

Abstract
Parkinson's disease (PD) is the second most common neurodegenerative disorder in the developed world, and is characterized by the loss of dopaminergic (DA) neurons in the substantia nigra (SN). Somatic mitochondrial DNA (mtDNA) deletions reach their highest concentration with age in the SN in humans, and may contribute to PD; yet whether mtDNA deletions cause DA neuron degeneration remains unclear. Inherited mutations of Twinkle helicase involved in mtDNA replication causes a dominant increase in mtDNA deletions in humans. We constructed a mouse model expressing mutant Twinkle in DA neurons. Mutant mice had an increase in age-related mtDNA deletions, reduction of DA neuron number in SN at 17–22 months and displayed abnormalities in rota-rod behavior. Functional analysis of midbrain indicated a slight reduction in mitochondrial state II respiration in mutants, but no decrease in maximal respiration. Also, Parkin expression was significantly decreased in DA neurons in the SN of 22-month-old mutant mice, and in PC12 cells after 48 h transfection of mutant Twinkle. Both confocal imaging and coimmunoprecipitation indicated interaction of Twinkle with Parkin in the mitochondria. Parkin overexpression rescued the reduction of proteasome activity caused by mutant Twinkle in PC12 cells. In addition, the autophagy marker LC3 was increased in the SN of 22-month transgenics, and this increase was similarly mutant Twinkle-dependent in PC12 cells. Collectively, our data demonstrate that mammalian Twinkle is important for mitochondrial integrity in DA neurons and provide a novel mouse model in which increased mtDNA deletions may lead to DA neuron degeneration and parkinsonism.
INTRODUCTION
Parkinson's disease (PD) is the second most common age-related neurodegenerative disorder. The pathological hallmark of PD is the accumulation of Lewy bodies, and the degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNC), yet the mechanism of selective DA neuron loss is still unclear. A growing number of clinical and experimental studies indicate that mitochondrial dysfunction contributes to the pathogenesis of PD (1,2). The defects of mitochondrial genes have been recognized in PD patients such as Complex I, Pink1 (PTEN-induced kinase) and Parkin (3–5). The exposure of humans and animals to complex I inhibitors causes parkinsonism (6), and the loss of Pink1 causes the recessive Park6 variant of PD (4). Mutations of Parkin are the most common cause of autosomal recessive PD (5), recent studies indicated that Parkin regulates mitochondrial dysfunction through either mitochondrial quality control or regulation of Paris (7).
Mitochondrial DNA (mtDNA) deletions have been shown to be important in neurodegeneration of PD (8). MtDNA deletions occur somatically in humans, increase with age and reach their highest level in the SN of older humans (9,10). High mtDNA deletion levels were detected in the SN of patients with PD (8), and deletions co-localized with cytochrome c oxidase -deficient neurons (8). Furthermore, defects of mitochondrial replicative DNA polymerase (POLG) accumulate high amounts of mtDNA deletions in mice (11), POLG mutations are a recognized cause of parkinsonism (12). However, it remains unclear how mtDNA deletions cause DA neuron degeneration in the SN of PD. Mitochondrial replication is important for mtDNA maintenance and integrity. Twinkle is a key helicase involved in mtDNA replication and critical for the maintenance of human mtDNA integrity (13), and dominant Twinkle mutations cause multiple mtDNA deletions and progressive mitochondrial myopathy in humans, and transgenic transformation of the same mutations into the mouse causes increased deletions and a mitochondrial myopathy (14). In addition, parkinsonism has been reported in Twinkle-Progressive External Ophthalmoplegia patients (15).
To further elucidate the role of DNA deletions in DA neurons pathophysiology and parkinsonism in vivo, our group has generated a mouse model expressing duplicate mutant Twinkle driven by the tyrosine hydroxylase (TH) promoter, assessed mtDNA deletions, mtDNA quantity and TH neurons of aging mice. We observed a significant effect of mutant Twinkle on mtDNA deletion accumulation and DA neuron loss.
RESULTS
Generation of TH-specific mutant Twinkle transgenic mice
We generated six transgenic lines expressing TH-specific mouse mutant Twinkle-Green Fluorescent Protein (GFP) with an in-frame duplication of amino acids 353–365 under the TH promoter (Fig. 1A), four of six lines have higher GFP copy number (Supplementary Material, Table S1). Twinkle DNA levels of four lines are 20-fold and 207-fold higher compared with the endogenous Twinkle in the SN of mouse brain, and the expression levels of Twinkle mRNA increased from 1.2-fold to 4-fold in SN (Fig. 1C and D), the highest in line 2. Therefore, we decided to use line 2 in this study. The expression of transgene in the SNC was also detected by immunostaining using anti-GFP antibody and anti-Twinkle antibody (Fig. 1B, Supplementary Material, Fig. S1).
Figure 1.
