

Abstract
Mitochondria continually undergo fusion and fission, and these dynamic processes play a major role in regulating mitochondrial function. Studies of several genes associated with familial Parkinson's disease (PD) have implicated aberrant mitochondrial dynamics in the disease pathology, but the importance of these processes in dopaminergic neurons remains poorly understood. Because the mitofusins Mfn1 and Mfn2 are essential for mitochondrial fusion, we deleted these genes from a subset of dopaminergic neurons in mice. Loss of Mfn2 results in a movement defect characterized by reduced activity and rearing. In open field tests, Mfn2 mutants show severe, age-dependent motor deficits that can be rescued with L-3,4 dihydroxyphenylalanine. These motor deficits are preceded by the loss of dopaminergic terminals in the striatum. However, the loss of dopaminergic neurons in the midbrain occurs weeks after the onset of these motor and striatal deficits, suggesting a retrograde mode of neurodegeneration. In our conditional knockout strategy, we incorporated a mitochondrially targeted fluorescent reporter to facilitate tracking of mitochondria in the affected neurons. Using an organotypic slice culture system, we detected fragmented mitochondria in the soma and proximal processes of these neurons. In addition, we found markedly reduced mitochondrial mass and transport, which may contribute to the neuronal loss. These effects are specific for Mfn2, as the loss of Mfn1 yielded no corresponding defects in the nigrostriatal circuit. Our findings indicate that perturbations of mitochondrial dynamics can cause nigrostriatal defects and may be a risk factor for the neurodegeneration in PD.
INTRODUCTION
Parkinson's disease (PD) is a neurodegenerative movement disorder characterized by resting tremor, rigidity, bradykinesia and postural instability. PD symptoms are classically attributed to dopamine depletion and the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc). However, additional neuronal circuits are affected, and non-motor symptoms are often present, suggesting a systemic pathology. There is compelling evidence that mitochondrial dysfunction is a primary event in the disease process. Decreased mitochondrial electron-transport chain activity has been detected in tissues from PD patients (1,2), and toxins that inhibit mitochondrial respiration can give rise to parkinsonism in humans and animal models (3,4). The most direct evidence, however, has come from the study of genes associated with familial forms of PD. Disruption of the PD-associated genes PINK1, Parkin, DJ-1 or LRRK2 results in mitochondrial defects and aberrant mitochondrial morphology (5–10).
Fusion and fission are fundamental processes that control the function of mitochondria (11). Mitofusins Mfn1 and Mfn2 are outer membrane GTPases that mediate the fusion of mitochondrial outer membranes. The optic atrophy protein OPA1 is necessary for the fusion of the mitochondrial inner membranes. The dynamin-related protein Drp1 mediates the opposing process of mitochondrial fission. Several neurodegenerative diseases, including PD, have been linked to perturbations in mitochondrial dynamics (12).
Recently, this link has been strengthened by the analysis of genes that cause familial forms of PD. The PINK1/Parkin pathway is profoundly affected by mitochondrial fusion and fission. In Drosophila, PINK1 and Parkin loss-of-function mutants have severe mitochondrial defects, characterized by swollen mitochondria with abnormal cristae (6–8,13). These mitochondrial perturbations lead to the dysfunction of the indirect flight muscles. The defects in mitochondria and muscle are efficiently rescued by the overexpression of Drp1 or the knockdown of Marf (a fly ortholog of mitofusin) or OPA1 (14–16). In cultured mammalian cells, the PINK1/Parkin pathway has been shown to promote the elimination of dysfunctional mitochondria by the autophagy machinery. This degradative process, termed mitophagy, is dependent on mitochondrial fission (17,18). Disruption of DJ-1 function causes mitochondrial fragmentation and enhanced sensitivity to oxidative stress (5,9,19,20). The overexpression of PINK1 and Parkin in DJ-1-deficient cells rescues the fragmented mitochondrial morphology, suggesting the regulation of mitochondrial dynamics by each pathway (5,20). Finally, PD-associated mutations of LRRK2 in cells induce mitochondrial fragmentation and increased localization of Drp1 to the mitochondria (10).
