

Abstract
The Nr4a family of transcription factors transactivate expression of the transcription factor Foxp3 and are essential for the generation of regulatory T cells. Mice deficient in all three members of the Nr4a family develop massive multiorgan inflammation.
Unrestrained activation and cytokine production by T cells can result in extensive autoimmune damage to host tissue. Specialized populations of regulatory T cells (Treg cells) that express the transcription factor Foxp3 provide an essential layer of defense against unwarranted T cell activation through their potent suppressor activity, which limits the proinflammatory properties of effector T cells1,2. Treg cells can develop in the thymus as natural Treg cells or in the periphery as inducible Treg cells and constitute a small proportion (~10%) of the total CD4+ T cell population. The importance of Treg cells in normal homeostasis of the immune system is underscored by the fact that in humans, naturally occurring mutations that impair Foxp3 function are associated with the development of IPEX syndrome (‘immune-mediated polyendocrinopathy X-linked’ syndrome), in which affected male children develop multiorgan inflammation and have a limited life expectancy1 Similarly, deletion of Foxp3 in mice causes massive multiorgan inflammation that results in perinatal death2. Consequently, considerable effort has been expended to define the mechanisms by which Foxp3 expression is induced in Treg cells. In this issue of Nature Immunology, Sekiya et al. report that the Nr4a family of nuclear receptors is required for Foxp3 expression and the generation of Treg cells and that mice lacking all three members of this family in T cells have a pathology that is even more pronounced than that of Foxp3-deficient scurfy mice3.
Sekiya et al. analyze mice lacking one or more Nr4a proteins in T cells3. They find that mice lacking individual Nr4a transcription factors—Nr4a1 (Nurr77), Nr4a2 (Nurr1) or Nr4a3 (Nor1)—do not have an overtly altered phenotype. However, combined deletion of two of the three family members, Nr4a1 and Nr4a3, is associated with a considerably greater abundance of thymic and peripheral Foxp3+ Treg cells, whereas triple deletion of all three members is much more deleterious. The lifespan of mice deficient in only Nr4a1 and Nr4a3 is similar to that of Foxp3-deficient scurfy mice (~30 days)4,5, whereas mice lacking all three Nr4a family members in T cells succumb within 21 days. These data demonstrate a strong and redundant requirement for Nr4a transcription factors in the generation of Foxp3+ Treg cells and hint at additional roles for Nr4a proteins in limiting T cell proinflammatory functions (Fig. 1).
Figure 1.
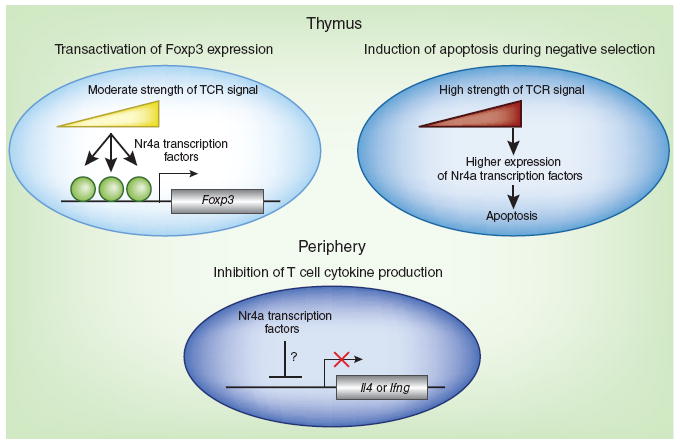
Pleiotropic roles of Nr4a transcription factors in limiting autoimmunity. Members of the Nr4a family of transcription factors serve important roles in limiting autoimmunity by at least three distinct mechanisms. In the thymus (top), Nr4a expression is directly proportional to the strength of TCR signaling: TCR signaling increases Nr4a expression in thymocytes, which leads to either transactivation of Foxp3 expression (left) or induction of apoptosis (right) during negative selection. Through mechanisms that remain to be characterized, members of the Nr4a family also suppress the production of IL-4 (encoded by Il4) and other cytokines (such as interferon-γ, encoded by Ifng) by T cells in the periphery (bottom).
