of and Src kinase recruitment to NO-sensitive guanylyl cyclase. J Biol Chem. 2005;280:33149–33156. doi: 10.1074/jbc.M507565200. [DOI] [PubMed] [Google Scholar]
- 61.Kharitonov VG, Russwurm M, Magde D, Sharma VS, Koesling D. Dissociation of nitric oxide from soluble guanylate cyclase. Biochem Biophys Res Commun. 1997;239:284–286. doi: 10.1006/bbrc.1997.7470. [DOI] [PubMed] [Google Scholar]
- 62.Russwurm M, Koesling D. NO activation of guanylyl cyclase. Embo J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winger JA, Derbyshire ER, Marletta MA. Dissociation of nitric oxide from soluble guanylate cyclase and heme-nitric oxide/oxygen binding domain constructs. J Biol Chem. 2007;282:897–907. doi: 10.1074/jbc.M606327200. [DOI] [PubMed] [Google Scholar]
- 64.Zhao H, Balbo A, Brown PH, Schuck P. The boundary structure in the analysis of reversibly interacting systems by sedimentation velocity. Methods. 2011;54:16–30. doi: 10.1016/j.ymeth.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koglin M, Behrends S. A functional domain of the alpha1 subunit of soluble guanylyl cyclase is necessary for activation of the enzyme by nitric oxide and YC-1 but is not involved in heme binding. J Biol Chem. 2003;278:12590–12597. doi: 10.1074/jbc.M212740200. [DOI] [PubMed] [Google Scholar]
- 66.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry. 2005;44:4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 67.Haase T, Haase N, Kraehling JR, Behrends S. Fluorescent fusion proteins of soluble guanylyl cyclase indicate proximity of the heme nitric oxide domain and catalytic domain. PLoS One. 2010;5:e11617. doi: 10.1371/journal.pone.0011617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Y, Brandish PE, Ballou DP, Marletta MA. A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc Natl Acad Sci USA. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin E, Berka V, Sharina I, Tsai AL. Mechanism of binding of NO to soluble guanylyl cyclase: implication for the second NO binding to the heme proximal site. Biochemistry. 2012;51:2737–2746. doi: 10.1021/bi300105s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amezcua CA, Harper SM, Rutter J, Gardner KH. Structure and interactions of PAS kinase N-terminal PAS domain: model for intramolecular kinase regulation. Structure. 2002;10:1349–1361. doi: 10.1016/s0969-2126(02)00857-2. [DOI] [PubMed] [Google Scholar]
- 71.Yoo BK, Lamarre I, Rappaport F, Nioche P, Raman CS, Martin JL, Negrerie M. Picosecond to Second Dynamics Reveals a Structural Transition in Clostridium botulinum NO-Sensor Triggered by the Activator BAY-41-2272. ACS Chem Biol. 2012 doi: 10.1021/cb3003539. [DOI] [PubMed] [Google Scholar]