

Abstract
Blastomyces dermatitidis , the etiologic agent of blastomycosis, belongs to a group of thermally dimorphic fungi that change between mold (22°C) and yeast (37°C) in response to temperature. The contribution of structural proteins such as septins to this phase transition in these fungi remains poorly understood. Septins are GTPases that serve as a scaffold for proteins involved with cytokinesis, cell polarity, and cell morphology. In this study, we use a GFP sentinel RNA interference system to investigate the impact of CDC3, CDC10, CDC12, and ASPE on the morphology and phase transition of B. dermatitidis. Targeting CDC3, CDC10, and CDC12 by RNA interference resulted in yeast with aberrant morphology at 37°C with defects in cytokinesis. Downshifting the temperature to 22°C promoted the conversion to the mold phase, but did not abrogate the morphologic defects. CDC3, CDC10, and CDC12 knockdown strains grew as mold with curved, thickened hyphae. Knocking down ASPE transcript did not alter morphology of yeast at 37°C or mold at 22°C. Following an increase in temperature from 22°C to 37°C, all septin knockdown strains were able to revert to yeast. In conclusion, CDC3, CDC10, and CDC12 septin-encoding genes are required for proper morphology of yeast and hyphae, but are dispensable for the phase transition.
Keywords: dimorphic fungi, septin, phase transition, morphogenesis
Introduction
Blastomyces dermatitidis, the etiologic agent of blastomycosis, belongs to a group of human pathogenic fungi that exhibit thermal dimorphism [1]. These organisms, which include Histoplasma capsulatum, Coccidioides spp., Paracoccidioides brasiliensis, Sporothrix schneckii, and Pencillium marneffei, undergo a reversible, temperature-dependent shift in their morphology known as phase transition [1]. In the environment (22°C), they grow as molds and produce infectious conidia. Following the disruption of soil, aerosolized mycelial fragments and conidia that are inhaled into the lungs of a mammalian host (37°C) convert into pathogenic yeasts (or spherules for Coccidioides spp.) to cause disease [1–3]. Collectively, the dimorphic fungi infect several million people each year, including persons with either intact or impaired immune defenses [4,5]. Clinical manifestations are heterogeneous and include subclinical illness, pneumonia, acute respiratory distress syndrome, and disseminated disease [1,5].
The conversion between mold and yeast is a complex process that requires the fungal cell to sense changes in temperature and undergo a radical rearrangement in morphology [1]. Our understanding of how the phase transition is regulated is limited to only a handful of genes including those that alter intracellular signaling or transcription including DRK1, RYP1-3, and SREB [6–10]. The impact of genes that encode ultrastructural proteins such as septins, remains largely unexplored in the endemic dimorphic fungi. Septins are GTP-binding proteins that form heteroligomeric complexes at sites of bud formation in yeast and growth at the hyphal tips in filamentous fungi [11–13]. Septin complexes serve as a scaffold to facilitate localization of proteins involved with cytokinesis and cell polarity [11]. In fungi such as Saccharomyces cerevisiae, septins also form a barrier to prevent back-diffusion of proteins from the daughter to the mother cell [11]. In several fungi including S. cerevisiae, Ustilago maydis, Cryptococcus neoformans, and Candida albicans, the impact of septins on morphology can be influenced by temperature [14–17]. The link between morphology and temperature involving septins has yet to be fully elucidated in the dimorphic fungi.
In this study, we use RNA interference to investigate the impact of septin-encoding genes CDC3, CDC10, CDC12, and ASPE on the morphology and phase transition of Blastomyces dermatitidis. This research expands upon our analysis of B. dermatitidis CDC11 on morphogenesis [12]. We found that B. dermatitidis strains knocked down for CDC3, CDC10, and CDC12 grow as misshapen yeast at 37°C and aberrant hyphae at 22°C. Knocking down ASPE transcript did not impact the morphology of yeast or hyphae. Knockdown strains did not exhibit defects in thermotolerance and were able to convert between mold and yeast.
Materials and methods
Strains and media
Transformants were generated from a reporter strain of B. dermatitidis that was engineered to express green fluorescent protein (GFP) [12]. This strain, T53-19 GFP, was derived from ER-3 (ATCC MYA-2586), which forms conidia but is hypovirulent in a murine model of infection [6]. GFP expression in the reporter is driven by the BAD1 promoter, which is expressed at 37°C, but not 22°C [12]. The H2AB promoter, which is active at 37°C and 22°C when analyzed by quantitative real-time PCR (data not shown), drives transcription of the RNA hairpin responsible for RNA interference. The GFP reporter strain and transformants were grown on 3M medium [18] or Histoplasma macrophage medium (HMM) [19], with or without supplementation with 100 μg/ml hygromycin B (AG Scientific, San Diego, CA) at 37°C and 22°C. Liquid HMM, with or without supplementation with 100 μg/ml hygromycin B, was used for the growth of yeast and mold phases at 37°C and 22°C, respectively. Agrobacterium tumefaciens strain LBA1100 harboring the Ti helper plasmid pAL1100 (gift from C. van den Hondel; Leiden University, The Netherlands) was maintained on Luria-Bertani (LB) medium supplemented with 0.1% glucose, spectinomycin 100 μg/ml, and kanamycin 100 μg/ml following transformation with a binary vector [20].
