

Abstract
Objective
To determine cardiovascular disease risk in a larger cohort of patients with glycogen storage disease (GSD) I through the use of noninvasive measures of arterial function and anatomy.
Study design
Carotid intima media thickness (IMT), radial artery tonometry, and brachial artery reactivity were performed in 28 patients with GSD I (13F/15M, mean age 23 years) and 23 control subjects (19F/4M, mean age 23 years).
Results
The primary outcome measure, mean left distal IMT was greater in the GSD cohort (0.500 ± 0.055 mm) than in the control group (0.457 ± 0.039 mm) (P = .002, adjusted for age, sex, and body mass index). Mean augmentation index measured by radial artery tonometry was higher in the GSD cohort (16.4% ± 14.0%) than in the control group (2.4% ± 8.7%) (P < .001). No significant difference was observed between mean brachial artery reactivity in the GSD cohort (6.3% ± 4.9% change) versus control subjects (6.6% ± 5.1% change) (P = .46).
Conclusions
GSD I is associated with arterial dysfunction evident by increased IMT and augmentation index. Patients with GSD I may be at increased risk for cardiovascular disease.
The glycogen storage diseases (GSDs) are a group of inherited enzymatic disorders involving the synthesis and breakdown of glycogen. GSD Ia is caused by deficiency in glucose-6-phosphatase, which catalyses the conversion of glucose-6-phosphate to glucose, and GSD Ib is caused by a defect in the transporter protein that shuttles glucose-6-phosphatase from the cytoplasm to the lumen of the endoplasmic reticulum.1 The metabolic consequences of GSD I include hypoglycemia, lactic acidemia, hyperuricemia, and dyslipidemia.
The dyslipidemia in GSD I is consistent with a proatherogenic profile characterized by hypercholesterolemia, hypertriglyceridemia, and reduced high-density lipoprotein (HDL).2 Nevertheless, the potential relationship between hyperlipidemia and cardiovascular disease (CVD) in patients with GSD remains unclear. Ubels et al3 detected no increase in cardiovascular risk in nine patients using non-invasive measures of vascular function. Similarly, Lee et al4 detected no vascular dysfunction in 6 patients. Given these observations, several mechanisms have been postulated to be protective against heart disease, including reverse cholesterol transport, increased antioxidant potential, or decreased platelet aggregation.5–7
Vascular endothelial dysfunction is a major promoter of atherogenesis and has been established as a precursor of cardiovascular disease.8 Carotid intima-media thickness (IMT), brachial artery reactivity (BAR), and radial artery tonometry (RAT) are noninvasive techniques commonly used as surrogate markers of CVD risk and to assess the efficacy of therapeutic interventions.9 Abnormalities in these surrogate markers have been observed in at-risk populations as young as 9 years of age.10,11
Because of improved management strategies and treatment options, patients with GSD now commonly reach adulthood. Despite the inability of previous studies to document increased CVD risk in patients with GSD, the potential contribution of hyperlipidemia to the development of atherosclerosis cannot be ignored. Herein, we present the results of noninvasive studies performed on the largest cohort of patients with GSD I to date.
METHODS
Patients with GSD and unrelated healthy control subjects were recruited to participate in a case-control study. No more than 1 patient with GSD was enrolled per family. These investigations were approved by the Institutional Review Board of the University of Florida and the Scientific Advisory Committee of the University of Florida General Clinical Research Center. Signed informed consent (and assent when appropriate) was obtained before commencement of the investigations.
Cases were recruited from the GSD Program at the University of Florida. All participants with GSD I who were 13 years or older were eligible to participate. The diagnosis of GSD was established biochemically or by liver biopsy, and GSD Ia or Ib was confirmed by mutation analysis.12 The clinical characteristics of the 28 patients with GSD (20 Ia, 8 Ib) and 23 control participants are summarized in Table I.
Table I.
