

Abstract
The pathogen Brucella melitensis secretes a Toll/interleukin-1 receptor (TIR) domain containing protein that abrogates host innate immune responses. In this study, we have characterized the biochemical interactions of Brucella TIR-like protein TcpB with host innate immune adaptor proteins. Using protein-fragment complementation assays based on Gaussia luciferase and green fluorescent protein, we find that TcpB interacts directly with MyD88 and that this interaction is significantly stronger than the interaction of TcpB with TIRAP, the only other adaptor protein that detectably interacts with TcpB. Surprisingly, the TcpB–MyD88 interaction depends on the death domain (DD) of MyD88, and TcpB does not interact with the isolated TIR domain of MyD88. TcpB disrupts MyD88DD-MyD88DD, MyD88DD-MyD88TIR and MyD88DD-MyD88 interactions but not MyD88–MyD88 or MyD88TIR-MyD88TIR interactions. Structural models consistent with these results suggest how TcpB might inhibit TLR signaling by targeting MyD88 via a DD–TIR domain interface.
Keywords: Death domain, TIR domain, Protein interactions, Host–pathogen, Protein complementation assays
1. Introduction
Pathogens use a variety of mechanisms to evade elimination by host immune responses [1]. A recently discovered mechanism of immune evasion is molecular mimicry of host proteins involved in Toll-like receptor (TLR) signaling [2], which plays an important role in innate immunity [3]. One example of such mimicry is the A46R protein of vaccinia virus, which contains a Toll/interleukin-1 (TIR) domain [4]. The TIR domain is a constituent component of all TLRs (human TLR1–10) [5] and five adaptor proteins involved in TLR signaling [6], including MyD88, TIRAP (TIR domain-containing adaptor protein), which is also called Mal (MyD88 adaptor-like protein), TRIF (TIR domain-containing adaptor protein inducing interferon-β), and TRAM (TRIF adaptor molecule). The A46R TIR protein has been shown to inhibit TLR signaling by interacting, directly or indirectly, with MyD88, TIRAP, TRIF and TRAM [7].
Proteins that contain TIR domains and contribute to immune evasion are also found in pathogenic bacteria [2]. One of these proteins, TcpB [8,9,28], is found in at least three Brucella species: Brucella melitensis, Brucella abortus and Brucella ovis. TcpB impairs TLR2- and TLR4-mediated NF-κB activation and inhibits the maturation of dendritic cells [8,9]. The mechanism of TcpB mediated immune evasion has yet to be fully elucidated, and there are conflicting reports about the host targets of TcpB. TcpB co-immunoprecipitates with endogenous and transfected MyD88 [8] and abrogates TIRAP induced NF-κB-activation [10]. Recently, it is reported that TcpB co-immunoprecipitates with co-transfected TIRAP, but not MyD88 [11].
In an attempt to better characterize the host targets of TcpB, we have used protein-fragment complementation assays (PCAs) based on split green fluorescent protein (GFP) [12] and Gaussia luciferase [13]. These assays allow us to probe for direct interactions of TcpB with host adaptor proteins, that classical biochemical assays such as co-immunoprecipitation are unable to distinguish due to co-associations between these adaptors. Since TcpB has been characterized as a TIR domain mimic that impairs TLR signaling, we reasoned that TcpB might participate in heterotypic TIR domain interactions with one or more of the TIR domain-containing adaptor proteins that mediate TLR signaling. We report that of all the TIR containing adaptor proteins tested, TcpB interacts directly with only MyD88 and TIRAP. The interaction with MyD88 is considerably stronger than that observed with TIRAP. Furthermore, TcpB–MyD88 interaction does not abrogate MyD88 homodimerization, which is required for downstream activation of IRAK kinases [26]. Besides the carboxy-terminal TIR domain, MyD88 also contains an amino-terminal death domain (DD) [14]. Here, we report that TcpB interacts strongly with the DD of MyD88. TcpB disrupts the dimerization of MyD88DD as well as an interaction between MyD88DD and MyD88TIR. This interaction between the DD and TIR domain of MyD88 has hitherto not been described. Structural models for complexes of MyD88DD, MyD88TIR and the isolated TIR domain of TcpB (TcpBTIR) suggest possible interfaces for these complex interactions. Finally, our results suggest that the unexplored interface of the DD and TIR domains of MyD88 may regulate MyD88 activity and that pathogens may target this interface to evade innate immunity.