Transformation of B. dermatitidis GFP reporter strain
Agrobacterium-mediated DNA transfer was used to transform T53-19 GFP reporter strain with vectors targeting CDC3, CDC10, CDC12, and ASPE for RNA inferference (RNAi). Control strains were transformed with pH2AB, which only targeted GFP (GFPi-only control), or pBTS4, which lacked an RNAi construct (non-RNAi vector control). For each septin gene, two separate, non-overlapping regions in the open reading frame (ORF) were targeted for RNAi (Fig. 1). Primer design was based on the sequenced genome of B. dermatitidis strain SLH14081 (www.broadinstitute.org). Primers used to amplify the septin loci targeted for RNAi were as follows:
Fig. 1.
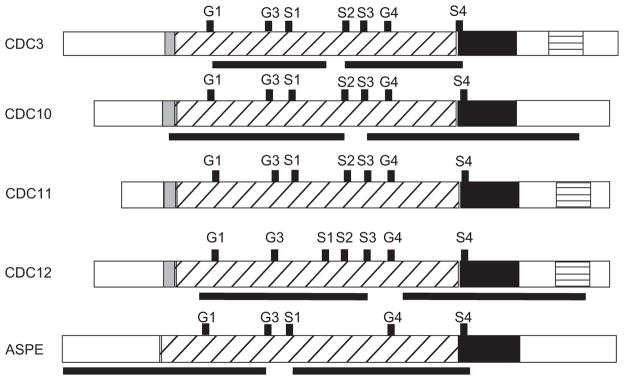
Septin regions targeted for RNA interference. Black bars below CDC3, CDC10, CDC12, and ASPE correspond to the regions targeted for RNA interference. In addition, conserved motifs (G1, G3, G4, S1-S4), GTPase domain, polybasic region, septin unique element, and coiled-coil domains are shown for all septins. The polybasic region is shaded gray. Diagonal lines represent the GTPase domain. The septin unique element is black. Horizontal lines indicate the coiled-coil domain.
CDC3 5′ locus forward (CDC3 5′ F) GGGGAC AAGTTTGTACAAAAA-AGCAGGCTtgttcgtcgcaagttgaccg and reverse (CDC3 5′ R) GGGGACC ACTTTGTACAAGAAAGCTGGGTTGGGTTGGATAAAGTAGACACAGGC;
CDC3 3′ locus forward (CDC3 3′ F) GGGGAC A AG T T T G TAC A A A A A AG C AG G C T T C GCTAAGGCTGATACTCTCACCG and reverse (CDC3 3′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTCGCTCTTCTTCCTGTTTGACCG;
CDC10 5′ forward (CDC10 5′ F) GGGGACAA GTTTGTACAAAAAAGCAGGCTAGATCGAGCGGAAACTACTGA and reverse (CDC10 5′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTG CTGAAAAGGCATTGAGAA;
CDC10 3′ locus forward (CDC10 3′ F) GGGGACA AGTTTGTACAAAAAAGCAGGCTTGATATCGTCGTC-CTGAAGAAAC and reverse (CDC10 3′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTGTCGGCTGCCAC-CTGAATGCG;
CDC12 5′ locus forward (CDC12 5′ F) GGGGACA AGTTTGTACAAAAAAGCAGGCTTAGCTCCCAAATCAACGGTATGC and reverse (CDC12 5′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTTATCCAGGGGCTTTAGGGTATGAC;
CDC12 3′ locus forward (CDC12 3′ F) GGGGAC AAGTTTGTACAAAAAAGCAGGCTGTTGCCAAAGCCGATACAC and reverse (CDC12 3′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTAAAGTTAGCATTGGCACAT;
ASPE 5′ locus forward (ASPE 5′ F) GGGGACA AGTTTGTACAAAAAAGCAGGCTCTCAAACCGCAAGACGAACA and reverse (ASPE 5′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTCACGCAGCTGCAGATCAACAACAT; and
ASPE 3′ locus forward (ASPE 3′ F) GGGGACA AGTTTGTACAAAAAAGCAGGCTTGCGCGACACTCACATTCACTGC and reverse (ASPE 3′ R) GGGGACCACTTTGTACAAGAAAGCTGGGTCTCCATCGCTCGTACCACACTTTC.
Following PCR amplification using Elongase (Invitrogen, Carlsbad, CA), the amplicons were separated using gel electrophoresis and purified using a QiaQuick PCR purification kit (Qiagen, Valencia, CA). Once sequences of the amplicons were confirmed, RNAi vectors were constructed using the Gateway cloning system (Invitrogen, Carlsbad, CA) as described by Krajaejun et al. [12].
A. tumefaciens transformation of B. dermatitidis T53-19 GFP reporter strain was performed as previously described [7]. In brief, 2 × 107 yeast cells were co-cultured with 6 × 108 A. tumefaciens, which harbored the RNAi vector, on Biodyne A membranes on induction medium with acetosyringone (IMAS). Following 72 h of incubation at 22°C, the membranes were transferred to 3M medium supplemented with 100 μg/ml hygromycin and 200 μM cefotaxime, and incubated at 37°C. Strains knocked down for CDC3, CDC10, CDC12, and ASPE were screened for loss of GFP intensity using VersaDoc imaging system (BioRad, Hercules, CA) and fluorescent microscopy (Olympus BX60). Control strains were screened for GFP expression (reporter strain, non-RNAi vector control) or loss of fluorescence (GFPi-only).