Clinical characteristics
| Sex (M/F) | Age (y) | BMI (kg/m2) | TG (mg/dL) | HDL (mg/dL) | LDL (mg/dL) | TC (mg/dL) | |
|---|---|---|---|---|---|---|---|
| GSD I | 15/13 | 23 (13–41) | 26 (20–39) | 432.0 ± 307.0 n = 27 |
29.0 ± 9.1 n = 27 |
63.7 ± 52.9 n = 17 |
194.4 ± 70.0 n = 27 |
| Control | 4/19 | 23 (13–30) | 23 (17–33) | 99.6 ± 51.5 n = 17 |
60.5 ± 18.5 n = 17 |
89.4 ± 27.2 n = 17 |
169.9 ± 32.5 n = 17 |
BMI, Body mass index; TG, triglycerides; HDL, high-density lipoprotein; LDL, low-density lipoprotein; TC, total cholesterol.
The data represent the means of each category. For the age and BMI, the range is expressed in parenthesis. Data are expressed as mean ± standard deviation for the lipid values. LDL could not be calculated for 10 GSD I subjects because of extremely high triglycerides.
Study Protocol
After obtaining informed consent from the participants or parents and assent from the children, a brief history and physical was performed. For 24 hours before testing, subjects were asked to abstain from caffeine. Baseline laboratory studies were obtained at 07:30 a.m. with documentation of blood pressure, height, and weight followed by a standardized 300-calorie breakfast with less than 30% fat. Additionally, patients with GSD took their morning cornstarch dose. After breakfast, participants remained sedentary for 2 hours. RAT was performed first with the protocol outlined by Haller et al.13 Carotid IMT and BAR were performed by a cardiology research technician using methods previously described.9,10
Statistical Analysis
The primary hypothesis (stated as null) was that the target distribution of IMT would be the same for GSD and control subjects. The study was powered for this objective, with secondary objectives requiring confirmation in a future study before declaring any associations as significant. The protocol goal was to recruit 67 GSD cases and 67 control subjects, which could detect a difference in mean IMT of 0.02 mm (half an anticipated standard deviation) at P = .05 two-sided and 80% power. After 1.5 years of recruitment, it became clear that the 2-year accrual goals could not be met, and, hence, the study was closed early. The closure decision was not based on any interim analysis of the data. Secondary analysis tested a similar hypothesis for BAR by vessel and site, and test for differences in the augmentation index (corrected to 75 beats per minute) and by lipid profiles. The primary hypothesis was tested using analysis of covariance with age, sex, and body mass index as covariates to compare cases vs. controls. The t-statistics generated from the analysis of covariance models determined the P values (2-sided), and 95% confidence intervals for the differences in means, adjusted for the covariates.
RESULTS
BAR and IMT were not performed in 1 patient in whom hypoglycemia developed, and RAT was not performed in 2 control subjects because the technician was absent. The major case-control comparisons appear in Table II. Mean left distal IMT was greater in the GSD I cohort than in the control group. Mean augmentation index measured by RAT was higher in the GSD cohort than in the control group. Mean brachial artery diameter was not significantly different in the GSD cohort than in the control group (Figure). Although specific results are not shown, the comparisons, restricted to GSD Ia versus control subjects yield the same qualitative conclusions.
Table II.
Comparison of results from noninvasive testing
| Mean | SD | N | P value | P value Adjusted for age, sex, and BMI | |
|---|---|---|---|---|---|
| Control | |||||
| AI75 | 2.38 | 8.72 | 21 | <.001 | <.001 |
| BAR_PCT | 6.6 | 5.1 | 23 | .84 | .46 |
| IMT | 0.457 | 0.039 | 23 | .0032 | .0020 |
| GSD | |||||
| AI75 | 16.4 | 14.0 | 27 | ||
| BAR_PCT | 6.3 | 4.9 | 27 | ||
| IMT | 0.500 | 0.055 | 28 | ||
All P values by 2-sided 2 sample t tests, unadjusted and adjusted for covariates. IMT is the Primary endpoint per protocol. Point estimate (95% confidence intervals for difference of means, adjusted for covariates).