These observations indicate that PD-related mutations and mitochondrial dynamics have a reciprocal relationship. PD-related mutations can perturb mitochondrial dynamics, and the consequences of these mutations can be modulated by mitochondrial dynamics. These results highlight the need to understand the function of mitochondrial dynamics in dopaminergic neurons. To address this issue, we have analyzed the consequences of removing the mitofusins from dopaminergic neurons. We find that the loss of Mfn2 results in a severe movement disorder attributed to the progressive degeneration of the nigrostriatal circuit.
RESULTS
Deletion of mitofusins from dopaminergic neurons
We utilized a genetic approach to assess the role of mitochondrial fusion in dopaminergic neurons. Mfn1 and Mfn2 conditional knockout mice were crossed to the Slc6a3-Cre driver (21), in which the endogenous dopamine transporter locus expresses Cre recombinase in the A8–A10 subset of dopaminergic neurons, including those of the SNc (Fig. 1A). Our mating scheme also incorporated a mito-Dendra2 Cre reporter for the dual purpose of labeling mitochondria and monitoring Cre-dependent excision. Mice with the disruption of Mfn1 show no phenotype up to 1 year of age (Supplementary Material, Fig. S1). In contrast, mice with the disruption of Mfn2 are hunched and hypoactive by 5 weeks of age when compared with wild-type or heterozygous Mfn2 littermates. These mice also exhibit kyphosis and reduced activity (Fig. 1B and Supplementary Material, Movie S1). Weight gain is stagnant after 4 weeks of age, resulting in an increasingly larger weight difference between mutant and control littermates (Fig. 1C).
Figure 1.

Growth defect in Mfn2 mutant mice. (A) Mating scheme. Slc6a3-Cre is controlled by the dopamine transporter locus and is expressed in a subset of dopaminergic neurons, particularly the SNc, VTA and retrorubral field (21). The cross also incorporated a cre-induced, mitochondrially targeted Dendra2 (termed PhAM). (B) Representative image of a 22-week-old Mfn2 mutant (KO) compared with a heterozygous littermate (Het). Note the small size and severe kyphosis of the Mfn2 mutant mice. (C) Plot showing weekly weight measurements. Each point represents the average weight ± SEM (n = 10–15 for each genotype and sex). Both male and female Mfn2 mutants are significantly smaller (P < 0.05, two-tailed Student's t-test) than control animals by 5 weeks of age.
In our initial cohort of Mfn2 mutants, many animals died at ∼6–7 weeks of age due to apparent malnutrition. Mutant animals (n = 15) died or were culled due to significant weight loss between 36 and 48 days of age. However, when supplied with hydrated gel packs and crushed pieces of regular chow on the cage floor, all mutant Mfn2 mice survive beyond 6 months, with a majority surviving past 1 year of age. These mice likely have difficulty accessing food and water in normal cages due to a severe rearing defect (see later). In this respect, our mice are distinct from some other dopamine depletion models that exhibit aphagia and adipsia even when food is placed nearby (22,23). To minimize possible secondary effects due to malnutrition, the phenotypic analyses reported below were performed using mutant and control mice provided with this dietary supplementation.
Movement disorder in Mfn2 mutants
Because initial observations suggested that Mfn2 mutant mice had reduced activity, we monitored their spontaneous movements in a longitudinal open field study. Mfn2 mutant mice show an age-dependent decline in locomotive activity (Fig. 2A). At 4–5 weeks, mutant animals travel only 68% of the distance traversed by wild-type control animals. This defect progresses over the next several weeks. By 8–11 weeks of age, the distance traveled by mutants reduces to 34% of wild-type controls (Fig. 2B). In contrast, Mfn2 heterozygous animals show normal locomotion. We compared the Mfn2 homozygotes to both wild-type controls and heterozygous controls carrying Slc6a3-Cre, because the knock-in Cre allele causes a slight, non-significant, decrease in dopamine transporter levels in the heterozygous state (21). We found that Mfn2 heterozygotes carrying Slc6a3-Cre are indistinguishable from wild-type controls in all the assays used in this study.
Figure 2.

Longitudinal analysis of locomotive activity in Mfn2 mutants. (A) Representative traces from open field analysis. The traces represent spontaneous movement in an open field during a 15-min observation period. The genotypes and ages of the mice are indicated. The open field analysis was quantified to obtain the (B) distance traversed, (C) average velocity, (D) rearing frequency and (E) immobile periods between activities. In all graphs, values from the heterozygous and homozygous animals were normalized to that of the wild-type controls, and error bars represent the propagated standard error. The Student's t-test was used to obtain P-values between Mfn2 mutants and wild-type controls (*P < 0.05; **P < 0.001; n = 6–10 animals per age and genotype).