Conidial Formation studies
To investigate for defects in spore production, yeast from control and septin RNAi strains were inoculated at a density of 2 × 107 yeast per plate, in duplicate, and incubated at 22°C for 14 days. Conidia were harvested from each plate in phosphate buffered saline by manual disruption [21] and counted using a hemocytometer. Control and knockdown strains formed conidia poorly on 3M medium, which was unexpected because B. dermatitidis usually produces abundant conidia on this medium [7]. Subsequent studies used Bird medium to promote conidial production [22].
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from control and knockdown strains harvested from 3M agar at 37°C as previously described by Gauthier et al. [7]. Following treatment with Turbo DNase (BioRad) and additional purification using an RNeasy kit (Qiagen), total RNA was converted to cDNA using iScript cDNA Synthesis Kit (Bio-Rad). PCR was performed with SsoFast EvaGreen Supermix (Bio-Rad), using a MyiQ real-time PCR detection system (Bio-Rad). Reactions were performed in triplicate. Bio-Rad iCycle conditions were as follows: 1 cycle 95°C × 30 sec, followed by 40 cycles at 95°C for 5 sec, 60°C for 10 sec. Primers used for qRT-PCR were CDC3 5′ forward CCCTCTTCAAGCAAAGGATTC and reverse GTTTTCGGCGATGGTTTC; CDC10 5′ forward AACGCTCAAATCAAGGACATC and reverse CGCAATG GTTCTCGTCTTC; CDC11 5′ forward GGTGGGATG GG GATACAAG and reverse ATCGCTATGTTAGGA AAGGTCTC; CDC12 5′ forward ACGCTTCACCGAAC AAGT and reverse TGTTTGATAGCTGCATGAGT; ASPE 5′ forward TGGAGGTCTTGACGCTTGAG and reverse CTTGGGCTTGGGCATCAG; alpha-tubulin forward GGTCACTACACCATCGGAAAG and reverse CTGGAG-GGACGAACAGTTG. Transcript abundance of CDC3, CDC10, CDC12, and ASPE were normalized relative to transcript abundance of alpha-tubulin. Following normalization, data was analyzed using 2 −ΔΔCt [23].
Cell staining
B. dermatitidis nuclei were stained using sytox green and visualized using fluorescent microscopy (Olympus BX60). Sytox staining was performed on fixed cells (2% paraformaldehyde for 1 h) as described by Sullivan et al. [20]. Cell wall and septal chitin were stained in yeast and mold cells using calcofluor white (Sigma-Aldrich, St. Louis, MO). Cells fixed in 2% paraformaldehyde were washed with phosphate buffered saline (PBS), stained with 0.025 mg/ml of calcofluor white (Sigma-Aldrich, St. Louis, MO) for 30 min at room temperature, and washed three times with PBS containing 0.2% BSA. Stained cells were visualized by fluorescent microscopy using an excitation filter with a range of 400–410 nm.
Statistical analysis
Analysis of variance (ANOVA) was used to compare knockdown septin strains with controls for quantitative abnormalities in morphology and transcript abundance. For analysis of growth rate, repeated measures ANOVA was used. Probability values were adjusted based on the Dunnett’s procedure or Tukey’s HSD (Honestly Significant Difference).
Results
Identification and characterization of septins in Blastomyces dermatitidis
Using tBLASTn to search the sequenced genomes of B. dermatitidis strains 26199 (http://genome.wustl.edu/genomes/fungi) and SLH14081 (www.broadinstitute.org), we identified septins from all 5 clades including CDC10 (clade 1A), CDC3 (clade 2A), CDC11 (clade 3), CDC12 (clade 4), and ASPE (clade 5) (Fig. 1) [24]. Similar to other filamentous fungi and pathogenic basidomycetes, the genome of B. dermatitidis lacked genes for accessory (SHS1/SEP7) and sporulation-regulated (SPR3, SPR8) septins, as found in S. cerevisiae, Candida spp., and Kluyveromyces lactis [24–28]. We have previously described the percent identity and similarity of the amino acid sequences of B. dermatitidis septin homologs to S. cerevisiae, Schizosaccharomyces pombe, C. albicans, and Aspergillus nidulans [12]. Subsequent to that publication, the genomes of several dimorphic fungi have been sequenced (www.broadinstitute.org). B. dermatitidis septins CDC3, CDC10, CDC11, and CDC12 show a high degree of amino acid identity and similarity with H. capsulatum, P. brasiliensis, Coccidioides immitis, and Coccidioides posadasii (Table 1).
Table 1.
Comparison of Blastomyces dermatitidis septins against other dimorphic fungi.
| B. dermatitidis Septin |
H. capsulatum
|
P. brasiliensis
|
||||
|---|---|---|---|---|---|---|
| HS | Identity | Similarity | HS | Identity | Similarity | |
| CDC3 | 896 | 98.0% | 99.0% | 903 | 93.7% | 95.7% |
| CDC10 | 671 | 97.0% | 98.0% | 664 | 95.0% | 97.0% |
| CDC11 | 681 | 99.7% | 100.0% | 663 | 97.0% | 99.0% |
| CDC12 | 747 | 96.7% | 97.6% | 748 | 97.0% | 97.0% |
| ASPE | 795 | 87.3% | 92.0% | 865 | 76.7% | 84.0% |
|
| ||||||
| B. dermatitidis Septin |
C. immitis
|
C. posadasii
|
||||
| HS | Identity | Similarity | HS | Identity | Similarity | |
|
| ||||||
| CDC3 | 760 | 92.0% | 95.0% | 848 | 92.0% | 95.0% |
| CDC10 | 633 | 92.2% | 94.0% | 646 | 94.0% | 95.0% |
| CDC11 | 612 | 91.8% | 95.5% | 623 | 91.0% | 95.0% |
| CDC12 | 594 | 88.5% | 92.0% | 721 | 94.0% | 96.0% |
| ASPE | 539 | 66.5% | 74.0% | 654 | 61.0% | 70.0% |
B. dermatitidis septin amino acid sequences were compared to H. capsulatum (strains G186R, H88, Nam1), P. brasiliensis (strains Pb01, Pb03, Pb18), C. immitis (strains H538, RS, RMSCC 3703, RMSCC 2394), and C. posadasii (strains Silveira, RMSCC 3488) using protein-protein blast from the Broad Institute (www.broadinstitute.org). HS refers to high score.