In support of that hypothesis, Sekiya et al. show that Nr4a-deficient T cells produce more interleukin 4 (IL-4) and interferon-γ in vitro than do Nr4a-sufficient T cells, although the inflammation observed in vivo is mainly of the T helper type 2 type3. They establish, through the use of mixed–bone marrow chimeras, that the exaggerated cytokine production is not secondary to the inflammation caused by loss of Treg cells but is a cell-intrinsic property of Nr4a-deficient T cells. Even in the presence of normal numbers of Foxp3+ T cells derived from wild-type bone marrow precursor cells, CD4+ T cells that develop from Nr4a-deficient precursors produce more IL-4. Thus, Nr4a transcription factors promote immunological homeostasis in at least two ways: by inducing Foxp3 expression in Treg cells and by limiting cytokine production by non-Treg cells (Fig. 1). The mechanisms by which members of the Nr4a family regulate IL-4 production in T cells in vivo remain to be explored. Do Nr4a transcription factors directly bind to the promoters of the genes encoding the transcription factor GATA-3 or IL-4 and repress their expression, or do they induce an unidentified negative regulator of IL-4 production? Future studies should clarify whether these transcription factors also serve an important role in CD4+ T cell differentiation in the periphery.
High-affinity interactions with T cell antigen receptors (TCRs) lead to negative selection and the elimination of most developing thymocytes. A small proportion of those cells upregulate Foxp3 expression, escape negative selection and develop into natural Treg cells2. The evidence suggests that members of the Nr4a family have an important role in both negative selection and the generation of natural Treg cells. In lethally irradiated mice reconstituted with a mixture of bone marrow precursor cells from both wild-type mice and mice triply deficient in all three members of the Nr4a family, a higher proportion of CD4+CD8+ double-positive and CD4+CD8– or CD4–CD8+ single-positive T cells arise from Nr4a-deficient precursor cells than from Nr4a-sufficient precursor cells in the thymus. Moreover, all three members of the Nr4a family are induced by stimulation of the TCR in naive CD4+ T cells3,6,7, and precursors of Foxp3–CD25+ Treg cells in the thymus have detectable expression of Nr4a1 protein3. Sekiya et al. propose that Nr4a transcription factors simultaneously promote Foxp3 expression and enhance cell death during negative selection (Fig. 1). Their observations are consistent with published studies demonstrating that forced expression of Nr4a1 and Nr4a3 augments apoptosis in double-positive thymocytes8,9. Studies with peptides that induce graded increases in TCR signaling will help clarify this issue.
One of the most exciting aspects of this study is the finding that ectopic overexpression of a single Nr4a family member is sufficient to drive Foxp3 expression in non-Treg cells3,6. The authors inject recipient mice deficient in recombination-activating gene 2 with bone marrow cells retrovirally transduced to express a tamoxifen-inducible version of Nr4a2 in which the ligand-binding domain is replaced with the estrogen receptor ligand-binding domain and a linked Thy-1.1 reporter. When they administer a moderate dose of tamoxifen to the recipient mice 42 days later, a substantial proportion of Thy-1.1+ cells express Foxp3. Notably, higher concentration of tamoxifen lead to substantial loss of Thy-1.1+ cells as well as Foxp3+ cells, consistent with the idea that whereas moderate induction of Nr4a promotes the generation of Treg cells, strong induction of Nr4a family members promotes negative selection and cell death (Fig. 1).