2. Materials and methods
2.1. Cells and reagents
Chinese hamster ovary (CHO) and human epithelial (HEK293) cells were cultured using ATCC recommended growth media and protocols. Cells were transfected with DNA using lipofectamine 2000™ transfection reagent (Invitrogen). The pUNO plasmids hMyD88, hTRAM, hTRIF, and hTIRAP were obtained from InvivoGen (San Diego, CA). Plasmids expressing N- and C-terminal fragments of humanized luciferase from Gaussia princeps, hGLuc1 and hGLuc2, were obtained from the Michnick laboratory [13]. These plasmids are derivatives of the pcDNA3.1 mammalian expression vector (Invitrogen). A plasmid expressing TcpB from B. melitensis was obtained from the Miethke laboratory [8].
2.2. Design of constructs
The plasmid pUNO–hMyD88 was subcloned to derive constructs encoding the DD of MyD88 (MyD88DD), beginning at N26MRV and ending at LLEL105. Likewise, the construct encoding the TIR domain of MyD88 (MyD88TIR), beginning at R160FDA and ending at KALS294 was generated. MyD88, MyD88DD, MyD88TIR, TIRAP, TRIF, TRAM, and TcpB plasmids were subcloned into pcDNA3.1–hGLuc plasmids using restriction sites (NotI and ClaI) to generate the following constructs: pcDNA3.1–protein–hGLuc1 (linking hGLuc1 to the C-terminus of protein), pcDNA3.1–hGLuc1–protein (linking hGLuc1 to the N-terminus of the protein), and pcDNA3.1–protein–hGLuc2 (linking hGLuc2 to the C-terminus of protein). In each construct, a 10-residue flexible polypeptide linker (GGGGSGGGGS) was introduced to connect the hGLuc fragment to the protein of interest.
2.3. Gaussia luciferase PCA in live cells
Plasmids harboring hGluc1 and hGluc2 fusions were co-transfected in a 1:1 ratio into CHO and HEK293 cells on tissue culture treated microtiter plates using lipofectamine 2000 transfection reagent (Invitrogen). Prior to assaying, cells were serum starved overnight (0.1% fetal bovine serum). Native coelenterazine (NanoLight Technology, Pinetop, AZ) was used at a final concentration of 20 μM. Signal intensities integrated over 10 s were measured using a Synergy 2 plate reader and Gen5 data analysis software (BioTek).
2.4. NF-κB-luciferase reporter assay
The GloResponse™ NF-κB RE-luc2PHEK293 cell line (Promega) that contains a luciferase gene under the control of a minimal TATA promoter with multiple NF-κB response elements was used to evaluate the effect of TcpB on MyD88 mediated NF-κ activation. Mammalian expression plasmids carrying MyD88, MyD88DD and MyD88TIR were singly transfected or co-transfected with TcpB expression plasmid in the reporter cells at 1:1 ratio. Transfected cells were split equally into two sets, and reporter gene activity was the assayed in one set using the luciferase assay reagent (Promega). This activity was normalized to the cell titer per assay sample in the second set using CellTiter-Glo Luminescent Cell Viability assay (Promega). Luminescence was measured using Synergy2 Microplate Reader (BioTek).
2.5. GFP PCA in live cells
Strand 11 (S11), a 15-residue fragment of GFP, and a flexible polypeptide linker (GGGGSGGGGS) were fused to the C-terminus of proteins of interest, including MyD88, MyD88DD and MyD88TIR. Plasmids derived from pcDNA3.1 harboring an S11 fusion protein and the GFP1–10 detector fragment of superfolder GFP, described previously [15], were co-transfected into CHO cells. Cells were trypsinized, counted and suspended on white Nunc Immuno Assay plates (Thermo Scientific), which are characterized by low background fluorescence. Green fluorescence from each well was measured using an FL600 Microplate Fluorescence Reader (BioTek) with excitation at 488 nm and emission at 530 nm.