Similar to travel distance, the speed of movement exhibited by Mfn2 mutant mice declines with age (Fig. 2C). We also observed a strong rearing defect in mutant Mfn2 mice that is present as early as 4 weeks of age (Fig. 2D and Supplementary Material, Movie S1). This postural defect likely contributes to the starvation and dehydration observed at 6 weeks when cages are not supplemented with food and gel packs on the floor. Consistent with the decreased locomotion, Mfn2 mutants spend twice as much time inactive at 6–7 weeks of age. By 8–11 weeks, this discrepancy increases to 6-fold (Fig. 2E). Of note, the locomotive defect is specific for Mfn2 mutants; Mfn1 mutants show no motor deficiency in the open field test (Supplementary Material, Fig. S1). Moreover, the double Mfn1/Mfn2 mutants do not have an exacerbated phenotype compared with Mfn2 mutants (Supplementary Material, Fig. S2). Overall, measurements from the open field test suggest that, beginning at 4–5 weeks, Mfn2 mutants exhibit progressive bradykinesia and a postural defect, both cardinal signs of PD.
Retrograde degeneration of SNc dopaminergic neurons
To determine whether the motor deficits in mutant animals are accompanied by a loss of dopaminergic innervation, we used tyrosine hydroxylase (TH) immunoreactivity to assess the nigrostriatal circuit. We first analyzed the striatum, the endpoint of the nigrostriatal pathway. Here, TH-staining marks the axon terminals derived from the SNc. In Mfn2 mutants, the striatum shows a 25% reduction in dopaminergic terminals at 3 weeks of age (Fig. 3A and C). Loss of TH immunoreactivity is detected first in the dorsolateral striatum (Fig. 3A, outlined region in a 3-week sample) and gradually encompasses the entire striatum by 11 weeks. Interestingly, the regional severity of striatal loss in Mfn2 mutants resembles the pattern described in PD patients (24,25). By 8–10 weeks, the depletion of dopaminergic terminals increases to 76% in Mfn2 mutant animals (Fig. 3C). In contrast, the projections to the nucleus accumbens (NAc) and olfactory tubercle (OT), which come from dopaminergic neurons in the ventral tegmental area (VTA), appear to be more protected. These dopaminergic terminals, which are part of the mesolimbic pathway, are moderately preserved at 11–14 weeks (Fig. 3A, outlined regions in the 11 and 14 week panels).
Figure 3.

Retrograde degeneration of SNc dopaminergic neurons. (A) Dopaminergic projections to the striatum (Str), NAc and OT. The diagram delineates these regions. Sections were stained with TH antibody to label dopaminergic projections (brown pigment). At 3 weeks of age, Mfn2 mutant animals show decreased TH-immunoreactive terminals in the dorsolateral striatum (outlined region). Later time points reveal widespread loss in the striatum. VTA projections to the NAc and OT are still present, albeit reduced, at 11 and 14 weeks (outlined regions in bottom two panels). (B) Dopaminergic neurons at the SNc and VTA. Sections of the midbrain were stained with TH and counterstained with Cresyl violet (blue) to identify dopaminergic neurons. At 11 and 14 weeks, the Mfn2 mutants exhibit reduced staining in the SNc, whereas the VTA is relatively preserved. The SNc and VTA regions are outlined. (C) Quantification of TH-staining. Measured values of the TH-positive signal from heterozygous and homozygous animals were normalized to wild-type controls. For each animal, three sections were measured. The Student's t-test was used to obtain P-values (*P < 0.05; **P < 0.001; n = 3–6), and the error bars represent the propagated error. (D) Quantitation of dopaminergic cell loss in the SNc. Counts from heterozygous and homozygous animals were normalized to wild-type controls (n = 3 for each genotype). For each animal, nine sections spanning the rostro-caudal extent of the midbrain were manually counted. Statistical analysis was performed as in (C). (E) Magnified images of (B) showing the loss of dopaminergic neurons and processes in Mfn2 mutant animals.