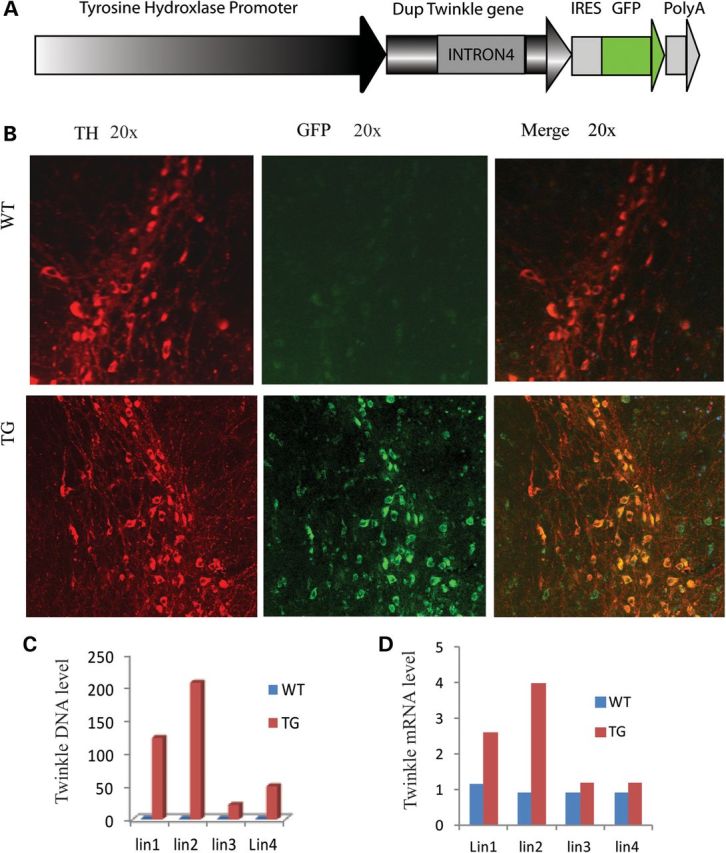
Expression of mutant Twinkle in SNC. (A) Twinkle IRESGFP driven by TH promoter. (B) Immunofluorescence confocal image. Mutant Twinkle-GFP is expressed in TH-positive neurons of SNC in TG. (C) DNA levels of transgene in SN of mice from four different lines of TG. SN tissues from 22-month-old mice were subjected to real-time PCR for Twinkle. (D) RNA levels of transgene in the SN of mice from four different lines of TG.
Alterations in behavior and TH-positive neurons in transgenic mice
Body weights of 11- and 23-month-old mutants were not significantly different from controls, and mutant longevity was similar to controls (Supplementary Material, Fig. S2C). There were no obvious ataxia or gait differences (Supplementary Material, Fig. S2A). The accelerating rota-rod test was used to evaluate motor function in 23-month-old mice. We observed that mutant mice had a shorter time to first fall compared with littermate controls (P = 0.05, one-tailed t-test) (Fig. 2E). Consistent with this, the number of total falls of transgenic mice (TG) is significantly increased (P = 0.027, one-tailed t-test) (Fig. 2F).
Figure 2.
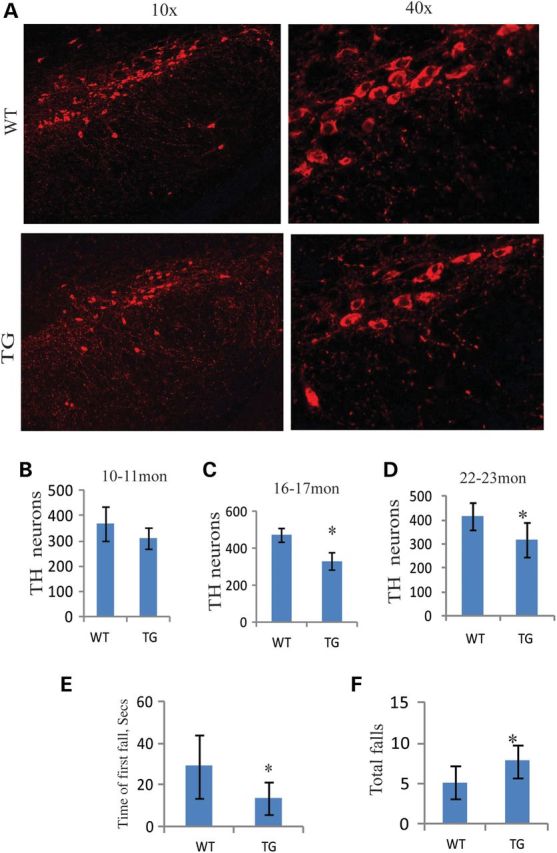
Behavior and TH-positive neuron cells in SNC of aging transgenic mice. (A and B) TH immunostaining in SNC of 22-month-old mice. (B–D) TH-positive cells were reduced with time in the SNC of transgenic mice compared with controls (10–11 months: WT = 3, TG = 3, P = 0.271, two-tailed; 16–17 months: WT = 3, TG = 3, P = 0.014, two-tailed; 22–23 months: WT = 4, TG = 4, P = 0.038, one-tailed). (E) The time of first fall was significantly short compared with control littermates at about 23 months (WT = 4, TG = 6, P = 0.05, one-tailed). (F) The number of total falls of transgenic mice are markedly increased (WT = 4, TG = 6, P = 0.027, one-tailed). *P≤ 0.05.
To determine whether rota-rod defects were attributable to DA neurodegeneration in SNC, we performed a systematic histological evaluation of TH-positive neuron populations in the SNC by TH immunostaining. TH-positive neuron cells were blindly counted in the SNC of mice at 10–11, 16–17 and 22–23 months using Nikon NIS-elements imaging software. Although there was no significant difference in TH neuron number in SNC of 10–11-month-old mice, there were significant decreases in 16–17 and 22–23-month-old mice (Fig. 2B–D). In addition, there was a trend for decrease in TH neuron number in the ventral tegmental area of 22-month-old mutant mice (P = 0.14, one-tailed t-test) (Supplementary Material, Fig. S2B).
Accumulation of mitochondrial deletions in the SN of mutant mice
Twinkle plays a critical role in mtDNA maintenance and replication. Humans bearing Twinkle mutations develop multiple mtDNA deletions in tissues. Mouse model studies indicate that mutation of Twinkle gene causes mtDNA deletions in brain tissue (14). Therefore, we detected DNA deletions of SN from mice at 1, 12, 18 and 22 months using long PCR and found that mean deletion bands increased with time. The differences over controls were significant at 18- and 22-month-old mice, the same time period at which TH neurons were decreased (Figs 2C–D and 3A). At 18–23 months, multiple deletion bands were observed in both mutant and control animals with 2 ng DNA. However, product bands were significantly increased in the SN of TG compared with controls (Fig. 3A and Supplementary Material, Fig. S7), and almost two times more than controls (Supplementary Material, Table S2). When the DNA template was reduced to 0.2 ng, we found that multiple deletion products still appear in TG, but not in controls, and the full-length, undeleted product was observed in controls (Supplementary Material, Fig. S7). Most products were located in the size category 4361–2322 kb, and this deletion pattern was similar to the brain mtDNA deletions caused by mutant Twinkle observed by others using these primers (14). Furthermore, we used quantitative real-time PCR (QPCR) to detect quantities of mtDNA deletions. Consistent with the approximate doubling observed by the band-counting method, mtDNA deletions in the SN of mutant mice were 1.57 times above controls by QPCR (Fig. 3B). The total mtDNA genomes of SN were also detected quantitatively using QPCR in 1-, 12-, 18- and 22-month-old mice. Although mtDNA copy number appeared to be reduced at all four age mutants, the only significant decrease was at 22 months (Fig. 3C). These data indicate that mtDNA deletions accumulate in the SN of older mutant mice. In addition, we also found that mtDNA deletions accumulate in PC12 cells after mutant Twinkle transfection (Supplementary Material, Fig. S2D).
