

Abstract
Dysosteosclerosis (DSS) is the form of osteopetrosis distinguished by the presence of skin findings such as red-violet macular atrophy, platyspondyly and metaphyseal osteosclerosis with relative radiolucency of widened diaphyses. At the histopathological level, there is a paucity of osteoclasts when the disease presents. In two patients with DSS, we identified homozygous or compound heterozygous missense mutations in SLC29A3 by whole-exome sequencing. This gene encodes a nucleoside transporter, mutations in which cause histiocytosis–lymphadenopathy plus syndrome, a group of conditions with little or no skeletal involvement. This transporter is essential for lysosomal function in mice. We demonstrate the expression of Slc29a3 in mouse osteoclasts in vivo. In monocytes from patients with DSS, we observed reduced osteoclast differentiation and function (demineralization of calcium surface). Our report highlights the pleomorphic consequences of dysfunction of this nucleoside transporter, and importantly suggests a new mechanism for the control of osteoclast differentiation and function.
INTRODUCTION
Dysosteosclerosis (DSS) is the form of osteopetrosis characterized by the additional features of platyspondyly, remarkable acquired metaphyseal osteosclerosis and red-violet macular atrophy of skin (1). Fewer than 25 cases have been reported since the first description by Ellis in 1934 (2). The features of DSS were later delineated by Spranger et al. (3) in 1968. Although the molecular basis is unknown, locus heterogeneity is suggested by consanguinity in some families (2,4) which supports autosomal recessive inheritance, and X-linked recessive transmission in another family with classical signs including dermatological findings (5).
RESULTS
We identified two unrelated children with DSS. Their clinical features are given in Table 1 and their radiographs are shown in Figure 1. They presented with fractures, cortical thickening and widening of the diaphysis of the long bones, cranial base sclerosis, broad ribs with sclerosis and platyspondyly. Subject 1 suffered from recurrent ear and skin infections between the ages of 8 and 11 months. Subject 2 presented with a pulmonary infection at two years of age and later necessitated surgical removal of her adenoids for breathing difficulties. Bone marrow biopsies were not performed in either child. Neither child had neurological symptoms or dental anomalies. Only subject 1 had skin findings, which were recurrent transient erythematous papules on the lower abdomen (1). Dual-energy X-ray absorptiometry for subject 1 showed a lumbar spine BMD Z-score of +8.1 at 2 years of age, and +11.9 at 5 years of age. Because she has short stature, these greatly elevated bone density scores are actually underestimates (6).
Table 1.
Summary of clinical and radiographic characteristics of children with DSS that we studied
| Subject 1 | Subject 2 | |
|---|---|---|
| References | Whyte et al. (1) | This publication |
| Consanguinity | − | + |
| Gender | F | F |
| Height | −2 SD | −2.8 SD |
| Head circumference | +4 SD | 91st percentile |
| Skin anomalies | + | − |
| Blue sclera | + | − |
| Recurrent infections | + | + |
| Laboratory anomalies | Increased erythrocyte sedimentation rate | Reactive lymphocytes on complete blood count, hypercalcemia |
| Serum glucose level | Normal | Normal |
| Cognition | Normal | Normal |
| Vision | Normal | Normal |
| Hearing | Normal | Normal |
| Hair and pilosity | Normal | Normal |
| Teeth anomalies | − | − |
| Frontal bossing | + | − |
| Cranial base sclerosis | + | + |
| Broad ribs with sclerosis | + | + |
| Platyspondyly | + | + |
| Pelvis, peripheral sclerosis | + | + |
| Diaphyseal widening and relative hypodensity | + | + |
| Cortical thickening (initially) | + | + |
| Fractures | + | + |
| Other skeletal features | Delayed closure of fontanelle | Genu valgum |
Figure 1.

Radiographs of patients. Images from subject 1 at 33 months (and 22 months for the pelvis) are from Whyte et al. (1), and have been reproduced with authorization from the publisher. For subject 2, radiographs were taken at 4 years of age. Note the sclerosis at the skull base, periphery of the iliac wings and metaphyses, platyspondyly, and in the long bones cortical thickening at the diaphysis, diaphyseal enlargement with relative radiolucency and a fracture in subject 2.