Analysis of the predicted amino acid sequences revealed that B. dermatitidis septins contained conserved motifs including septin unique elements, sep motifs, polybasic regions, guanine nucleotide binding regions, and coiled-coil domains (Fig. 1). Septin unique elements were identified near the C-terminus for all predicted B. dermatitidis septins. The function of this 53 amino acid motif, which is conserved among septin proteins, remains unknown [28]. Similar to septin unique element, B. dermatitidis septins possessed Sep1–4 motifs, which are conserved in 86–98% of septins [24]. With the exception of a homolog of ASPE, all B. dermatitidis septins had an N-terminal polybasic region, which promotes the binding of septins to plasma membrane phosphoinositides [29]. As expected, B. dermatitidis septins contained domains involved with guanidine nucleotide binding, including G1 (GxxxxKG [S/T]), G3 (DxxG), and G4 (NKxD). G1 and G3 domains are predicted to bind the phosphates of GTP, whereas G4 impacts binding specificity [30]. Three B. dermatitidis septins, CDC3, CDC11, and CDC12, were predicted to have a C-terminal coiled-coil domain by the multicoil program [31]. The coiled-coil domain facilitates septin-protein interactions and promotes some septin–septin interactions (i.e. Cdc3-Cdc11 in S. cerevisiae) [29].
RNAi of CDC3, CDC10, and CDC12 in B. dermatitidis results in misshapen yeast at 37°C
To investigate the function of CDC3, CDC10, CDC12, and ASPE on B. dermatitidis morphology, we knocked down the expression of these genes using a GFP sentinel RNAi system. In this system, RNAi simultaneously reduces transcription of target genes (i.e., CDC3) and GFP. The loss of GFP fluorescence indicates successful knockdown of the target gene [12]. For each septin gene, two non-overlapping regions were targeted for RNAi (Fig. 1). Strains knocked down for CDC3, CDC10, and CDC12 displayed a loss of GFP fluorescence and grew as large, misshapen yeast at 37°C (Fig. 2A). In contrast, ASPE knockdown strains and control strains (GFP reporter, non-RNAi vector control, GFPi-only) grew as broad-based budding yeast (Fig. 2A). Quantitative analysis of 3–5 independent knockdowns revealed that the majority of cells (55–85%) for CDC3, CDC10, and CDC12 RNAi strains grew as aberrant yeast (bean-shaped, large ovoid forms, and elongated) when compared to control and ASPE RNAi strains (adjusted p-value < 0.05; Figs. 2A, 3).
Fig. 2.

Phenotype of CDC3, CDC10, CDC12, & ASPE knockdown strains at 37°C and 22°C. (A) T53-19 GFP (reporter) and non-RNAi vector (NRV) control strains grew as broad-based budding yeast with intense green fluorescence. GFP RNAi only (GFPi) control displayed normal yeast morphology, but reduced GFP intensity. CDC3, CDC10, and CDC12 knockdown strains grew as large, misshapen yeast with reduced GFP intensity. ASPE-GFP knockdown strains had reduced GPF intensity and similar morphology to the control strains. Scale bar is 10 μm. Cells were grown on 3M agar at 37°C. (B) Following a downshift in temperature from 37°C to 22°C, control strains grew as thin, smooth, branched hyphae. In contrast, CDC3, CDC10 and CDC12 knockdown strains grew as curved mold with a knobby appearance. ASPE RNAi strains have a similar morphology as the control strains. Corresponding fluorescent images are not shown for hyphal cells because GFP, which functions as a reporter, is under control of the BAD-1 promoter and is not expressed at 22°C. The H2AB promoter, which drives transcription of the hairpin responsible for RNA interference is active at both 37°C and 22°C. Scale bar is 10 μm. Cells were grown on 3M agar at 22°C.
Fig. 3.

Quantification of abnormal yeast morphology in septin knockdown strains. The majority of yeast cells for B. dermatitidis strains knocked down for CDC3, CDC10, and CDC12 had abnormal morphology (55–85%) when compared to control strains (<15%). This difference was statistically significant (adjusted P value <0.05; represented by an asterisk). ASPE knockdown strains grew as normal shaped yeast similar to controls. At least 400 cells were analyzed per strain. Numbers below the histogram bars represent the different transformants that were analyzed. 5′ and 3′ refer to the gene region targeted for RNA interference. GFP is the GFP reporter strain (T53-19 GFP). GFPi refers to the control strain knocked down for GFP only and NRV refers to the non-RNAi vector control.
Quantitative real-time PCR (qRT-PCR) demonstrated reduction of transcript abundance for CDC3, CDC10, CDC12, and ASPE (Fig. 4). Using the 2 −ΔΔCt method, there was a 73% reduction in transcript abundance of CDC3 for CDC3 RNAi, 22.2% reduction of CDC10 for CDC10 RNAi, 54.3% reduction of CDC12 for CDC12 RNAi, and 43.4% reduction of ASPE for ASPE RNAi strains when compared to controls (Fig. 4). Although the morphologic appearance of CDC3, CDC10, and CDC12 RNAi strains were similar, knockdown of non-targeted septins did not occur (Fig. 4).