Figure 3.
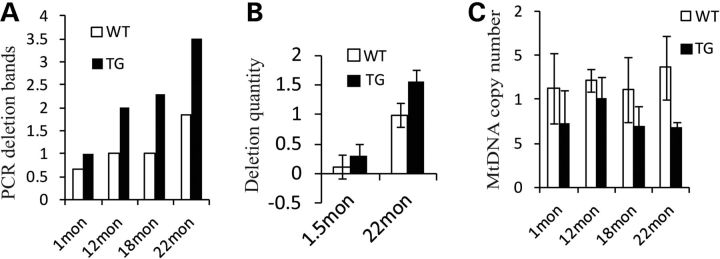
Accumulation of mitochondrial deletions in substantia nigra (SN) of aging transgenic mice. (A) Average long PCR deletion bands of mouse SN at 1, 12, 18, and 22 months (1 month: WT = 6, TG = 8, P = 0.8511; 12 months: WT = 3, TG = 5, P = 0.7171; 18 months: WT = 2, TG = 6, P = 0.025, two-tailed; 22 months: WT = 6, TG = 6, P = 0.04, one-tailed). (B) Quantification of deletions (WT = 3, TG = 3, P = 0.022, two-tailed). (C) mtDNA copy number in the SN of different age mice (1 month: WT = 5, TG = 6, P = 0.0638, one-tailed; 12 months: WT = 3, TG = 3, P = 0.1358; 18 months: WT = 2, TG = 5, P = 0.165; 22 months: WT = 3, TG = 3, P = 0.034, two-tailed). *P≤ 0.05, w = WT, t = TG.
Impaired mitochondrial basal respiration in the midbrain of young Twinkle mutant mice
It is very difficult to isolate sufficient mitochondria from the SN to do bioenergetic analysis, so mitochondria were isolated from the midbrain of transgenics and controls (see Methods). We observed that mitochondrial basal respiration (state 2 respiration) was significantly decreased in the midbrain of mutant mice at 1–3 months for complex I (P = 0.05, one-tailed t-test), whereas complex II activity was not significantly decreased (Fig. 4A). No other respiratory parameters were significantly different. Since the decrease in O2 consumption was exclusively in through mitochondria-fed complex I substrates, we presume that there is a mild (20%) complex I defect which is observed only in state 2 (basal) respiration. However, the maximal (state3) mitochondrial respiratory rates observed upon addition of ADP were not different between the mutants and controls (Fig. 4B), which show that there is no difference in mitochondrial capacity.
Figure 4.

Impaired mitochondrial basal respiration in the midbrain of twinkle mutant mice. (A) State 2 activities are decreased in 1–3 month-old mutant mice for complex I (WT = 3, TG = 6, P = 0.05, one-tailed), whereas complex II activities have no obvious difference (WT = 3, TG = 6, P = 0.3, one-tailed). (B) State 3 respiratory activities are similar between controls and transgenic mice for complex I and complex II. (C) State 4 respiratory activities for complex I are slightly lower in mutant mice (WT = 3, TG = 6, P = 0.12, one-tailed), no difference for complex II. (D) Uncoupled respiration activities for complex I and complex II in midbrain. (E) PC12 cells transfected with Twinkle mutant (MUT) have very lower basal respiratory compared with cells transfected with Twinkle WT, three independent experiments (P = 0.002, two-tailed). (F) Uncoupled respiration activities were baselined and normalized in PC12 cells (P = 0.12, two-tailed). *P≤ 0.05.
Next, we transfected mutant Twinkle plasmids into PC12 cells and fed complex I substrates, glutamate/malate and observed basal oxygen consumption rate (OCR). Consistent with the in vivo results, we observed that mutant Twinkle PC12 cells have slightly lower basal respiratory capacity compared with cells transfected with wild-type (WT) Twinkle (P = 0.002, two-tailed t-test) (Fig. 4E) and a non-significant trend for decreased uncoupled respiration in mutants (Fig. 4F). Thus, Twinkle mutations cause a mild complex I defect in basal state 2 respiration, with no alteration in the maximal respiratory rate (Fig. 4E).
The reduction of Parkin expression in SNC of mutant Twinkle mice
Parkin is ubiquitously expressed in neurons, and mutations in Parkin are the most common cause of recessive familial PD. Experimental studies suggest that Parkin is involved in the maintenance of mitochondrial function and protect mitochondrial genomic integrity (16,17). Therefore, we measured Parkin expression in the SNC of mice at 22–23 months. We observed significantly reduced Parkin expression in the SNC of mutant mice compared with controls (Fig. 5A), and the mean intensity of Parkin expression was decreased in mutant mice (P = 4.54E-06, two-tailed t-test), Figure 5B. In addition, we observed an insignificant trend for reduction in Parkin expression in the midbrain of 1-month-old mutant mice (P = 0.06, one-tailed t-test) (Supplementary Material, Fig. S5). Furthermore, we explored Parkin expression in PC12 cells with mutant Twinkle transfection and found that mutant Twinkle transfection results in the reduction of Parkin expression in PC12 cells after 48 h (P = 0.019, two-tailed t-test) (Fig. 5C and D). These data suggest that mutant Twinkle causes deficiency of Parkin expression in the mouse brain.