We performed whole-exome sequencing to identify the genetic basis of DSS. Our study was approved by the Institutional Review Board of the Baylor College of Medicine. As shown in Table 2, after discrete filtering against dBSNP129 for variants with known minor allele frequencies <5% (according to the 1000 Genomes Pilot project), only 303 genes had rare or novel protein-impacting variants in both subjects. As an initial strategy, we kept genes in which variants corresponded to an autosomal recessive model (i.e. homozygous or compound heterozygote variants). There were 132 such overlapping genes. Given the high number of false-positive INDEL calls, we preferentially explored genes that showed at least one single nucleotide polymorphism (SNP). As shown in Table 2, only 17 genes shared by the two subjects showed rare or novel protein-impacting variants. Variants were visualized and compared with 12 other exomes of subjects with unrelated conditions. After keeping only high-quality variants (e.g. removing variants in repeat regions or variants seen in unrelated conditions that are likely false positives), and considering variants predicted to be pathogenic, only SLC29A3 variants remained. Subject 1 is heterozygous for p.Ser203Pro (c.607T>C) and p.Arg386Gln (c.1157G>A). Subject 2, whose parents are second cousins, is homozygous for p.Thr449Arg (c.1346C>G). The mutated residues are highly conserved among mammalian species, and we confirmed the variants by Sanger sequencing (Fig. 2). The heterozygous variants in subject 1 were also confirmed to be on separate alleles (p.Ser203Pro from the mother and p.Arg386Gln from the father).
Table 2.
Number of variants/genes identified
| Subject 1 | Subject 2 | Shared | |
|---|---|---|---|
| Total variants | 3 629 754 | 3 259 789 | |
| Variants after base quality filtering | 1 334 715 | 1 262 398 | |
| Novel or rare variants (dbSNP129/1000GP queried) | 1 018 297 | 886 839 | |
| Non-synonymous/indel/splice-site variants | 811 | 871 | 303 |
| Genes fitting autosomal recessive model (homozygous or two heterozygous variants) | 190 | 217 | 132 |
| Genes with at least one single nucleotide variant | 11 | 11 | 11a |
| Genes after removal of genes with high mutation rates | 2 | 2 | 2b |
| Genes with plausible function/disease association | 1 | 1 | 1c |
aHLA-A, BC139719, SLC29A3, HRNR, FAM22A, HSP90AB3P, FMN2, BCLAF1, RIMBP3, LILRB3, GSPT1.
bBCLAF1, SLC29A3.
cSLC29A3.
Figure 2.
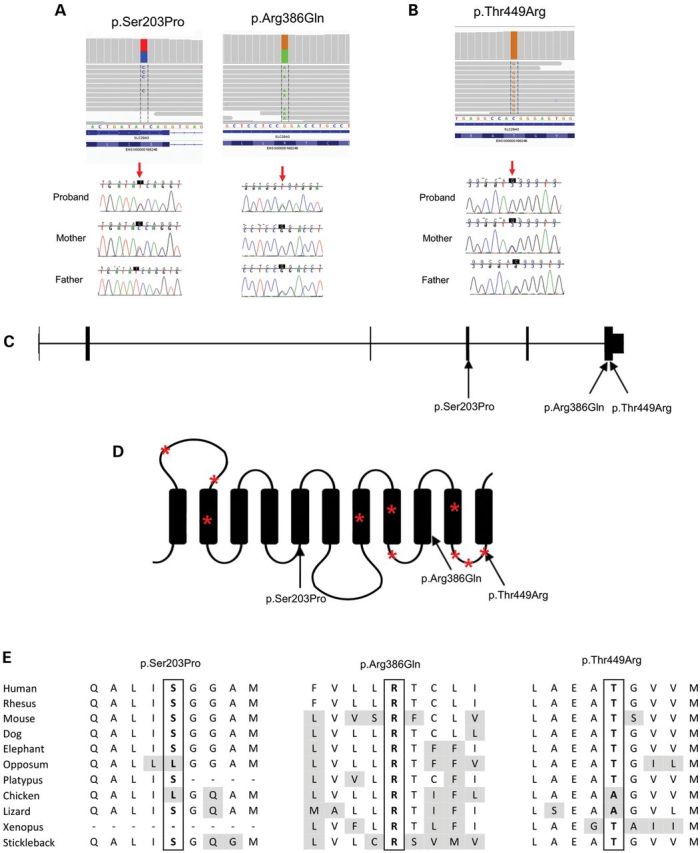
Mutation details. Images from the exome alignments and the confirmation by Sanger sequencing for subjects 1 (A) and 2 (B). Location of mutations in the SLC29A3 gene (C) and protein (D). Asterisks denote known mutations in histiocytosis–lymphadenopathy plus syndrome [see mutation database at http://www.lovd.nl and reference (24) for more details]. (E) Amino acid conservation of the mutated residue across species.