2.6. Microscopy and image analysis
Cells were fixed with 2% paraformaldehyde and imaged using an Olympus camera attached to a Zeiss Axiovert S100 microscope. Images were captured at a resolution of 600 dpi with 200 ms exposures and analyzed using Image-Pro Plus software version 6.2 (Media Cybernetics).
2.7. Homology modeling and docking
We used available structures for the DD and TIR domain of MyD88 (PDB# 3MOP [16] and 2Z5V [17]). The structure of TcpBTIR is unknown but the amino acid sequence is similar (56% identity) to the sequence of a TIR domain in Paracoccus for which a structure is available (PDB# 3H16 [18]). This structure was used as a template. To model the main chain structure of the target TIR domain, the template and target sequences were aligned, and for each subsequence aligned without insertions or deletions, the structure of the template subsequence was assigned to the target subsequence. For insertions, an algorithm described previously to model loops [19,20] was employed. This algorithm works well for loop structures with fixed ends. To account for side chains, we initially assumed that the amino acid residues in the target occupy the same space as the corresponding residues in the template. This assumption is most appropriate for residues that maintain the structural integrity of a compact protein and for residues involved in protein–protein interfaces. Structures for complexes of domains were obtained through a described procedure [20] that combines a reduced-coordinate rigid-body docking procedure (α-carbon atoms only) and an all-atom annealing procedure involving molecular dynamics/molecular mechanics simulations with AMBER [21]. PSIPRED [27] was used for secondary structure prediction.
3. Results
3.1. Split-luciferase complementation assays for detection of TcpB interactions with MyD88, MyD88DD, MyD88TIR, TIRAP, TRIF and TRAM
Co-immunoprecipitation assays are heavily reliant on antibody epitopes, and are unable to distinguish direct interactors from co-associated-proteins. To bypass this dependence, we used a split-luciferase complementation assay [13] to probe for direct interactions of TcpB with host adaptor proteins involved in TLR signaling (Fig. 1). We initially probed only for interactions with full-length MyD88, TIRAP, TRIF and TRAM but later probed for interactions with each of the isolated protein interaction domains of MyD88, the death domain (MyD88DD) and the TIR domain (MyD88TIR). Towards this goal, we generated mammalian expression plasmids encoding the fusion proteins indicated in Fig. 1A, each with a fragment of humanized Gaussia luciferase (hGLuc1 or hGLuc2). Bioluminescence analyses of luciferase activity reveal that TcpB interacts with MyD88, TIRAP and MyD88DD but not MyD88TIR, TRIF or TRAM (Fig. 1B). The interaction with MyD88 is strongest (mean induction 277% greater than baseline luminescence measured for cells transfected with hGLuc1–TcpB alone with a standard deviation of 36%). The interaction with TIRAP is weakest (mean induction 32 + 13% above baseline). The interaction with the isolated DD of MyD88 is intermediate (mean induction 49 + 13% above baseline). These results provide evidence for direct interactions of TcpB with adaptor proteins MyD88 and TIRAP.
Fig. 1.
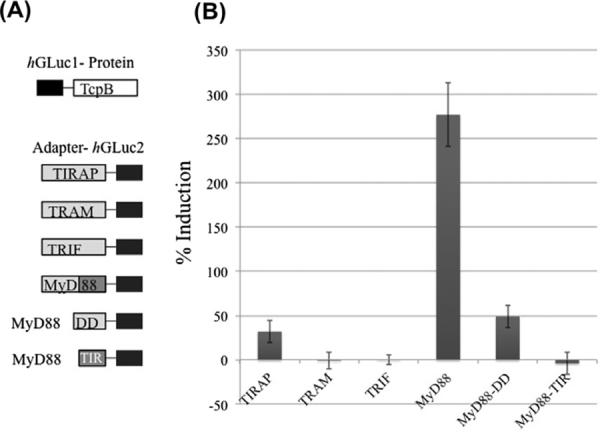
Direct interaction of TcpB with adaptor proteins. HEK293 cells transfected with hGLuc1–TcpB alone and in combination with TIRAP–hGLuc2, TRAM–hGLuc2, TRIF–hGLuc2, MyD88–hGLuc2, MyD88DD–hGLuc2 or MyD88TIR–hGLuc2 (A). Percent induction of luminescence is calculated compared to baseline luminescence from hGLuc1–TcpB alone expressing cells (B). Data shown is a mean of three experiments performed in triplicate. Error bars indicate standard deviation.