Figure 5.

Mutant Twinkle reduces Parkin expression in SNC. (A) Parkin and TH immunostaining in SNC. Parkin expression was decreased in the SNC of transgenic mice. (B) The mean intensity of Parkin expression in SNC (WT = 3, TG = 3, P = 4.54–06, two-tailed). (C and D) Western blot analysis. Twinkle mutant resulted in the reduction of Parkin expression in PC12 cells after 48 h transfection, three independent experiments (P = 0.019, two-tailed), CTL: only transfection reagents, no plasmid, WT: transfection with Twinkle WT plasmids. MUT: transfection with Twinkle duplicate plasmids. *P≤ 0.05.
Twinkle interacts with Parkin
Both Twinkle and Parkin play a critical role in regulating mitochondrial function. Therefore, we searched for a link between Twinkle and Parkin. Flag-tagged Twinkle and Myc-tagged Parkin were co-transfected into Cos7 cells after 24 h transfection, confocal imaging indicated that Twinkle colocalizes with Parkin in mitochondria (Fig. 6A and B). Furthermore, Myc-tagged Parkin and Flag-tagged Twinkle coimmunoprecipitate in 293T cells after 24 h transfection. Flag antibody immunoprecipitates Myc-Parkin, and anti-Myc antibody immunoprecipitates Flag-Twinkle, which strongly indicates that Twinkle directly interacts with Parkin in mitochondria (Fig. 6C and D).
Figure 6.

Twinkle interacts with Parkin. (A) Confocal image. Twinkle WT colocalized with Parkin in cytoplasm in Cos7 cells after 24 h transfection. (B) Twinkle duplicate colocalized with Parkin in cytoplasm in Cos7 cells after 24 h transfection. (C) Myc-Parkin and flag-Twinkle WT were co-transfected in 293T cells for 24 h. Cell lysates were immunoprecipitated with anti-flag antibody, followed by immunoblotting with anti-flag and anti-myc antibody, arrows indicate that flag pulls down Myc, IgG as control. (D) Cell lysates were immunoprecipitated with anti-myc antibody, followed by immnoblotting with anti-flag and anti-myc antibody, arrows indicate that Myc pulls down Flag.
Mutant Twinkle decreases proteasome activity
Parkin is an E3 ubiquitin ligase promoting the degradation of multiple proteins via the ubiquitin-proteasome system (UPS), which may digest aggregation-prone proteins. Studies indicate that Parkin is critical for the activation of the proteasome (18). Therefore, proteasome activity was measured. First, we transfected mutant Twinkle into PC12 cells, after 48 h transfection proteasome activity was significantly reduced (Fig. 7A). These results were consistent with that of our previous work, that mtDNA deletions caused a decrease in proteasome activity (19). Interestingly, when we co-transfected Parkin plasmids with mutant Twinkle into PC12 cells, the proteasome activity was restored (Fig. 7B), demonstrating a functional rescue. Although the absolute mean control proteasome activity was decreased ∼10% in transgenic midbrains, these differences were not statistically significant (Fig. 7C).
Figure 7.

Mutant Twinkle reduces proteasome activity. (A) PC12 cells were transfected with Twinkle duplicate plasmids and Twinkle WT plasmids, respectively, for 48 h. Proteasome activities were significantly reduced in PC12 cells transfected with Twinkle duplicate, four independent experiments, P < 0.001. (B). Duplicate Twinkle plasmids were co-transfected with Parkin plasmids or empty vector into PC12 cells. WT Twinkle plasmids were co-transfected with empty vector into PC12 cells as controls. Proteasome activities were decreased in PC12 cells with Twinkle duplicate and empty vector compared with Twinkle WT and empty vector (P < 0.05). However, proteasome activities are increased in PC12 cells with Twinkle duplicate and Parkin plasmids compared with Twinkle duplicate and empty vector plasmids (P < 0.05). (C) Proteasome activities in midbrain of 1- and 23-month mice (1 month: WT = 3, TG = 3, P = 0.39, one-tailed; 23month: WT = 4, TG = 4, P = 0.29, one-tailed). *P≤ 0.05.
Mutant Twinkle induces autophagy in SNC
Studies indicate that autophagy plays a critical role in neurodegeneration (20). Both mitochondrial dysfunction and proteasome activity reduction can increase autophagy (21,22). The autophagy protein LC3 is a widely used marker for induction of autophagy. We observed that LC3 expression was significantly increased in SNC of Twinkle mutant (MUT) mice at 22 months (P = 0.00097) (Fig. 8A). Furthermore, LC3-II was observed in PC12 cells transfected with mutant Twinkle, but not in cells transfected with WT Twinkle (Fig. 8C–E). Consistent with these results, LC3-II was increased in neuronal cultures of midbrain from Parkin null mice (23). Apoptosis is another important factor in neurodegeneration and we investigated apoptosis in both the mutants and controls, and no obvious difference was found in the SNC of old TG (Supplementary Material, Fig. S3). These data suggest that mutant Twinkle induce TH neurodegeneration through autophagy, not apoptosis.
Figure 8.

Mutant Twinkle induces autophagy in the SNC. (A) Immunohistochemistry of LC3 in the SNC. (B) LC3 mean density was increased in the SNC (WT = 4, TG = 4, P = 0.000977). (C and D) LC3-II was found in PC12 cells transfected with Twinkle duplicate after 48 h, not in cells transfected with Twinkle WT. *P≤ 0.05.
To determine whether increased autophagy was the result of increased oxidative stress, we measured MnSOD and CuSOD protein expression in 22-month-old mice (Supplementary Material, Fig. S4A), but did not detect any significant or obvious differences. Consistent with these data, using the Mitosox molecular probe to measure reactive oxygen species (ROS), we did not observe any increase in ROS production in mutant Twinkle-transfected PC12 cells (Supplementary Material, Fig. S4B).