To assess whether SLC29A3 is expressed in osteoclasts, we performed immunohistochemistry in mouse bones. As shown in Figure 3, there is a strong punctate pattern of SLC29A3 in the cytoplasm of multinucleated cells, i.e. osteoclasts, lining the bone (Fig. 3A). We also isolated peripheral blood monocytes from the two DSS subjects described, differentiated them in vitro and performed a demineralization assay. Consistent with the ‘osteoclast-poor’ bone biopsy findings previously demonstrated in subject 1 (1), osteoclastic differentiation was impaired in the DSS samples (Fig. 3B and C), and the ability of the differentiated osteoclasts to demineralize a crystalline calcium phosphate surface was also markedly reduced (Fig. 3D–F).
Figure 3.
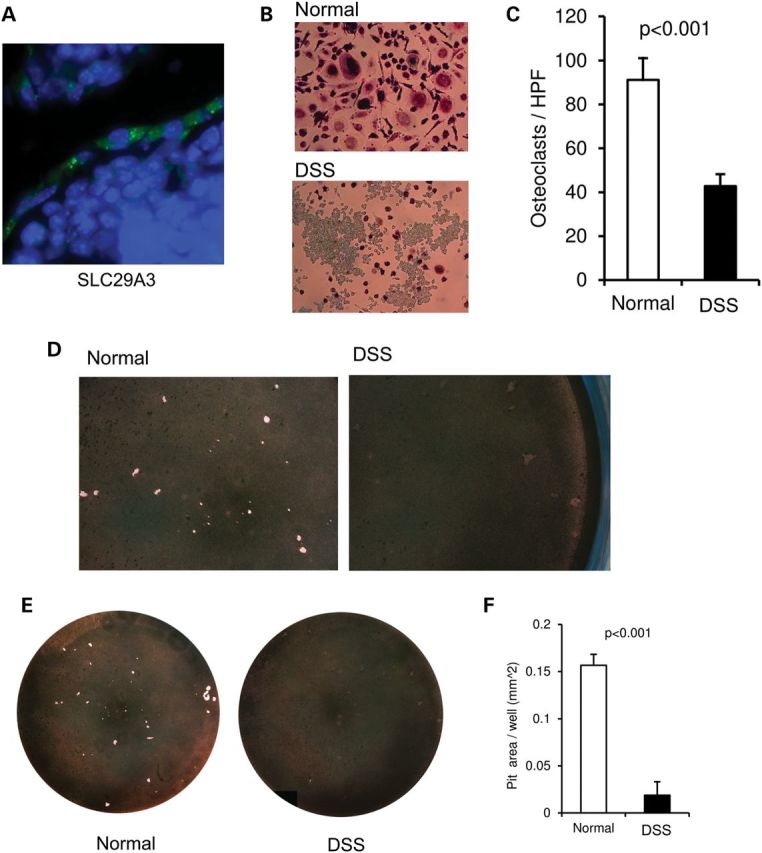
(A) SLC29A3 expression in mouse osteoclasts. Nuclei stained by DAPI are in blue and SLC29A3 is in green. (B) Tartrate-resistant acid phosphatase (TRAP) staining of osteoclasts after 21 days of differentiation of human monocytes with RANKL and MCSF (105 cells/well in a 96-well plate). (C) Quantification of osteoclasts based on TRAP staining. Experiments were done in triplicate with an N of 2 per group (two affected and two unaffected); unaffected unrelated individuals were used as controls. (D–F) Osteoclastic function assessed by the ability of the in vitro-differentiated human osteoclasts to demineralize inorganic crystalline calcium phosphate coated on a polystyrene 96-well plate. Magnified images are shown in (D), the complete wells in (E) and a quantification of the demineralized surface in (F).