Fig. 4.

Quantitative real-time PCR analysis. qRT-PCR demonstrated a reduction in transcript abundance for CDC3, CDC10, CDC12, and ASPE for their respective knockdown strains when compared to GFP reporter and non-RNAi vector (NRV) control strains. NRV control had similar transcript levels to the reporter strain. Quantitative RT-PCR data was analyzed using the 2 −ΔΔCt method and values are expressed as fold change in gene transcript relative to the GFP reporter strain. Ct values were normalized to α-tubulin with ΔCt = (Ct of gene of interest) − (Ct of α-tubulin). ΔΔCt = (ΔCt of NRV or knockdown strain) − (ΔCt of GFP reporter). ANOVA was used to analyze transcript abundance between septin knockdown strains and controls. Transcript reduction in knockdown strains was not statistically significant when compared to controls; however, the sharp differences in morphology between knockdown and controls indicated that septin transcript in knockdown strains fell below a threshold required for normal cellular function.
The large size of the misshapen yeast along with a slow growth rate at 37°C (Fig. 5) suggested a defect in cytokinesis in knockdown strains when compared to controls. In yeast, nuclear and cellular divisions are often coordinated and disruption of septins in U. maydis, C. neoformans, and S. cerevisiae can result in accumulation of nuclei [14,16,32]. We used sytox green to stain and quantify B. dermatitidis nuclei under fluorescent microscopy. Strains knocked down for CDC3, CDC10, CDC12, and ASPE demonstrated a similar number of nuclei as reporter, non-RNAi vector control, GFPi-only controls (range 2–8 nuclei/cell). To assess for mislocalization of septa and alterations in chitin deposition, we stained control and knockdown strains with calcofluor white, which binds septal and cell wall chitin molecules. In the septin RNAi strains, cell wall chitin distribution appeared normal and misplaced septa were not present (data not shown).
Fig. 5.

Growth curve for septin knockdown strains. At 37°C, yeast strains knocked down for CDC3, CDC10, and CDC12 grew substantially slower than T53-19 GFP reporter and non-RNAi vector control strains. ASPE knockdown yeast grew faster than other septin RNAi strains, but slower than control strains. By day 2 in liquid culture, all septin knockdown strains demonstrated a statistically significant reduction in growth when compared to the reporter and non-RNAi controls (adjusted P value < 0.05). Yeast cultures were grown in liquid HMM medium over a period of 6 days and growth was measured daily by absorbance at 600 nm. Numbers next to CDC3, CDC10, CDC12, and ASPE represent the strains analyzed. A representative experiment is shown.
RNAi of CDC3, CDC10, and CDC12 alters hyphal morphology but not the phase transition
Because the interruption of septin function can impact cell morphology in a temperature-dependent manner[15,16,29], we investigated how B. dermatitidis septins affect morphogenesis at 22°C, as well as the phase transition. The H2AB promoter, which drives transcription of the RNAi hairpin, is active at 37°C and 22°C. Following a downshift in temperature from 37°C to 22°C, B. dermatitidis control and septin-RNAi strains fully converted to mold at 22°C (Fig. 2B). Knocking down CDC3, CDC10, CDC12, and ASPE did not delay the phase transition as all strains began the conversion within 24 h of 22°C incubation. Control and ASPE knockdown strains grew as thin, smooth, branching hyphae, whereas strains knocked down for CDC3, CDC10, and CDC12 had curved, thick, knobby appearing hyphae (Fig. 2B). Despite abnormal hyphal appearance for CDC3, CDC10, and CDC12 knockdown strains, chitin distribution by calcoflour staining was similar to control strains (data not shown). Previous investigation of B. dermatitidis CDC11 demonstrated that knocking down this septin resulted in reduced production of conidia on 3M medium at 22°C. Following 14 days of incubation at 22°C on 3M medium, CDC3, CDC10, CDC12, and ASPE knockdown strains, along with control strains (reporter, non-RNAi vector control, GFPi-only), formed conidia poorly. Subsequent experiments on Bird medium improved sporulation efficiency but production of conidia was similar for septin RNAi and control strains (data not shown).
The ability for B. dermatitidis to convert from infectious mold to pathogenic yeast at 37°C is essential for virulence [6]. Proper septin function in U. maydis and C. neoformans contributes towards survival at elevated temperatures [15,16]. To investigate the role of B. dermatitidis septins on the conversion from mold to yeast, we increased the temperature from 22°C to 37°C. Knocking down CDC3, CDC10, CDC12, and ASPE did not delay the conversion to yeast when compared to controls, with all strains converting within 4 days of the upward shift in temperature. CDC3, CDC10, and CDC12 RNAi strains still grew as misshapen yeast. ASPE RNAi strains grew a broad-based budding yeast, similar to control strains.