DISCUSSION
Twinkle and mitochondrial defects
We constructed the mouse model expressing mutant Twinkle, i.e. ‘twinkle-dup’ driven by the TH promoter. These mice developed progressive TH neuron loss in SNC at 16–17 and 22–23 months, as well as a motor abnormality at 22–23 months. Furthermore, mtDNA deletions were significantly increased over controls in the SN at 18 and 22 months. In agreement with these results, previous mouse models expressing mutant Twinkle from its natural promoter cause an increase in multiple mtDNA deletions in muscle and brain tissue at 22 months (14,24). These data suggest, but do not prove a link between mtDNA deletion formation and progressive TH neuron degeneration. Mitochondrial integrity plays an important role in mitochondrial respiratory function, other studies indicated that the defects of mitochondrial transcripts markedly reduced mitochondrial respiratory function in DA and precipitated a parkinsonian phenotype in MitoPark mice (25). In contrast to that severe model in which mitochondrial transcripts go to zero, we observed a mild defect in complex I basal respiration in the midbrain of mutant Twinkle mice (but none in maximal mitochondrial respiration), this mild defect was confirmed in mutant Twinkle-transfected PC12 cells (Fig. 4). Others demonstrated that globally expressed mutant twinkle resulted in decreased activities of mitochondrial respiratory chain enzymes in mouse muscle (14). In our hands, a mild mitochondrial deficit occurred in the midbrain of 1–2 month-old mutant mice, which had low mtDNA deletions and normal behavior. This could suggest that respiratory chain dysfunction precedes all other events as was reported in the global Twinkle mutant mouse model by Tyynismaa et al.(14). However, loss of TH neurons occurred at 16–17 and 22–23 months, which overlaps with the occurrence of a significant rise in mtDNA deletions, and decrease in Parkin (which is thought to be involved in mitochondrial quality control). In contrast, mice with global defects in mitochondrial respiration (one example is the NDUFS4 knockout) had neither increased deletions nor SN neurodegeneration.
Parkin and mitochondrial integrity
Parkin mutations are the most common cause of autosomal recessive PD. An impressive amount of evidences indicate that Parkin is involved in the maintenance of mitochondrial function and mitochondrial quality control, and protects mtDNA from oxidative damage and mutation (16,26,27). Twinkle is responsible for mtDNA replication as well as for repair. Our data demonstrate that mutant Twinkle significantly reduces Parkin expression in the SNC of 22-month-old mice and PC12 cells (Fig. 5). Moreover, Twinkle protein directly interacts with Parkin protein in mitochondria (Fig. 6). Supporting this, endogenous cellular Parkin is physically associated with mtDNA in brain tissue, and Parkin expression stimulates the replication of mtDNA (16). Furthermore, Parkin can rescue the mitochondrial dysfunction induced by the deletion of mitochondrial genes (26,28). Recent studies showed that Parkin is involved in mitochondrial quality control (26,28), and is a regulator of Paris which regulates mitochondrial number through PGC-1alpha, NRF1 (7). Therefore, our results reinforce that there is an interaction of Twinkle and Parkin, and provide a support for link between mitochondrial replication and Parkin function.
In addition, Parkin is an E3 ubiquitin ligase, a component of the UPS, and critical for the activation of proteasome (18). We found that mutant Twinkle reduced proteasome activity, which is reversed by Parkin overexpression in PC12 cells, providing definitive evidence for functional interaction between Twinkle and Parkin. The UPS has been shown to play a critical role in the pathogenesis of PD, proteasome inhibitors induce a sustained DA neuron degeneration and decrease motor activities in animal models (29). However, Parkin−/− mice do not have any motor behavior abnormality and DA neuron loss (30), this difference with mutant Twinkle mice suggests that Twinkle functions upstream of Parkin to maintain mitochondrial integrity via other pathways in addition to Parkin.
Autophagy and neurodegeneration
Autophagy is essential for the survival of neural cells, and is also thought to be a precursor of cell death (31). An increased number of autophagic vacuoles have been found in PD patients and the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-injected PD mouse model (32). It has been suggested that late-stage neuronal cell loss generally occurs via autophagy (33). We found that LC3 was remarkably increased in the SNC of aging TG and PC12 cells transfected by mutant Twinkle (Fig. 8). Recent studies indicate that mitochondrial dysfunction induces cell survival-promoting autophagy (34). Mitochondrial dysfunction is associated with increased oxidative stress and the activation of mitochondrial permeability transition, which trigger the degradation of impaired mitochondria and increase autophagy activity (21). In addition, studies indicate that once UPS is inhibited, autophagy is upregulated (35,36). Parkin has been found to associate with impaired mitochondria, and Parkin-marked mitochondria are subsequently cleared by autophagy. Moreover, Parkin induces ubiquitination, which is required for efficient clearance of impaired mitochondria (37). Studies indicate that LC3-II/I are increased in neuronal cultures of the midbrain from Parkin null mice (23).
In summary, these data demonstrate a new mouse model of SN-specific accumulation of mtDNA deletions for PD and suggest that downstream of mutant Twinkle are a mild respiratory dysfunction, Parkin reduction, increased mtDNA deletions and autophagy increase, which together cause DA neuron degeneration and movement defects (Fig. 9). It is likely that we will have a robust model of accumulation of mtDNA deletions in the SN through crossing mutant Twinkle mice with Parkin knockout mice, these crosses are underway.
Figure 9.

The mechanism of mutant Twinkle causing DA neuron degeneration. The mutations of Twinkle cause mitochondrial respiratory dysfunction, reduce mtDNA copy number and Parkin expression, increase mtDNA deletions and autophage, together these factors may lead to the degeneration of DA neurons in mice. Direct regulation is indicated by solid arrows. Direct and indirect regulation is indicated by dotted arrows.