Sekiya et al. use chromatin immunoprecipitation of endogenous Nr4a proteins to show that all three Nr4a family members bind the Foxp3 promoter in stimulated naive T cells. Thus, all three Nr4a transcription factors are potentially able to activate Foxp3 expression directly. Nr4a1 shows the most binding in chromatin-immunoprecipitation assays, most probably because Nr4a1 mRNA is induced more than Nr4a2 or Nr4a3 mRNA after stimulation of the TCR in naive CD4+ T cells. Nr4a1 also has the highest expression of all members of Nr4a family in Treg cells, with ~10- and ~30-fold more Nr4a1 mRNA than Nr4a2 mRNA and Nr4a3 mRNA, respectively. Paradoxically, however, Nr4a2 has a much greater ability than Nr4a1 to induce Foxp3 expression after ectopic expression in naive T cells in vitro, despite the fact that loss of Nr4a2, either alone or in combination with Nr4a1 or Nr4a3, is not associated with loss of Foxp3 expression or the development of autoimmunity in mice. These differences are unlikely to reflect differences in DNA binding; replacing the DNA-binding domain of Nr4a2 with that of Nr4a1 does not affect the ability of the chimeric protein to transactivate Foxp3 expression. One clue to this paradox is provided by the authors’ observation that tamoxifen-inducible versions of the three Nr4a proteins are equivalent in their ability to induce Foxp3 when retrovirally expressed in naive T cells. Together the data suggest that the considerable redundancy among the three members of the Nr4a family observed in vivo requires additional factors operating on individual Nr4a family members during the development of Treg cells. For example, the various members of the Nr4a family could undergo specific post-translational modifications that render them unable to interact with specific transcriptional regulatory partners in the nucleus to regulate Foxp3 expression.
The unique ability of Nr4a transcription factors to directly upregulate Foxp3 expression makes them attractive targets for the treatment of autoimmune and inflammatory diseases. Unfortunately, structural analyses suggest that these transcription factors are probably not ligand regulated10, which would eliminate the simplest strategy for manipulation of Nr4a function—that is, the identification of synthetic or naturally occurring ligands (agonists or antagonists) that could potentially be used therapeutically. The ligand-binding pocket of Nr4a2 is occupied by the side chains of bulky hydrophobic residues, and its ligand-binding domain adopts a constitutively active conformation, in part because of a salt bridge and aromatic stacking interactions between two adjacent helices10. Moreover, Nr4a proteins have a charged interaction surface rather than the hydrophobic coactivator-binding region present in other nuclear receptors11 and thus they probably interact with unique transcriptional partners. An oncogenic, constitutively active version of the receptor tyrosine kinase Ret destabilizes the active conformation of Nr4a2, presumably through tyrosine phosphorylation of Nr4a2 itself, and inhibits its transcriptional activity10. Together these data suggest that identifying molecular partners that influence the transcriptional activity of individual members of the Nr4a family might facilitate the design of novel therapeutic strategies aimed at manipulating Foxp3 expression in the context of diverse clinical situations, including autoimmune and inflammatory diseases, transplant medicine and cancer.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Ziegler S. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 2.Josefowicz SZ, Lu L-F, Rudensky AY. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sekiya M, et al. Nat Immunol. 2013;14:230–237. doi: 10.1038/ni.2520. [DOI] [PubMed] [Google Scholar]
- 4.Lyon MF, Peters J, Glenister PH, Ball S, Wright E. Proc Natl Acad Sci USA. 1990;87:2433–2437. doi: 10.1073/pnas.87.7.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godfrey VL, Wilkinson JE, Russell LB. Am J Pathol. 1991;138:1379–1387. [PMC free article] [PubMed] [Google Scholar]
- 6.Sekiya T, et al. Nat Commun. 2011;2:269. doi: 10.1038/ncomms1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moran AE, et al. J Exp Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng LE, Chan FK, Cado D, Winoto A. EMBO J. 1997;16:1865–1875. doi: 10.1093/emboj/16.8.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou T, et al. J Exp Med. 1996;183:1879–1892. doi: 10.1084/jem.183.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, et al. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]