Figure.
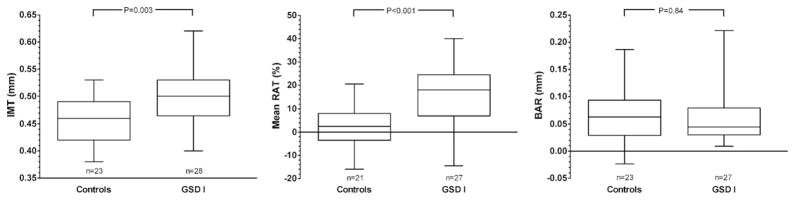
Box-and-whisker plot for IMT, RAT, and BAR. Boxes represent the 25th and 75th percentiles, and the median is represented in each box. The range of observations is depicted by the whiskers.
DISCUSSION
Cardiovascular disease is the leading cause of death in the United States, and hyperlipidemia is a known risk factor. Nonetheless, patients with GSD and hyperlipidemia were believed to be exempt from the consequences of this risk. This conclusion, however, was based on limited data. We have determined, using the largest number of patients with GSD studied to date, that GSD is associated with arterial dysfunction. Both carotid IMT and RAT measurement indicate arterial dysfunction in contrast to the previous studies.
Before this study, arterial dysfunction had not been found in patients with GSD, and researchers postulated 3 mechanisms that could protect patients with GSD from CVD. Increased antioxidant potential in GSD was proposed by Fathi and Marwick9 on the basis of the calculation of an antioxidant formula–total radical-trapping antioxidant parameter (TRAP). The 4 contributors to plasma TRAP are uric acid, ascorbate, alpha-tocopherol, and sulfhydryl groups of proteins. Fathi and Marwick9 found that TRAP levels were elevated in the GSD Ia group only as a result of elevated uric acid levels in these patients. Hyperuricemia occurs in 89% of patients with GSD because of both increased production via shunting through the pentose phosphate pathway and decreased elimination at the kidney where lactate competes for the renal anion transporter. Recent evidence has confirmed an oxidant role for uric acid, making hyperuricemia predictive for CVD.14 Thus the high frequency of hyperuricemia in GSD would seem to negate increased antioxidant potential as a protective mechanism.
The second proposed protective mechanism similarly lacks support. The concept of decreased platelet aggregation in the study by Muhlhausen et al7 was based on evidence of a relative von Willebrand deficiency. Patients were noted to have a bleeding diathesis, and von Willebrand levels were lower than population norms. Although hypercoagulable disorders are strongly associated with increased CVD,15 evidence to support a decreased risk of CVD from von Willebrand disease is lacking. Although patients with GSD exhibit a bleeding diathesis when in poor metabolic control or acutely ill, with improved dietary control, measurements of bleeding time and platelet aggregation normalize.16
The third mechanism, increased reverse cholesterol transport, is supported by the up-regulation of cellular cholesterol transporters. Cellular cholesterol efflux is mediated by 2 transport proteins—scavenger receptor class B type 1 (SR-B1) and ATP-binding cassette transporter A1 (ABCA1). Reverse cholesterol transport is the pathway by which peripheral cell cholesterol is returned through the plasma to the liver for excretion.17 The efflux of cholesterol from cells and its incorporation primarily into HDL, as well as lipoproteins and apolipoproteins, is believed to be the initial step in reverse cholesterol transport. HDL cholesterol levels are inversely correlated with the incidence of coronary artery disease, and decreased HDL cholesterol concentrations are the most frequent abnormality seen in premature CVD.18 Even though HDL levels are typically low in GSD Ia, both mice and human beings with GSD Ia demonstrate increased cellular cholesterol efflux. The mechanisms differ between species. In mice, preliminary studies demonstrate that both SRB1 and ABCA1 transporters are increased in contrast to human beings in whom only SRB1 is increased.5 The effect of multiple cardiovascular risk factors on vascular function may be attenuated by increased reverse cholesterol transport.