MATERIALS AND METHODS
Generation of TH-specific mouse mutant Twinkle transgenic mice
Mouse Twinkle DNA with an in-frame duplication of amino acids 353–365 a kind gift from the Suomalainen laboratory (14), was introduced into pRTH/EGFP plasmids gifted by Kobayashi's laboratory (38) and expressed under TH promoter (Fig1A). Microinjection of DNA fragment into oocytes was performed by the mouse biology program of the University of California, Davis. Mice were subsequently crossed with C57BL/6J mice for colony maintenance. Mouse genotyping was done by detecting Twinkle and GFP using mouse tails with Twinkle-F 5′-GCGAGGGCTGTGGAGACAGA-3′, Twinkle-R5′CGGCGACACTGGCATCTCTAA-3′primers, GFP-F5′-aagttcatctgcaccaccg-3′, GFP-R 5′-tgctcaggtagtggttgtcg-3′ primers. All experiments were performed according to a protocol approved by the University of California, Davis institutional animal care and use committee.
Rota-Rod and gait analysis
Rota-rod treadmill for mice 7600 was used. On the first day, mice were trained at 10 am, at a speed of 16 rpm; the animal ran on the cylinder for 120 s or until it fell off. On the second day, testing was undertaken at the same time of day as the first training trial, the animal remained on the cylinder at 24 rpm for 120 s or until it fell off. The time of first fall and number of total falls were recorded. There were three trials for every mouse with 2 min breaks between the intervals.
Gait analysis was performed at 22-month-old mice. First, animals were trained to walk through a narrow alley. Each animal's forelimbs and hindlimbs are brushed with nontoxic paint and they were made to walk over the paper placed along the alley floor. Stride length was determined by measuring the distance between paw prints. Stride lengths at the beginning and end of the alley were not counted (39).
Immunostaining, TUNEL assay and confocal image
Mice were perfused with PBS (phosphate buffered saline), followed by 4% paraformaldehyde (PFA); mouse brains were fixed in 4% PFA overnight at 4°C, then were placed in 30% sucrose overnight and embedded in optical cutting temperature (OCT) compound. For counting TH-positive cells in mouse SNC, 20 µm coronal series sections were cut from about Bregma −2.75 mm to −1.28 mm according to mouse brain atlas: C57BL/6J Coronal in mouse brain library (http://www.mbl.org/atlas170/atlas170_frame.html). TH staining was performed on five consecutive non-serial sections, each spaced out 100 µm. TH neurons in SNC were blindly counted using Nikon NIS-elements imaging software. Immunofluorescence studies were performed as described previously (40). Alexa488 and Alexa594 fluorescence-conjugated secondary antibodies (Invitrogen) were used for visualization of the signal. Total nuclei were visualized with 4′,6-diamidino-2-phenylindole staining. Immunohistochemistry studies were performed using the Envision+ system (DAKO) following manufacturer's instructions. The primary antibodies used in this study include antibodies TH (Santa Cruz), Twinkle (Santa Cruz), GFP (Abcam), LC3 (Santa Cruz) and Parkin (Assay Biotechnology). Samples were examined using a Nikon Eclipse 80i fluorescent microscope. Positive signal mean intensity and mean density were measured by Nikon NIS-elements imaging software. TUNEL assays were performed using the ‘Dead End Colormetric TUNELsystem” (Promega) following the manufacturer's instructions.
For colocalization between Twinkle and Parkin, flag-tagged Twinkle plasmids (13) and myc-tagged Parkin plasmids (Addgene ID 17612) (41) were transfected into COS7 cells cultured for 24 h, fixed in 4% PFA for 15 min at 4°C and permeabilized with 0.2% Triton X-100 for 5 min, and then washed three times in PBS followed by blocking in fresh 1% bovine serum albumin (BSA) for 30 min. Next, cells were incubated with anti-flag mAb and anti-myc mAb at 4°C overnight. Alexa488 and Alexa594 secondary antibodies were used. All samples were imaged using an Olympus confocal microscope.
Long PCR, real-time PCR and quantitative mitochondrial DNA
Mice were decapitated, brains were quickly removed, frozen in OCT and placed at −80°C. Cryostat sections (20–30 µm) were cut, and SN were microdissected from 6 to 10 piece sections by using a tiny blade. Total DNA was isolated from the SN using a DNA mini kit (Qiagen, cat. No. 51304). Mouse mtDNA was amplified from 2 ng or 0.2 ng total DNA with primers 1953–1924 and 2473–2505 of mouse mtDNA (14) using LA Taq polymerase (TAKARA, Japan), PCR condition: 1 cycle of 94°C for 1 min, 35 cycles 98°C for 10 s, 58°C for 30 s, 68°C for 5 min, 1 cycle of 72°C for 10 min.
Real-time PCR was used to assay Twinkle expression level as described in 42, and primers are forward 5′-TCTTAACTCAGAGCTTTGTCCGT-3′ and reverse 5′-GTACACAAGTCCAGGGCATAC-3′. For mtDNA deletions by real-time PCR, we used 5549-forward and 15501-backward primers to amplify mtDNA with 10 kb deletions, and amplified the undeleted region of mtDNA ND1 as control using 2785-forward and 2869-backward primers (24). Reactions were carried out in triplicate in 10 µl volume containing 2 ng template DNA on Lightcycler 480. Melting curve and agarose-gel electrophoresis were used to evaluate specificity of each reaction. Deletion levels were normalized by ND1 using the ΔCt method (43).
Mouse mitochondrial and nuclear copy number analysis was performed as previously described (42). Briefly, genomic DNA preparations (also containing mtDNA) were quantitatively PCR on Lightcycler 480. ND1 and cystic fibrosis transmembrane receptor (CFTR) were used for mitochondrial and nuclear copy number analysis, respectively. The primers used were: ND1 forward, 5′-cat gat cta gga ggc tgc tga cct c-3′; reverse, 5′- cgt tta cct tct ata agg cta tga-3′ and CFTR forward, 5′-atg cag cct ttg gtg aaa cag-3′; reverse, 5′-ctg tga cac gtg tgc ttt cag-3′.
Dissection of midbrain with substantia nigra
Mice were decapitated, brains quickly removed and chilled on ice. According to the mouse brain atlas, C57BL/6J Coronal in mouse brain library (http://www.mbl.org/atlas170/atlas170_frame.html), we cut brain from about Bregma −3 mm to −1.5 mm, dissected the midbrain containing whole SN except cortex and hippocampus (Supplementary Material, Fig. S6), the tissue obtained from individual mouse was analyzed for respiratory function, proteasome activity and western blot.