3.2. Expression and folding of MyD88, MyD88DD and MyD88TIR
Given that TcpB binds MyD88 strongly (Fig. 1), the finding that TcpB does not interact with the isolated TIR domain of MyD88 was surprising. Using a previously reported split-GFP complementation assay [22], we tested the expression and folding of full-length MyD88 and its two domains in mammalian cells. In this assay, a short non-fluorescent peptide tag, strand 11 (S11) from superfolder GFP, a robustly folded version of GFP, and a flexible polypeptide linker are fused to a protein of interest, and the fusion protein is co-expressed with a non-fluorescent detector GFP fragment (GFP1–10), which is missing S11. Green fluorescence upon co-expression is indicative of expression and folding of the S11-tagged protein [15]. Cells transfected with a plasmid encoding MyD88-S11, MyD88TIR-S11 or MyD88DD–S11 exhibit green fluorescence upon co-transfection with the plasmid encoding GFP1–10 (Fig. 2A). The intensity of fluorescence observed for MyD88-S11 is stronger than that observed for MyD88DD–S11 or MyD88TIR–S11, which suggests that the full-length MyD88 is more stable than either of its individually expressed domains. Consistent with previously published immunocytochemical results [23], we detect MyD88 and MyD88TIR in both the cytoplasm and the nucleus. In contrast, the isolated DD of MyD88 is more excluded from the nucleus, and its overexpression shows early signs of apoptosis.
Fig. 2.
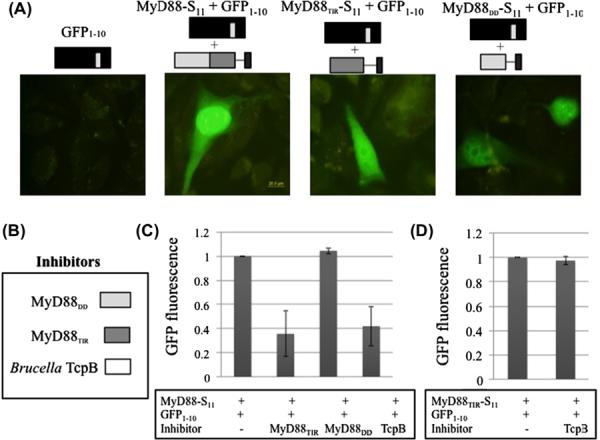
Expression and folding of MyD88 and its domains. (A) CHO cells were singly or co-transfected with GFP1–10, and MyD88-S11, MyD88TIR-S11 or MyD88DD-S11, as indicated. Fluorescence micrographs of cells were visualized using FITC channel excitation at 488 nm and emission at 515 nm filters with 100× magnification. Scale bar is 20 μm. (B, C) CHO cells were transfected in triplicate with MyD88-S11 and GFP1–10, with and without MyD88TIR, MyD88DD and TcpB. GFP fluorescence was quantified as indicated in materials and methods. (D) CHO cells were transfected with MyD88TIR-S11 and GFP1–10, with and without TcpB. Mean GFP fluorescence + the standard deviation from three experiments is indicated.
GFP1–10 complementation of an S11-tagged protein can be inhibited by aggregation of the S11-tagged protein [30]. Interference by a binder that makes an S11-peptide less accessible to GFP1–10 is also detectable as a loss of green fluorescence. TcpB and MyD88TIR both inhibit GFP1–10 complementation of MyD88-S11 (Fig. 2C). Thus MyD88TIR and TcpB interact with MyD88 via a similar interface, while MyD88DD interacts via a different interface. TcpB does not abrogate the complementation of S11 with GFP1–10 non-specifically (Fig. 2D).