DISCUSSION
Variants in SLC29A3 are known to cause pigmented hypertrichosis with insulin-dependent diabetes (PHID) and H syndrome (skin hyperpigmentation and hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism and low height) (7,8), with the addition of two related conditions, Faisalabad histiocytosis and sinus histiocytosis with massive lymphadenopathy. All four disorders were recently suggested to be aggregated under the term histiocytosis–lymphadenopathy plus syndrome [see OMIM (Online Mendelian Inheritance in Man) no. 602782 and references therein]. There is some clinical overlap between PHID and H syndrome, and in some families both conditions manifest (9). Furthermore, there is phenotypic variability as mildly affected individuals have also been described (10). Table 3 shows the overlap between PHID, H syndrome and DSS. Although PHID and H syndrome do not manifest osteopetrosis, affected individuals can have camptodactyly, clinodactyly, short stature and hallux valgus. In one family where one child was diagnosed with H syndrome and another more severely affected child was diagnosed with PHID and brachydactyly, a skeletal survey in the latter was noted only to show osteopenia in the metacarpals and metatarsals (9).
Table 3.
Comparison of clinical features among different SLC29A3-related conditions
| Pigmented hypertrichosis with insulin-dependent diabetes (PHID) | H syndrome | Dysosteosclerosis | |
|---|---|---|---|
| Skin | Patches and plaques of hyperpigmented and hypertrichotic skin, lymphohistiocytic infiltration | Patches and plaques of hyperpigmented and hypertrichotic skin, lymphohistiocytic infiltration | Patches of hyperpigmented skin. Hypertrichosis is rare (25) |
| Craniofacial | Rarely: hypertelorism, flat and broad nasal bridge, upturned nares, prominent maxillary bone and gingival hypertrophy (9) | Exophthalmos | Frontal bossing, mid-face hypoplasia |
| Other skeletal defects | Camptodactyly, clinodactyly, short stature | Camptodactyly, short stature, hallux valgus | Short stature, sclerotic platyspondyly, metaphyseal sclerosis, diaphyseal widening and demarcated osteosclerosis |
| Hearing | Deafness | Otosclerosis | |
| Bone marrow | The bone marrow: non-clonal myeloproliferative process. Numerous monocytes and histiocytes and moderate myelofibrosis | Red cell aplasia due to myelofibrosis in one patient | Myelofibrosis and anemia are rare but have been reported in other families (2,26) |
| Liver | Hepatosplenomegaly | Hepatosplenomegaly | Normal |
| Heart | Pulmonary stenosis, pericarditis | Pulmonary stenosis, patent ductus arteriosus with pulmonary hypertension, pericarditis | Dysplastic pulmonary valves, pulmonary hypertension |
| Endocrine | Diabetes, hypogonadism | Diabetes, hypogonadism | Normal |
| Other | Lymphadenopathy, histiocyte infiltration | Azoospermia | Regression, compression of CNS and cranial nerves |
| SLC29A3 mutations | p.Met116Arg, p.Glu444X, p.Tyr314fs, p.Gly437Arg, p.Thr449Arg (homozygous) | p.Arg134Cys, p.Leu349SerfsX56, p.Arg363Gln, p.Arg363Trp, p.Gly427Ser, p.Gly437Arg | Subject 1: p.Ser203Pro, p.Arg386Gln; subject 2: p.Thr449Arg (homozygous) |