Discussion
Septin proteins have been identified in mammals, nematodes, insects, microsporidia, and fungi [24]. The genomes of most animals contain multiple septin-encoding genes including two in Caenorhabditis elegans (UNC-59, UNC-61), five in Drosphila melanogaster (SEP1, 2, 4, 5 and PNUT), and 14 for Homo sapiens (SEPT1-14) [33–35]. Similarly, fungi have multiple septin-encoding genes [24]. In B. dermatitidis, we have identified five septin genes including CDC3, CDC10, CDC11, CDC12, and ASPE. Homologs to SHS1/SEP7, as well as sporulation-specific septins (SPR3, SPR28) were not present in the B. dermatitidis genome. Comparative analysis of B. dermatitidis, H. capsulatum, Coccidioides spp., and P. brasiliensis septins revealed a high degree of conservation among the endemic dimorphic fungi. All B. dermatitidis septin proteins possessed GTP binding motifs (G1, G3, and G4) and a septin unique element. Three B. dermatitidis septins (CDC3, CDC11, CDC12) were predicted to have coiled-coil domains at the C-terminal region. The presence of a coiled-coil domain in CDC11 was unusual because the majority of CDC11 homologs (82.7%) analyzed by Pan and colleagues lacked this domain [24]. ASPE, which was first identified in A. nidulans, has been primarily described in filamentous fungi [13,36] and in B. dermatitidis lacks the N-terminal polybasic region, which is similar to ASPE homologs in other fungi.
RNA interference of B. dermatitidis CDC3, CDC10, and CDC12 resulted in misshapen yeast cells with bean-like, irregular ovoid, and elongated morphologies. These abnormalities were similar to B. dermatitidis CDC11 knockdown strains [12]. This finding was not expected because the phenotype is often specific for the mutated septin gene in most fungi [14,17,37]. Although this similarity was unanticipated, it is not unprecedented. U. maydis SEP1-4 null mutants have nearly identical morphologic abnormalities at 28°C [15]. The overlap in phenotypes for B. dermatitidis strains knocked down for CDC3, CDC10 and CDC12 is unlikely to be related to inadvertent RNAi of non-targeted septin genes. Analysis of transcript abundance by qRT-PCR demonstrated only the specific septin targeted by RNAi had reduced transcription. Interestingly, a reduction in CDC10 transcript was associated with an increase in transcription of CDC3, CDC11, CDC12, and ASPE. This may suggest a compensatory response in which ternary complexes (i.e., CDC3-CDC11-CDC12) are formed in response to altered stoichiometry of proteins in the septin filament [38].
Fungal septins are involved with the formation of the yeast bud and cytokinesis [14,37,39]. B. dermatitidis knocked down for CDC3, CDC10, and CDC12 yeast were defective in budding and cytokinesis. The aberrant ovoid, bean-shaped, and elongated cells appear as though they are attempting to undergo budding, but the ‘daughter cells’ fail to undergo proper development, which is likely related to impaired cell division. The large size of CDC3, CDC10, and CDC12 knockdown strains and slow growth rate suggest defective cytokinesis. Chitin staining did not reveal misplaced septa, which has been observed in U. maydis septin mutants [15].
Many pathogenic fungi are capable of yeast and filamentous growth. The impact of septins on the conversion from yeast to hyphal growth is often species specific and heterogenous. U. maydis SEP1-4 null mutants have defective filamentation under in vitro inducing conditions (1% corn oil or 1% Tween 40), but are able to form filaments during plant infection [15,32]. C. neoformans CDC3 Δ and CDC12 Δ fail to develop hyphae on V8 medium, but have filamentous growth on MS medium [16]. For C. albicans, both CDC10 Δ and CDC11 Δ yeast convert to hyphae under inducing conditions (20% serum, 37°C) [17]. We found that B. dermatitidis yeast knocked down for CDC3, CDC10, CDC12, or ASPE convert to mold at environmental temperature (22°C), but the morphologies of CDC3, CDC10, and CDC12 knockdown strains were aberrant and consisted of curved hyphae with a knobbed appearance. This aberrant pattern of curved filamentous growth has also been observed in C. albicans and U. maydis null mutants [17,32,40]. The knobs may represent sites where hyphal branches started, but failed to develop.
The effect of septin gene mutations on morphology and growth can be impacted by temperature. Deletion of U. maydis SEP1-4 results in cell lysis at 34°C, while at lower temperatures the cells are swollen with misplaced septa (28°C) or have a normal cigar shape (22°C) [15]. Similarly, septin genes in C. neoformans (CDC3, CDC11, CDC12) and S. cerevisiae (CDC10, CDC11) influence thermotolerance, with the deletion of these genes resulting in aberrant morphology and cell death at 37°C [16,29]. For C. albicans, abnormal yeast morphology was observed only at elevated temperatures for CDC10 Δ null mutants [17]. In contrast, the morphologic abnormalities in B. dermatitidis CDC3, CDC10, and CDC12 knockdown strains were present at both elevated and low temperatures. Moreover, the conversion from mold to pathogenic yeast at 37°C was unaffected in septin RNAi strains. Our results suggest that septin genes in B. dermatitidis influence morphogenesis at core human body and environmental temperatures, but are not required for the phase transition.
Although knocking down B. dermatitidis septins CDC3, CDC10, and CDC12 impact morphology, there are some limitations in this study. First, RNA interference does not completely abrogate transcript abundance, which may have hindered our ability to detect subtle phenotypic differences between the knockdown strains. We selected RNA interference over gene deletion because of the low rate of homologous recombination in B. dermatitidis (0.04%) and the high probability that one or more septin genes would be essential [7,12,17,41]. There was a 22–78% decrease in transcript abundance in septin knockdown strains when compared to controls. Despite transcript reduction not achieving statistical significance, the morphology and growth of multiple CDC3, CDC10, and CDC12 RNAi strains was markedly different (adjusted P value < 0.05) when compared to controls (reporter, non-RNAi vector, GFPi-only). This indicated that septin transcript abundance fell below a threshold required for normal cellular function. Moreover, when there was a loss of RNAi in a septin knockdown strain, GFP fluorescence increased and there was reversion to normal yeast morphology (data not shown). Collectively, our data supports the impact of CDC3, CDC10, CDC12 on morphology and growth of B. dermatitidis. Second, virulence testing in a murine model of infection was precluded due to the slow growth rate of knockdown strains at 37°C in the yeast form. Septins in other fungi such as C. albicans and C. neoformans can impact virulence [16,32].