The higher mean augmentation index and carotid IMT demonstrated in this study provides a link between hyperlipidemia and vascular dysfunction. Further studies are needed, however, to demonstrate that the vascular differences are clinically significant, and that proactive management to lower lipid concentrations will improve clinical outcome. Hyperlipidemia in GSD I occurs in the setting of glycogenolysis. The inability of glucose-6-phosphate to be released as glucose results in shunting of glycogen breakdown products through the glycolytic pathway, which increases formation of acetyl CoA and synthesis of fatty acids and cholesterol in the liver.19 With improvement in metabolic control, lipids normalize in GSD I, and aggressive treatment of GSD is therefore indicated when hyperlipidemia is present. If hypertriglyceridemia persists after maximizing control, pharmacologic therapy can be considered. Three classes of medication are appropriate for treatment of hypertriglyceridemia with or without concomitant hypercholesterolemia: fibric acid derivatives, niacin, and omega-3 fatty acids. Statins, which primarily lower low-density lipoprotein cholesterol, also lower triglycerides as much as 50% and thus can be used in GSD-related hyperlipidemia when low-density lipoprotein is also elevated. Concurrent use of fibrates and statins is associated with increased creatinine kinase, risk of rhabdomyolysis, and myoglobinuria.20
Acknowledgments
The authors would like to thank the nurses on the GCRC along with Dr. Karen Hamilton and Mr. John Larocca for technical help performing the carotid IMT and BAR testing.
Support for this project was provided by the Association for Glycogen Storage Disease, Scott Miller Glycogen Storage Disease Research Fund, the National Center for Research Resources General Clinical Research Center grant M01 RR 00082, National Institutes of Health, and NIH Mentored Career Award K23 RR 017560 (DAW).
Glossary
- BAR
Brachial artery reactivity
- CVD
Cardiovascular disease
- GSD
Glycogen storage disease
- HDL
High-density lipoprotein
- IMT
Intima media thickness
- RAT
Radial artery tonometry
- TRAP
Total radical-trapping antioxidant parameter
Footnotes
The authors declare no conflicts of interest.
References
- 1.Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 2.Levy E, Letarte J, Lepage G, Thibault L, Roy CC. Plasma and lipoprotein fatty acid composition in glycogen storage disease type I. Lipids. 1987;22:381–5. doi: 10.1007/BF02537265. [DOI] [PubMed] [Google Scholar]
- 3.Ubels FL, Rake JP, Slaets JP, Smit GP, Smit AJ. Is glycogen storage disease 1a associated with atherosclerosis? Eur J Pediatr. 2002;161:S62–4. doi: 10.1007/s00431-002-1006-9. [DOI] [PubMed] [Google Scholar]
- 4.Lee PJ, Celermajer DS, Robinson J, McCarthy SN, Betteridge DJ, Leonard JV. Hyperlipidaemia does not impair vascular endothelial function in glycogen storage disease type 1a. Atherosclerosis. 1994;110:95–100. doi: 10.1016/0021-9150(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen AD, Pan CJ, Weinstein DA, Chou JY. Increased scavenger receptor class B type 1-mediated cellular cholesterol efflux and antioxidant capacity in the sera of glycogen storage disease type Ia patients. Mol Genet Metab. 2006;89:233–8. doi: 10.1016/j.ymgme.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Wittenstein B, Klein M, Finckh B, Ullrich K, Kohlschutter A. Plasma antioxidants in pediatric patients with glycogen storage disease, diabetes mellitus, and hypercholesterolemia. Free Radic Biol Med. 2002;33:103–10. doi: 10.1016/s0891-5849(02)00863-8. [DOI] [PubMed] [Google Scholar]