One of the mutations we identified (p.Thr449Arg) has previously been shown to impair nucleoside transport (11). Also, in the homozygous state, like in subject 2, the variant caused PHID. Furthermore, one of the heterozygous mutations in subject 1 (p.Arg386Gln) causes H syndrome when homozygous in a Syrian family (12). The reasons for the pleiotropism and variability of SLC29A3-related diseases are not known; however, modifier genes could possibly be involved. We queried the list of rare and novel variant changes in genes known to be important for osteoclastogenesis and osteoclast function, using Uniprot (13) and Genedistiller (14), but we did not identify such likely candidates apart from the CLCN7 missense variant of unknown significance previously published for subject 1 and her father (1). Good coverage was obtained for known osteopetrosis genes in both subjects and no other notable variants were identified. Future investigation of additional DSS patients will allow the assessment of the role of known osteopetrosis genes or other modifier genes in the presentation of DSS. SLC29A3 encodes a nucleoside transporter localized in lysosomes and is highly expressed in some white blood cells such as histiocytes and macrophages. Mice deficient in SLC29A3 have significant lysosomal dysfunction in macrophages (15); proper lysosomal function is also crucial for osteoclasts. We propose that SLC29A3 could be important for osteoclast differentiation and activity.
In summary, we describe two unrelated children with DSS associated with autosomal recessive inheritance of variants in SLC29A3. While we have unraveled a single locus for this disease that has potential genetic heterogeneity, investigation of future cases will be critical to better understand the pathophysiology of DSS. The importance of SLC29A3 for lysosomes, its expression in osteoclasts and the reduced number of osteoclasts both in vivo and in vitro in our patients suggest that this protein may impact osteoclast differentiation and function.
MATERIALS AND METHODS
Whole-exome sequencing
Exomes were captured on Nimblegen's SeqCap EZ V2.0 library (Roche NimbleGen, Madison, WI, USA) and sequencing was conducted on Illumina HiSeq 2000 (Illumina, San Diego, CA, USA). Sequence reads were aligned to the hg18 iteration of the reference human genome using Burrows–Wheeler Aligner (v 0.5.9) (16) and realigned using the Genome Analysis Toolkit (GATK) for base quality score recalibration, indel (insertion or deletion) realignment and duplicate removal (17,18). Both samples achieved over 90% targeted bases at 20× coverage. SNPs were called using Sequence Alignment/Map tools (SAMtools) Pileup (version 0.1.17) (19) and short indels were called using SAMtools (19), Atlas-INDEL (20) and GATK (17). INDELs were overlapped using a three of three concordance criteria and included in our variant list. Variants were annotated with ANNOVAR (ANNOtation of genetic VARiants) (21); protein-impacting variants that were rare (minor allele frequency <5%) or novel were preferentially explored. Gene candidates were then assessed using databases such as database for non-synonymous SNPs’ functional predictions, which assesses the functional impact and the conservation of the mutations (22), Uniprot for the function of the proteins (13), Nextprot for the expression pattern, Mouse Genome Informatics and OMIM for the phenotypes in mice and humans and, finally, Genedistiller2 for a combination of some of the above databases. The accession numbers for the human SLC29A3 gene are as follows: NCBI Gene: 55315; GenBank transcript: NM_018344; CCDS protein: 7310.
In vitro differentiation of monocytes
Peripheral blood mononuclear cells were isolated using a double-gradient technique (Sigma Histopaque 1077 and 1119 using the manufacturer's instructions), and monocytes were selected and differentiated for 21 days as described previously (23), then stained for tartrate-resistant acid phosphatase (TRAP assay kit, Sigma). The demineralization assay was performed by differentiating monocytes on a Corning Osteo Assay Surface in a 96-well plate and then performing Von Kossa staining.