3.3. Luciferase–protein complementation assays for detection of dimerization interactions among MyD88, MyD88DD, and MyD88TIR
MyD88 and its isolated domains have been reported to form homodimers [24] and higher-order oligomers [25]. We used the Gaussia luciferase-based PCA to characterize the self-interactions of MyD88 so that we could directly determine if these interactions can be inhibited by TcpB. Using plasmids for expression of the fusion proteins illustrated in Fig. 3A, we systematically assayed each of the nine possible pairs for interaction. In all cases, transfection of a single plasmid yielded a background level of luminescence, similar to that of untransfected cells. As expected based on earlier reports [24], we detected MyD88–MyD88, MyD88DD–MyD88, MyD88TIR–MyD88, MyD88DD–MyD88DD, and MyD88TIRȃMyD88TIR interactions (Fig. 3A). Among these interactions, the MyD88DD–MyD88DD interaction is strongest, and the MyD88TIR–MyD88TIR interaction is weakest. Surprisingly, we also detected a MyD88DD–MyD88TIR interaction, which is stronger than the MyD88TIR–MyD88TIR interaction and weaker than the MyD88DD–MyD88DD interaction. An interaction between a DD and a TIR domain has not previously been observed.
Fig. 3.
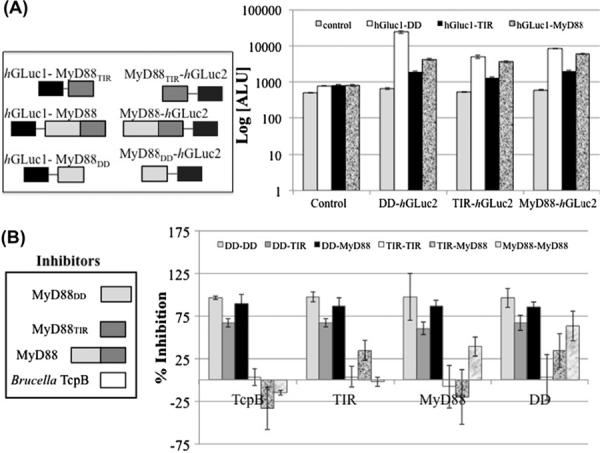
(A) Dimerization of MyD88 and domains. HEK293 cells were singly- or co-transfected with hGLuc1-tagged MyD88, MyD88DD or MyD88TIR, and MyD88, MyD88DD or MyD88TIR tagged-hGLuc2, as indicated, in a 96-well format. Cells were serum starved in phenol-red free media, coelenterazine was added to the cells, and luminescence was measured (Log [arbitrary luminescence units (ALU)]), and represent mean of five experiments in triplicate. Cells expressing hGluc1–DD are represented with white bars; hGluc1–MyD88TIR with black bars; and hGluc1–MyD88 with gray marble bars. The control on the x-axis indicates the background luminescence from cells not containing any hGluc2 construct. The corresponding MyD88DD, MyD88TIR and MyD88-tagged-hGluc2 is represented on the horizontal axis. (B) Effect of TcpB on MyD88 Interactions. Untagged MyD88DD, MyD88TIR, MyD88 and TcpB were co-expressed in these cells. Luminescence from singly-transfected cells (with hGLuc1–protein or protein–hGLuc2) was used as background. Percent inhibition of interaction was calculated by measuring inhibition of mean luminescence from cells co-expressing TcpB, MyD88TIR, full-length MyD88 or MyD88DD compared to controls (no inhibitors). Each experiment was performed in triplicate. The % inhibition of DD–DD, DD–TIR domain, DD–MyD88, TIR domain–TIR domain, TIR domain–MyD88 and MyD88–MyD88 interactions are reported in order (light gray, darker gray, black, white, gray marble and white marble respectively). The corresponding inhibitors are shown on the x-axis.