- 7.Muhlhausen C, Schneppenheim R, Budde U, Merkel M, Muschol N, Ullrich K, et al. Decreased plasma concentration of von Willebrand factor antigen (VWF:Ag) in patients with glycogen storage disease type Ia. J Inherit Metab Dis. 2005;28:945–50. doi: 10.1007/s10545-005-0184-9. [DOI] [PubMed] [Google Scholar]
- 8.Russo G, Leopold JA, Loscalzo J. Vasoactive substances: nitric oxide and endothelial dysfunction in atherosclerosis. Vasc Pharmacol. 2002;38:259–69. doi: 10.1016/s1537-1891(02)00250-1. [DOI] [PubMed] [Google Scholar]
- 9.Fathi R, Marwick TH. Noninvasive tests of vascular function and structure: why and how to perform them. Am Heart J. 2001;141:694–703. doi: 10.1067/mhj.2001.114972. [DOI] [PubMed] [Google Scholar]
- 10.van Dam MJ, de Groot E, Clee SM, Hovingh GK, Roelants R, Brooks-Wilson A, et al. Association between increased arterial-wall thickness and impairment in ABCA1-driven cholesterol efflux: an observational study. Lancet. 2002;359:37–42. doi: 10.1016/S0140-6736(02)07277-X. [DOI] [PubMed] [Google Scholar]
- 11.Slyper AH. Clinical review 168: what vascular ultrasound testing has revealed about pediatric atherogenesis, and a potential clinical role for ultrasound in pediatric risk assessment. J Clin Endocrinol Metab. 2004;89:3089–95. doi: 10.1210/jc.2003-030644. [DOI] [PubMed] [Google Scholar]
- 12.Elpeleg ON. The molecular background of glycogen metabolism disorders. J Pediatr Endocrinol Metab. 1999;12:363–79. doi: 10.1515/jpem.1999.12.3.363. [DOI] [PubMed] [Google Scholar]
- 13.Haller MJ, Samyn M, Nichols WW, Brusko T, Wasserfall C, Schwartz RF, et al. Radial artery tonometry demonstrates arterial stiffness in children with type 1 diabetes. Diabetes Care. 2004;27:2911–7. doi: 10.2337/diacare.27.12.2911. [DOI] [PubMed] [Google Scholar]
- 14.Lippi G, Montagnana M, Franchini M, Favaloro EJ, Targher G. The paradoxical relationship between serum uric acid and cardiovascular disease. Clin Chim Acta. 2008;392:1–7. doi: 10.1016/j.cca.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 15.McDonald L, Edgill M. Changes in coagulability of the blood during various phases of ischaemic heart-disease. Lancet. 1959;1:1115–8. doi: 10.1016/s0140-6736(59)90706-8. [DOI] [PubMed] [Google Scholar]
- 16.Marfaing-Koka A, Wolf M, Boyer-Neumann C, Meyer D, Odievre M, Labrune P. Increased levels of hemostatic proteins are independent of inflammation in glycogen storage disease type Ia. J Pediatr Gastroenterol Nutr. 2003;37:566–70. doi: 10.1097/00005176-200311000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Fielding CJ, Fielding PE. Cellular cholesterol efflux. Biochem Biophys Acta. 2001;1533:175–89. doi: 10.1016/s1388-1981(01)00162-7. [DOI] [PubMed] [Google Scholar]
- 18.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 19.Bandsma RH, Prinsen BH, de van Der Velden MS, Rake JP, Boer T, Smit GP, et al. Increased de novo lipogenesis and delayed conversion of large VLDL into IDL particles contribute to hyperlipidemia in Glycogen Storage Disease type 1a. Pediatr Res. 2008;63:702–7. doi: 10.1203/PDR.0b013e31816c9013. [DOI] [PubMed] [Google Scholar]
- 20.Shek A, Ferrill MJ. Statin-fibrate combination therapy. Ann Pharmacother. 2001;35:908–17. doi: 10.1345/aph.10315. [DOI] [PubMed] [Google Scholar]