Immunohistochemistry
Mouse limbs were fixed in 4% PFA overnight, washed with PBS, decalcified with 14% EDTA, pH 7.2 for 7 days, transferred to 25% sucrose for 2 h, then a 15% sucrose and 50% OCT solution for 2 h and finally in 100% OCT for 1 h. The limbs were then frozen in OCT on dry ice for cryosectioning. For immunohistochemistry, sections were rinsed with PBS and blocked with 3% Donkey Serum in 0.1% BSA and 0.1% Triton. Slides were then incubated overnight with a goat polyclonal antibody raised against a peptide mapping at the N-terminus of ENT3 of mouse origin [Santa Cruz ENT3 (N-20): sc-48150 antibody] at a dilution of 1:1000 in blocking buffer (a control without primary antibody was used). After rinsing, the slides were incubated for 1 h in Alexa Fluor® 594 donkey anti-goat IgG (Invitrogen, 1:600 final dilution in blocking buffer). After further rinsing, slides were mounted with ProLong® Gold antifade reagent with DAPI for imaging. Slides were viewed with a Zeiss Axioplan 2 fluorescence microscope.
FUNDING
This work was supported by the National Institutes of Health (P01 HD070394 and P01 HD22657 to B.H.L., R01 DK067145 to S.M.), by Shriners Hospitals for Children, by the Rolanette and Berdon Lawrence Bone Disease Program of Texas and by the Howard Hughes Medical Institutes Med in Grad Initiative. P.M.C. is supported by the Clinician-Scientist Training Award of the Canadian Institutes of Health Research.
ACKNOWLEDGEMENTS
We thank the families for participating in this study. We thank Alyssa Tran, for help enrolling subjects.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Whyte M.P., Wenkert D., McAlister W.H., Novack D.V., Nenninger A.R., Zhang X., Huskey M., Mumm S. Dysosteosclerosis presents as an ‘osteoclast-poor’ form of osteopetrosis: comprehensive investigation of a 3-year-old girl and literature review. J. Bone Miner. Res. 2010;25:2527–2539. doi: 10.1002/jbmr.131. doi:10.1002/jbmr.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis R.W. Osteopetrosis: (Section for the Study of Disease in Children); Proc. R. Soc. Med.; 1934. pp. 1563–1571. [PMC free article] [PubMed] [Google Scholar]
- 3.Spranger J., Albrecht C., Rohwedder H.J., Wiedemann H.R. Dysosteosclerosis—a special form of generalized osteosclerosis. Fortschr. Geb. Rontgenstr. Nuklearmed. 1968;109:504–512. doi:10.1055/s-0029-1228480. [PubMed] [Google Scholar]
- 4.Lemire E.G., Wiebe S. Clinical and radiologic findings in an adult male with dysosteosclerosis. Am. J. Med. Genet. A, 2008;146A:474–478. doi: 10.1002/ajmg.a.32182. doi:10.1002/ajmg.a.32182. [DOI] [PubMed] [Google Scholar]
- 5.Pascual-Castroviejo I., Casas-Fernandez C., Lopez-Martin V., Martinez-Bermejo A. X-linked dysosteosclerosis. Four familial cases. Eur. J. Pediatr. 1977;126:127–138. doi: 10.1007/BF00442194. [DOI] [PubMed] [Google Scholar]
- 6.Zhang F., Whyte M.P., Wenkert D. Dual-energy X-ray absorptiometry interpretation: a simple equation for height correction in preteenage children. J. Clin. Densitom. 2012;15:267–274. doi: 10.1016/j.jocd.2012.01.004. doi:10.1016/j.jocd.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Cliffe S.T., Kramer J.M., Hussain K., Robben J.H., de Jong E.K., de Brouwer A.P., Nibbeling E., Kamsteeg E.J., Wong M., Prendiville J., et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum. Mol. Genet. 2009;18:2257–2265. doi: 10.1093/hmg/ddp161. doi:10.1093/hmg/ddp161. [DOI] [PubMed] [Google Scholar]