In conclusion, we have identified five septin genes in B. dermatitidis that are homologs of CDC3, CDC10, CDC11, CDC12, and ASPE. All septin proteins contained the expected motifs but CDC11 was unusual because it was predicted to contain a coiled-coil domain, which is uncommon for this septin. Decrease of CDC3, CDC10, and CDC12 transcript levels by RNA interference in yeast at 37°C resulted in impaired cytokinesis, cell morphology, and growth. A downshift in temperature to 22°C did not alleviate morphologic abnormalities and promoted the conversion of yeast to morphologically aberrant mold. These findings were similar to what we had previously observed in CDC11 knockdown strains [12]. This was unanticipated because the phenotypes of septin mutants are often gene specific and can be influenced by temperature. Knocking down ASPE did not impact the morphology of yeast or mold. Finally, B. dermatitidis septins do not contribute to thermotolerance and their role in cytokinesis is not required for the conversion between yeast and mold.
Acknowledgments
Funding for this research was from the National Institutes of Health (5K08AI071004).
We thank Jens Eickhoff for his assistance with the statistical analysis.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Gauthier GM, Klein BS. Insights into fungal morphogenesis and immune evasion. Microbe. 2008;3:416–423. doi: 10.1128/microbe.3.416.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wheat LJ, Slama TG, Eitzen HE, et al. A large urban outbreak of histoplasmosis: clinical features. Ann Intern Med. 1981;94:331–337. doi: 10.7326/0003-4819-94-3-331. [DOI] [PubMed] [Google Scholar]
- 3.Schneider E, Hajjeh RA, Spiegel RA, et al. A coccidioidomycosis outbreak following the Northridge, Calif, earthquake. JAMA. 1997;277:904–908. [PubMed] [Google Scholar]
- 4.Klein BS, Tebbets B. Dimorphism and virulence in fungi. Curr Opin Microbiol. 2007;10:314–319. doi: 10.1016/j.mib.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gauthier GM, Safdar N, Klein BS, Andes DR. Blastomycosis in solid organ transplant recipients. Transpl Infect Dis. 2007;9:310–317. doi: 10.1111/j.1399-3062.2007.00227.x. [DOI] [PubMed] [Google Scholar]
- 6.Nemecek JC, Wuthrich M, Klein BS. Global control of dimorphism and virulence in fungi. Science. 2006;312:583–588. doi: 10.1126/science.1124105. [DOI] [PubMed] [Google Scholar]
- 7.Gauthier GM, Sullivan TD, Gallardo SS, et al. SREB, a GATA transcription factor that directs disparate fates in Blastomyces dermatitidis including morphogenesis and siderophore biosynthesis. PLoS Pathog. 2010;6:e1000846. doi: 10.1371/journal.ppat.1000846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen VQ, Sil A. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc Natl Acad Sci USA. 2008;105:4880–4885. doi: 10.1073/pnas.0710448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webster RH, Sil A. Conserved factors Ryp2 and Ryp3 control cell morphology and infectious spore formation in the fungal pathogen Histoplasma capsulatum. Proc Natl Acad Sci USA. 2008;105:14573–14578. doi: 10.1073/pnas.0806221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todd RB, Greenhalgh JR, Hynes MJ, Andrianopoulos A. TupA, the Penicillium marneffei Tup1 homologue, represses both yeast and spore development. Mol Microbiol. 2003;48:85–94. doi: 10.1046/j.1365-2958.2003.03426.x. [DOI] [PubMed] [Google Scholar]
- 11.Longtine MS, Bi E. Regulation of septin organization and function in yeast. Trends Cell Biol. 2003;13:403–409. doi: 10.1016/s0962-8924(03)00151-x. [DOI] [PubMed] [Google Scholar]
- 12.Krajaejun T, Gauthier GM, Rappleye CA, Sullivan TD, Klein BS. Development and application of a green fluorescent protein sentinel system for identification of RNA interference in Blastomyces dermatitidis illuminates the role of septin in morphogenesis and sporulation. Eukaryot Cell. 2007;6:1299–1309. doi: 10.1128/EC.00401-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juvvadi PR, Fortwendel JR, Rogg LE, Steinbach WJ. Differential localization of septins during growth of the human fungal pathogen Aspergillus fumigatus reveal novel functions. Biochem Biophys Res Commum. 2011;415:238–243. doi: 10.1016/j.bbrc.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartwell LH. Genetic control of the cell division cycle in yeast IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:245–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- 15.Alverez-Tabares I, Perez-Martin J. Septins from the phytopathogenic fungus Ustilago maydis are required for proper morphogenesis but dispensable for virulence. PLoS One. 2010;5:e12933. doi: 10.1371/journal.pone.0012933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozubowski L, Heitman J. Septins enforce morphogenetic events during sexual reproduction and contribute to virulence of Cryptococcus neoformans. Mol Microbiol. 2010;75:658–675. doi: 10.1111/j.1365-2958.2009.06983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warenda AJ, Konopka JB. Septin function in Candida albicans morphogenesis. Mol Biol Cell. 2002;13:2732–2746. doi: 10.1091/mbc.E02-01-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McVeigh I, Morton K. Nutritional studies of Histoplasma capsulatum. Mycopath Mycol Appl. 1965;25:294–308. doi: 10.1007/BF02049917. [DOI] [PubMed] [Google Scholar]