Next, we tested the ability of TcpB as well as MyD88, MyD88DD, and MyD88TIR, to inhibit the MyD88 interactions (Fig. 3B). TcpB inhibits MyD88DD–MyD88DD interaction, MyD88DD–MyD88TIR interaction, and MyD88DD–MyD88 interaction. In contrast, TcpB has a negligible effect on MyD88TIR–MyD88TIR interaction and MyD88–MyD88 interaction, and interestingly, TcpB has a modest positive effect on MyD88TIR–MyD88 interaction. Thus, TcpB does not significantly perturb homodimerization of full-length MyD88 but it disrupts homo- and heterotypic interactions mediated by the DD of MyD88. Notably, only MyD88DD and MyD88 can inhibit the homodimerization of full-length MyD88 (Fig. 3B).
3.4. Host effects of TcpB and structural models of interactions
We investigated how TcpB alone or together with MyD88, MyD88DD or MyD88TIR affects NF-κB activation (Fig. 4). Expression of full-length MyD88 alone or MyD88DD alone enhanced the activity of NF-κB. The expression of TcpB suppressed the activity of NF-κB, and co-expression of either full-length MyD88 or MyD88TIR with TcpB did not relieve suppression. In contrast, co-expression of MyD88DD with TcpB, attenuated the TcpB-mediated suppression of NF-κB activity. We note that the positive effects of MyD88DD and MyD88 on NF-κB activity are comparable, whereas the effects of MyD88DD and MyD88 on TcpB-mediated suppression of NF-κB activity are quite distinct, and may reflect the effects of the TIR-DD interaction in full length MyD88.
Fig. 4.
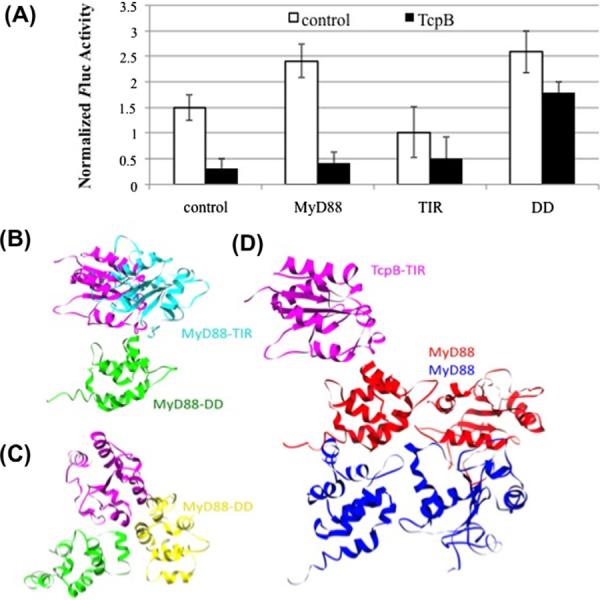
NF-κB response and model of TcpB interaction with MyD88. (A) GloResponse NF-κB RE-luc2PHEK293 cells were singly- or co-transfected with MyD88, MyD88DD or MyD88TIR, and TcpB as indicated. Luminescence measurements from cells were normalized to cell titer. Cells mock-transfected or singly-transfected with MyD88TIR, MyD88DD and MyD88 are indicated as controls. Means and standard deviations from a representative of three experiments performed in triplicate are shown. (B, C) Model for the binding of TcpB to MyD88DD and full length MyD88. The binding of TcpB to MyD88DD prevents the formation of a MyD88 DD–TIR domain heterodimer. TcpBTIR is shown in magenta, MyD88TIR is shown in cyan, MyD88DD is shown in green and yellow. (D) Model for the interaction of the TcpB TIR domain with a MyD88 homodimer. TcpBTIR is shown in magenta, and the two MyD88 molecules in the homodimer are shown in red and blue. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
We modeled interactions of MyD88TIR and TcpB with MyD88DD. Structural models for various TcpBTIR-containing complexes (Fig. 4B–D) indicate that TcpBTIR or MyD88TIR can be docked to MyD88DD via a variety of interfaces because of favorable charge interactions. TcpBTIR and MyD88TIR have net positive charges of +15 and +7, respectively, and MyD88DD has a net negative charge of −3. The potential TcpBTIR–MyD88DD and MyD88TIR–MyD88DD interfaces are overlapping. This explains the ability of the stronger TcpBTIR–MyD88DD interaction to block the MyD88TIR–MyD88DD interaction. The TcpBTIR–MyD88DD interaction can also block self-interaction of the isolated DD of MyD88. Significantly, the TcpBTIR–MyD88DD interaction cannot by itself inhibit homodimerization of full-length MyD88, as both the DD and TIR domain of MyD88 contribute to dimerization.