- 8.Molho-Pessach V., Lerer I., Abeliovich D., Agha Z., Abu Libdeh A., Broshtilova V., Elpeleg O., Zlotogorski A. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am. J. Hum. Genet. 2008;83:529–534. doi: 10.1016/j.ajhg.2008.09.013. doi:10.1016/j.ajhg.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiegel R., Cliffe S.T., Buckley M.F., Crow Y.J., Urquhart J., Horovitz Y., Tenenbaum-Rakover Y., Newman W.G., Donnai D., Shalev S.A. Expanding the clinical spectrum of SLC29A3 gene defects. Eur. J. Med. Genet. 2010;53:309–313. doi: 10.1016/j.ejmg.2010.06.012. doi:10.1016/j.ejmg.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Jonard L., Couloigner V., Pierrot S., Louha M., Gherbi S., Denoyelle F., Marlin S. Progressive hearing loss associated with a unique cervical node due to a homozygous SLC29A3 mutation: a very mild phenotype. Eur. J. Med. Genet. 2011;55:56–58. doi: 10.1016/j.ejmg.2011.06.009. doi:10.1016/j.ejmg.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Kang N., Jun A.H., Bhutia Y.D., Kannan N., Unadkat J.D., Govindarajan R. Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J. Biol. Chem. 2010;285:28343–28352. doi: 10.1074/jbc.M110.109199. doi:10.1074/jbc.M110.109199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farooq M., Moustafa R.M., Fujimoto A., Fujikawa H., Abbas O., Kibbi A.G., Kurban M., Shimomura Y. Identification of two novel mutations in SLC29A3 encoding an equilibrative nucleoside transporter (hENT3) in two distinct Syrian families with H syndrome: expression studies of SLC29A3 (hENT3) in human skin. Dermatology. 2012;224:277–284. doi: 10.1159/000338886. doi:10.1159/000338886. [DOI] [PubMed] [Google Scholar]
- 13.Uniprot_Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–75. doi: 10.1093/nar/gkr981. doi:10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seelow D., Schwarz J.M., Schuelke M. GeneDistiller—distilling candidate genes from linkage intervals. PLoS One. 2008;3:e3874. doi: 10.1371/journal.pone.0003874. doi:10.1371/journal.pone.0003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu C.L., Lin W., Seshasayee D., Chen Y.H., Ding X., Lin Z., Suto E., Huang Z., Lee W.P., Park H., et al. Equilibrative nucleoside transporter 3 deficiency perturbs lysosome function and macrophage homeostasis. Science. 2012;335:89–92. doi: 10.1126/science.1213682. doi:10.1126/science.1213682. [DOI] [PubMed] [Google Scholar]
- 16.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. doi:10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. doi:10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. doi:10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. doi:10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Havlak P., Chen R., Durbin K.J., Egan A., Ren Y., Song X.Z., Weinstock G.M., Gibbs R.A. The Atlas genome assembly system. Genome Res. 2004;14:721–732. doi: 10.1101/gr.2264004. doi:10.1101/gr.2264004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. doi:10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human non-synonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabokbar A., Athanasou N.S. Generating human osteoclasts from peripheral blood. Methods Mol. Med. 2003;80:101–111. doi: 10.1385/1-59259-366-6:101. [DOI] [PubMed] [Google Scholar]
- 24.Bolze A., Abhyankar A., Grant A.V., Patel B., Yadav R., Byun M., Caillez D., Emile J.F., Pastor-Anglada M., Abel L., et al. A mild form of SLC29A3 disorder: a frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS One. 2012;7:e29708. doi: 10.1371/journal.pone.0029708. doi:10.1371/journal.pone.0029708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy C., Maroteaux P., Kremp L., Courtecuisse V., Alagille D. A new bone syndrome with skin anomalies and neurologic disorders. Arch. Fr. Pediatr. 1968;25:893–905. [PubMed] [Google Scholar]
- 26.Sener R.N., Yalman O., Cetingul N., Tutuncuoglu S., Kavakli K., Ustun E.E. Dysosteosclerosis: clinicoradiologic findings including brain MRI. Comput. Med. Imaging Graph. 1997;21:355–357. doi: 10.1016/s0895-6111(97)00030-x. doi:10.1016/S0895-6111(97)00030-X. [DOI] [PubMed] [Google Scholar]