- 19.Worsham PL, Goldman WE. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J Med Vet Mycol. 1988;26:137–143. [PubMed] [Google Scholar]
- 20.Sullivan TD, Rooney PJ, Klein BS. Agrobacterium tumefaciens integrates transfer DNA into a single chromosomal site of dimorphic fungi and yields homokaryotic progeny from multinucleate yeast. Eukaryot Cell. 2002;1:895–905. doi: 10.1128/EC.1.6.895-905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonough ES, Wisniewski TR, Penn LA, Chan DM, McNamara WI. Preliminary studies on conidial liberation of Blastomyces dermatitidis and Histoplasma capsulatum. Sabouraudia. 1976;14:199–204. [PubMed] [Google Scholar]
- 22.Metzenberg RL. Bird medium: an alternative to Vogel medium. Fung Gen Newslet. 2004;51:19–20. [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;24:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Pan F, Malmberg RL, Momany M. Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol Biol. 2007;7:103. doi: 10.1186/1471-2148-7-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mino M, Tanaka K, Kamei T, et al. Shs1p: a novel member of septin that interacts with Spa2p, involved with polarized growth in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1998;251:732–736. doi: 10.1006/bbrc.1998.9541. [DOI] [PubMed] [Google Scholar]
- 26.Ozsarac N, Bhattacharyya M, Dawes IA, Clancy MJ. The SPR3 gene encodes a sporulation-specific homologue of the yeast CDC3/10/11/12 family of bud neck microfilaments and is regulated by ABFI. Gene. 1995;164:157–162. doi: 10.1016/0378-1119(95)00438-c. [DOI] [PubMed] [Google Scholar]
- 27.De Virgilio C, Demarini DJ, Pringle JR. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiology. 1996;142:2897–2905. doi: 10.1099/13500872-142-10-2897. [DOI] [PubMed] [Google Scholar]
- 28.Versele M, Gullbrand B, Shulewitz MJ, et al. Protein–protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:4568–4583. doi: 10.1091/mbc.E04-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casamayor A, Synder M. Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization and function. Mol Cel Biol. 2003;23:2767–2777. doi: 10.1128/MCB.23.8.2762-2777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Momany M, Pan F, Malmberg RL. Evolution and conserved domains of the septins. In: Hall PA, Russell SHE, Pringle JR, editors. The Septins. 1. New York: Wiley-Blackwell; 2008. pp. 35–45. [Google Scholar]
- 31.Wolf E, Kim PS, Berger B. Multicoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyce KJ, Chang H, D’Souza CA, Kronstad JW. An Ustilago maydis septin is required for filamentous growth in culture and for full symptom development on Maize. Euk Cell. 2005;4:2044–2056. doi: 10.1128/EC.4.12.2044-2056.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TQ, Sawa H, Okano H, White JG. The C. elegans septin genes, unc-59 and unc-61, are required for normal postembryonic cytokineses and morphogenesis, but have no essential function in embryogenesis. J Cell Sci. 2000;113:3825–3837. doi: 10.1242/jcs.113.21.3825. [DOI] [PubMed] [Google Scholar]
- 34.Adam JC, Pringle JR, Peifer M. Evidence of functional differentiation among Drosophila septins in cytokinesis and cellularization. Mol Biol Cell. 2000;11:3123–3135. doi: 10.1091/mbc.11.9.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakahira M, Macedo JN, Seraphim TV, et al. A draft of the human septin interactome. PLoS One. 2010;5:e13799. doi: 10.1371/journal.pone.0013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momany M, Zhao J, Lindsey R, Westfall PJ. Characterization of the Aspergillus nidulans septin (asp) gene family. Genetics. 2001;157:969–977. doi: 10.1093/genetics/157.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.An H, Morrell JL, Jennings JL, Link AJ, Gould KL. Requirements of fission yeast septins for complex formation, localization, and function. Mol Biol Cell. 2004;15:5551–5564. doi: 10.1091/mbc.E04-07-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farkasovsky M, Herter P, Beater V. Wittinghofer. Nucleotide binding and filament assembly of recombinant yeast septin complexes. Biol Chem. 2005;386:643–656. doi: 10.1515/BC.2005.075. [DOI] [PubMed] [Google Scholar]
- 39.Roncero C, Sanchez Y. Cell separation and the maintenance of the cell integrity during cytokinesis in yeast: the assembly of septum. Yeast. 2010;27:521–530. doi: 10.1002/yea.1779. [DOI] [PubMed] [Google Scholar]
- 40.Warenda AJ, Kauffman S, Sherrill TP, Becker JM, Konopka JB. Candida albicans septin mutants are defective for invasive growth and virulence. Infect Immun. 2003;71:4045–4051. doi: 10.1128/IAI.71.7.4045-4051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Momany M, Hamer JE. The Aspergillus nidulans septin encoding gene, aspB, is essential for growth. Fungal Genetic Biol. 1997;21:92–100. doi: 10.1006/fgbi.1997.0967. [DOI] [PubMed] [Google Scholar]