4. Discussion
The direct interaction of host adaptor protein MyD88 with Brucella protein TcpB depends on the DD in MyD88, but not the TIR domain. Additionally, the death and TIR domains of MyD88 interact. TcpB inhibits this interaction as well as self-interaction of the isolated DD. However, TcpB does not abrogate homodimerization of full-length MyD88. Interaction of DD with TcpB can modulate the effects of TcpB on NF-κB activation. Additionally, TcpB can affect host signaling through other mechanisms [11], indeed, we detect a direct, albeit weak interaction between TcpB and TIRAP.
We show evidence for a novel interaction between the two domains of MyD88, the DD and the TIR domain. In earlier work, the isolated TIR domain of MyD88 has been shown to bind to full length MyD88 [24,29], but not the DD alone [24] in a yeast two-hybrid assay. The role of appropriate post-translational modifications of these domains in these interactions remains completely unexplored. However, in the context of a folded small protein containing these two domains, it is highly likely that there are protein interfaces mediated by the two domains. The intramolecular interaction between the DD and TIR domain in MyD88, may serve to regulate interactions of MyD88 with binding partners. The targeting of this interaction by TcpB suggests a functional significance that needs further elucidation.
Consistent with previous reports [26], we find that overexpression of full-length MyD88 or the MyD88DD enhances the activity of NF-κB, and that TcpB by itself suppresses NF-κB [11]. It is significant however that neither full-length MyD88 nor MyD88TIR relieve suppression of NF-κB by TcpB, while DD has a significant effect. Biochemically, these results provide nice insight into the mechanisms of TcpB interaction with MyD88. The cellular context of these interactions remains baffling, since neither the isolated DD nor the TIR domains have been reported in cells.
In summary, the TIR domain mimic TcpB from Brucella interacts with MyD88 through the DD of MyD88, without inhibiting MyD88 homodimerization. Interaction of TcpB with MyD88 has the potential to disrupt a novel intramolecular interaction between the death and TIR domains of MyD88. The functional significance of the DD-TIR domain interface within MyD88 is unknown, but this interface is specifically targeted by a bacterial protein implicated in immune evasion, suggesting that future studies of the DD-TIR domain interface are warranted and may yield new insights into regulation of MyD88 and TLR signaling.
Acknowledgments
We thank Dr. Thomas Miethke for TcpB cDNA, Dr. Stephen W. Michnik for hGLuc1 and hGLuc2 constructs, and Dr. Goutam Gupta for helpful discussions. This work was supported by Transformational Medical Technologies Program Contract IACRO B0844971 (to A.C.) from the Department of Defense Chemical and Biological Defense Program through the Defense Threat Reduction Agency (http://www.dtra.mil/), Department of Energy Contract DE-AC52-06NA25396 (to Los Alamos National Security, LLC), and National Institutes of Health Grant R01 GM076570 from NIGMS (to W.S.H.), and Grant P41 RR01315 to the National Flow Cytometry Resource.