Isolation of mitochondria and measurement of respiratory function
Mitochondrial preparations were obtained from the midbrain as prescribed (44). The midbrains of 1–3 month mice were dissected in cold isolation buffer and homogenized in 12% percoll solution, then layered slowly onto the percoll density gradient (26% percoll + 40% percoll). After centrifugation at 30 700g for 5 min, the interface between 26 and 40% percoll layers was transferred to a new tube, 300 µl isolation buffer resuspended mitochondrial pellet, and the protein concentration was estimated by the Bradford method using BSA as a standard. Using a Seahorse XF24 analyzer, we assay isolated mitochondrial respiratory function following Seahorse mitochondrial assay's protocols. Briefly, loaded the XF sensor cartridge injection ports with ADP (A), oligomycin (B), FCCP (C), antimycin (D) or rotenone(D), dilute isolated mitochondria in 1× MAS3 to yield a final concentration of 200 µg of mitochondrial protein/ml, transfer 50 µl of diluted mitochondria into each well of a V7 XF24 tissue culture plate, added 450 µl of 1.1× initial media conditions with succinate and rotenone or pyruvate and malate, followed directions on the instrument controller to measure the plate containing the mitochondria. For PC12 cell OCR (basal oxygen consumption) measurement, following Seahorse XF24 bioenergetic profile experiment's protocols, PC12 cells transfected with duplicate Twinkle or WT Twinkle plasmids (14) are seeded in XF24 cell culture plates with 40 000 cells per well in 250 µl growth medium containing sodium pyruvate and glutamax, assayed in XF24 24 h after seeding. Compounds loaded into injector ports are 10 µg/ml Oligomycin (A), 0.3 µm FCCP (B) and 1.0 µm rotenone (C) in DMEM assay media, OCR is normalized by individual well protein.
Proteasome activity assay and Western blot
The midbrain of 1–month-old mice and PC12 cells were homogenized or lysed in 1× cell lysis buffer (cell signaling, #9803) with protease inhibitor (Roche) and phenylmethanesulfonyl fluoride (Sigma). The protein content was measured using the Bradford method with BSA as a standard. According to 20 s proteasome activity assay kit (cat. No. APT280) protocols, 10 µg protein was added to 100 µl of the assay reagent containing inhibitor lactacystin and/or luminogenic proteasome substrate in a 96-well plate. Samples were incubated at 37°C for 1 h and measured in a fluorometer using 380/460 nm filter. Each sample's fluorescence value was calculated by taking into account the test sample value subtracted from its inhibitor value, then normalized by the amount of the 20S proteasome. Western blot analysis was performed as described previously (45). The primary antibodies included antibodies recognizing Parkin (Assay Biotechnology), LC3 (Santa Cruz), MnSOD (Stressgen Bioreagents), CuSOD (Stressgen Bioreagents), VDAC (cell signaling) and 20S proteasome (Santa Cruz).
Coimmunoprecipitation
HEK293T cells were co-transfected with flag-tagged Twinkle plasmids and myc-tagged Parkin plasmids and cultured for 24 h. The cells were lysed with IP Lysis/wash buffer (Thermo scientific) with 1 mm phenylmethylsulfonyl fluoride and complete protease inhibitor cocktail (Roche). The whole cell lysates were incubated with anti-flag-m2 resin (Sigma), anti-c-myc agarose (Sigma) at 4°C, rotated overnight and washed four times with lysis buffer. Precipitates were analyzed by western blot using anti-flag mAb and anti-myc mAb.
ROS measurement
Molecular probe MitoSOX™ (Invitrogen) is used to measure ROS. Briefly, PC12 cells transfected by Twinkle duplicate or Twinkle WT were harvested after 48 h, resuspended in PBS(4 × 105 cells/ml) with 20 mm glucose and 5 µm MitoSOX, incubated at 37°C for 2 h and then measured in a fluorometer using a 530/620 nm filter.
Data analysis
Data obtained are presented as a mean from ≥3 mice per group and statistical significance is determined using the t-test. A single asterisk indicates P≤0.05 and the error bars in each figure represent standard deviations.
SUPPLEMENTARY MATERIAL
FUNDING
This work was mainly supported by a grant from the Cortopassi family foundation, and also received support from RO1NS077777.