References
- [1].Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- [2].Cirl C, Miethke T. Microbial Toll/interleukin 1 receptor proteins: a new class of virulence factors. Int. J. Med. Microbiol. 2010;300:396–401. doi: 10.1016/j.ijmm.2010.04.001. [DOI] [PubMed] [Google Scholar]
- [3].Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- [4].Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- [6].O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- [7].Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cirl C, Wieser A, Yadav M, Duerr S, Schubert S, Fischer H, Stappert D, Wantia N, Rodriguez N, Wagner H, Svanborg C, Miethke T. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat. Med. 2008;14:399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]
- [9].Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, Muller A, Lapaque N, Demaria O, Alexopoulou L, Comerci DJ, Ugalde RA, Pierre P, Gorvel JP. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4:e21. doi: 10.1371/journal.ppat.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR domain-containing protein mimics properties of the Toll-like receptor adaptor protein TIRAP. J. Biol. Chem. 2009;284:9892–9898. doi: 10.1074/jbc.M805458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, Nishikawa T, Park S-G, Roop RM, II, Ghosh S. Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter. MAL, J. Immunol. 2010;184:956–964. doi: 10.4049/jimmunol.0902008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- [13].Remy I, Michnick SW. A highly sensitive protein–protein interaction assay based on Gaussia luciferase. Nat. Methods. 2006;3:977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- [14].Hofmann K, Tschopp J. The death domain motif found in Fas (Apo-1) and TNF receptor is present in proteins involved in apoptosis and axonal guidance. FEBS Lett. 1995;371:321–323. doi: 10.1016/0014-5793(95)00931-x. [DOI] [PubMed] [Google Scholar]
- [15].Cabantous S, Waldo GS. In vivo and in vitro protein solubility assays using split GFP. Nat. Methods. 2006;3:845–854. doi: 10.1038/nmeth932. [DOI] [PubMed] [Google Scholar]
- [16].Lin SC, Lo YC, Wu H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. USA. 2009;106:10260–10265. doi: 10.1073/pnas.0812956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chan SL, Low LY, Hsu S, Li S, Liu T, Santelli E, Le Negrate G, Reed JC, Woods VL, Jr., Pascual J. Molecular mimicry in innate immunity: crystal structure of a bacterial TIR domain. J. Biol. Chem. 2009;284:21386–21392. doi: 10.1074/jbc.C109.007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tung C-S, Joseph S, Sanbonmatsu KY. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat. Struct. Biol. 2002;9:750–755. doi: 10.1038/nsb841. [DOI] [PubMed] [Google Scholar]
- [20].Tung C-S, Walsh DA, Trewhella J. A structural model of the catalytic subunit-regulatory subunit dimeric complex of the cAMP-dependent protein kinase. J. Biol. Chem. 2002;277:12423–12431. doi: 10.1074/jbc.M110298200. [DOI] [PubMed] [Google Scholar]
- [21].Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Jr., Weiner PK. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984;106:765–784. [Google Scholar]
- [22].Cabantous S, Rogers Y, Terwilliger TC, Waldo GS. New molecular reporters for rapid protein folding assays. PLoS One. 2008;3:e2387. doi: 10.1371/journal.pone.0002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jaunin F, Burns K, Tschopp J, Martin TE, Fakan S. Ultrastructural distribution of the death-domain-containing MyD88 protein in HeLa cells. Exp. Cell Res. 1998;243:67–75. doi: 10.1006/excr.1998.4131. [DOI] [PubMed] [Google Scholar]
- [24].Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- [25].Into T, Inomata M, Niida S, Murakami Y, Shibata K. Regulation of MyD88 aggregation and the MyD88-dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J. Biol. Chem. 2010;285:35759–35769. doi: 10.1074/jbc.M110.126904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Loiarro M, Gallo G, Fanto N, De Santis R, Carminati P, Ruggiero V, Sette C. Identification of critical residues of the MyD88 death domain involved in the recruitment of downstream kinases. J. Biol. Chem. 2009;284:28093–28103. doi: 10.1074/jbc.M109.004465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- [28].Newman RM, Salunkhe P, Godzik A, Reed JC. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect. Immun. 2006;74:594–601. doi: 10.1128/IAI.74.1.594-601.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li C, Zienkiewicz J, Hawiger J. Interactive sites in the MyD88 Toll/interleukin (IL) 1 receptor domain responsible for coupling to the IL1b signaling pathway. J. Biol. Chem. 2005;280:26152–26159. doi: 10.1074/jbc.M503262200. [DOI] [PubMed] [Google Scholar]
- [30].Chun W, Waldo GS, Johnson GVW. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3b activation and caspase 3 cleavage. J. Neurochem. 2007;103:2529–2539. doi: 10.1111/j.1471-4159.2007.04941.x. [DOI] [PubMed] [Google Scholar]