ACKNOWLEDGEMENTS
We are grateful to Anu Suomalainen and Dr Henna Tynnisma for mouse-mutant Twinkle plasmids, to Hans Spelbrink for flag-tagged twinkle plasmids (13), and to Kazuto Kobayashi for pRTH/EGFP plasmids. This work is dedicated to the memory of Kara Brewer.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Gautier C.A., Kitada T., Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl Acad. Sci. USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abou-Sleiman P.M., Muqit M.M., Wood N.W. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 3.Parker W.D., Jr., Boyson S.J., Parks J.K. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann. Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 4.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G., et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 5.Serikawa T., Shimohata T., Akashi M., Yokoseki A., Tsuchiya M., Hasegawa A., Haino K., Koike R., Takakuwa K., Tanaka K., et al. Successful twin pregnancy in a patient with parkin-associated autosomal recessive juvenile parkinsonism. BMC Neurol. 2011;11:72. doi: 10.1186/1471-2377-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 7.Shin J.H., Ko H.S., Kang H., Lee Y., Lee Y.I., Pletinkova O., Troconso J.C., Dawson V.L., Dawson T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T., et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 9.Cortopassi G.A., Shibata D., Soong N.W., Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl Acad. Sci. USA. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soong N.W., Hinton D.R., Cortopassi G., Arnheim N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992;2:318–323. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- 11.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly Y.M., Gidlof S., Oldfors A., Wibom R., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 12.Luoma P., Melberg A., Rinne J.O., Kaukonen J.A., Nupponen N.N., Chalmers R.M., Oldfors A., Rautakorpi I., Peltonen L., Majamaa K., et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 13.Spelbrink J.N., Li F.Y., Tiranti V., Nikali K., Yuan Q.P., Tariq M., Wanrooij S., Garrido N., Comi G., Morandi L., et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 14.Tyynismaa H., Mjosund K.P., Wanrooij S., Lappalainen I., Ylikallio E., Jalanko A., Spelbrink J.N., Paetau A., Suomalainen A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl Acad. Sci. USA. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baloh R.H., Salavaggione E., Milbrandt J., Pestronk A. Familial Parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch. Neurol. 2007;64:998–1000. doi: 10.1001/archneur.64.7.998. [DOI] [PubMed] [Google Scholar]
- 16.Rothfuss O., Fischer H., Hasegawa T., Maisel M., Leitner P., Miesel F., Sharma M., Bornemann A., Berg D., Gasser T., et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009;18:3832–3850. doi: 10.1093/hmg/ddp327. [DOI] [PubMed] [Google Scholar]
- 17.Glauser L., Sonnay S., Stafa K., Moore D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011;118:636–645. doi: 10.1111/j.1471-4159.2011.07318.x. [DOI] [PubMed] [Google Scholar]
- 18.Um J.W., Im E., Lee H.J., Min B., Yoo L., Yoo J., Lubbert H., Stichel-Gunkel C., Cho H.S., Yoon J.B., et al. Parkin directly modulates 26S proteasome activity. J. Neurosci. 2010;30:11805–11814. doi: 10.1523/JNEUROSCI.2862-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alemi M., Prigione A., Wong A., Schoenfeld R., DiMauro S., Hirano M., Taroni F., Cortopassi G. Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic. Biol. Med. 2007;42:32–43. doi: 10.1016/j.freeradbiomed.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 21.Cotan D., Cordero M.D., Garrido-Maraver J., Oropesa-Avila M., Rodriguez-Hernandez A., Gomez Izquierdo L., De la Mata M., De Miguel M., Bautista Lorite J., Rivas Infante E., et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011;25:2669–87. doi: 10.1096/fj.10-165340. [DOI] [PubMed] [Google Scholar]
- 22.Archer C.R., Koomoa D.L., Mitsunaga E.M., Clerc J., Shimizu M., Kaiser M., Schellenberg B., Dudler R., Bachmann A.S. Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochem. Pharmacol. 2010;80:170–178. doi: 10.1016/j.bcp.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 23.Casarejos M.J., Solano R.M., Rodriguez-Navarro J.A., Gomez A., Perucho J., Castano J.G., Garcia de Yebenes J., Mena M.A. Parkin deficiency increases the resistance of midbrain neurons and glia to mild proteasome inhibition: the role of autophagy and glutathione homeostasis. J. Neurochem. 2009;110:1523–1537. doi: 10.1111/j.1471-4159.2009.06248.x. [DOI] [PubMed] [Google Scholar]
- 24.Fukui H., Moraes C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009;18:1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A., et al. Progressive Parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl Acad. Sci. USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by Parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 27.Deng H., Dodson M.W., Huang H., Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl Acad. Sci. USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H., Zhou X., Liu X., Yang L., Chen Q., Zhao D., Zuo J., Liu W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem. Biophys. Res. Commun. 2011;410:114–120. doi: 10.1016/j.bbrc.2011.05.116. [DOI] [PubMed] [Google Scholar]
- 29.Greenwood S.M., Bushell T.J. Astrocytic activation and an inhibition of MAP kinases are required for proteinase-activated receptor-2-mediated protection from neurotoxicity. J. Neurochem. 2010;113:1471–1480. doi: 10.1111/j.1471-4159.2010.06737.x. [DOI] [PubMed] [Google Scholar]
- 30.Stichel C.C., Zhu X.R., Bader V., Linnartz B., Schmidt S., Lubbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum. Mol. Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- 31.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 32.Pan T., Kondo S., Le W., Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 33.Takacs-Vellai K., Bayci A., Vellai T. Autophagy in neuronal cell loss: a road to death. Bioessays. 2006;28:1126–1131. doi: 10.1002/bies.20489. [DOI] [PubMed] [Google Scholar]
- 34.Apostolova N., Gomez-Sucerquia L.J., Gortat A., Blas-Garcia A., Esplugues J.V. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology. 2011;54:1009–19. doi: 10.1002/hep.24459. [DOI] [PubMed] [Google Scholar]
- 35.Iwata A., Riley B.E., Johnston J.A., Kopito R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 36.Massey A.C., Kaushik S., Sovak G., Kiffin R., Cuervo A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA. 2006;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J.Y., Nagano Y., Taylor J.P., Lim K.L., Yao T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsushita N., Okada H., Yasoshima Y., Takahashi K., Kiuchi K., Kobayashi K. Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J. Neurochem. 2002;82:295–304. doi: 10.1046/j.1471-4159.2002.00972.x. [DOI] [PubMed] [Google Scholar]
- 39.Fleming S.M., Salcedo J., Fernagut P.O., Rockenstein E., Masliah E., Levine M.S., Chesselet M.F. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J. Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song L., Fassler R., Mishina Y., Jiao K., Baldwin H.S. Essential functions of Alk3 during AV cushion morphogenesis in mouse embryonic hearts. Dev. Biol. 2007;301:276–286. doi: 10.1016/j.ydbio.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y., Gao J., Chung K.K., Huang H., Dawson V.L., Dawson T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl Acad. Sci. USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoenfeld R., Wong A., Silva J., Li M., Itoh A., Horiuchi M., Itoh T., Pleasure D., Cortopassi G. Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion. 2010;10:143–150. doi: 10.1016/j.mito.2009.12.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goydos J.S., Gorski D.H. Vascular endothelial growth factor C mRNA expression correlates with stage of progression in patients with melanoma. Clin. Cancer Res. 2003;9:5962–5967. [PubMed] [Google Scholar]
- 44.Sims N.R., Anderson M.F. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 2008;3:1228–1239. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
- 45.Shan Y., Napoli E., Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]