Moving upstream in the nigrostriatal circuit, we counted the number of TH-immunoreactive neurons in the SNc. In contrast to the striatum, there is no notable loss of TH-positive neurons in the SNc at either 3 or 8–9 weeks (Fig. 3B and D). The earliest time point with neuronal loss occurs at 10–12 weeks, with a 52% decrease in TH-immunopositive neurons. Further degeneration followed at subsequent ages (Fig. 3D). Additionally, the neurons remaining in Mfn2 mutants appear to have smaller cell bodies as well as diminished neuronal processes (Fig. 3E). Partial loss of neurons was also observed at the VTA but not to the extent of the SNc.
Because degenerating neurons can lose the expression of neuronal markers, the loss of TH immunoreactivity does not necessarily indicate neuronal loss. To directly evaluate the loss of neurons, we performed Nissl staining of the SNc (Fig. 4). Normal staining was observed at 3 weeks of age, but by 11 weeks and beyond, the SNc showed a reduced density of Nissl-stained cells. Taken together, these results indicate two pertinent features of neurodegeneration in this mouse model. First, multiple types of dopaminergic neurons have a requirement for Mfn2, but the nigrostriatal circuit exhibits enhanced vulnerability compared with the mesolimbic pathway. Likewise, SNc neurons in PD patients are more severely affected than the VTA population (26,27). Second, the degeneration of Mfn2-deficient dopaminergic neurons occurs in a stepwise manner. The initial defects appear at the axon terminals, followed 1–2 months later by the degeneration of the cell bodies.
Figure 4.

Loss of dopaminergic neurons in the SNc. Coronal midbrain slices were analyzed by fluorescent Nissl staining to highlight neurons. (A) The SNc is indicated by the outlined oval. Decreased neuronal density in the SNc is apparent by 11 weeks in the Mfn2-null mutant. Scale bar is 100 μm. (B) High-magnification images of Nissl-stained cells in the SNc. Scale bar is 20 μm.
Mitochondrial fragmentation and depletion in dopaminergic neurons
We previously generated a Cre reporter of mitochondrial dynamics that targets the photo-convertible fluorescent protein Dendra2 to the mitochondrial matrix (28). The expression of mito-Dendra2 relies on the Cre-mediated excision of an upstream loxP-flanked termination signal. In our mating scheme (Fig. 1A), mito-Dendra2 expression depends on the Slc6a3-Cre driver, thereby allowing us to visualize mitochondria within the affected neurons, a key benefit in the densely populated midbrain. We have also established a slice culture system to assess mitochondrial dynamics in Mfn2-null dopaminergic neurons. The organotypic culture system has been extensively used for the long-term assessment of neuronal function and development in vitro because it preserves the cyto-architecture and circuitry between multiple brain regions (29,30). To best preserve the nigrostriatal connections, we sectioned the brains at an angle previously characterized to retain these projections (31,32).
In slice cultures from wild-type and heterozygous brains, we found that Slc6a3-Cre/mito-Dendra2 expression is specific for dopaminergic neurons, as evidenced by its restriction to cells with TH immunoreactivity (Fig. 5A). The mitochondria in control heterozygous slices have a mixed morphology profile, consisting of both tubular structures in proximal processes and short puncta in distal projections. In Mfn2-null slices, we found swollen and fragmented mitochondria in the soma and proximal processes. Consistent with our histological analysis (Fig. 3E), the Mfn2 mutant neurons have fewer and thinner processes extending from the cell body (Fig. 5A). In addition, the Mfn2 mutant cultures contain many neurons that express mito-Dendra2 but lack or have reduced TH signal. The failure to maintain TH expression suggests that these neurons are at an intermediate stage of degeneration (Fig. 5A, starred neurons). We also noted a severe depletion of mitochondria in neuronal processes both proximal and distal to dopaminergic cell bodies (Fig. 5B). Mutant slices show a 70% reduction in mitochondrial mass after normalizing to mito-Dendra2-positive cell bodies (Fig. 5C). In contrast, slices from Mfn1-null mice showed normal mitochondrial morphology and density (Supplementary Material, Fig. S3A and B).
Figure 5.

Mitochondrial fragmentation and depletion in slice cultures of Mfn2 mutants. Cre-mediated expression of mito-Dendra2 labels dopaminergic neurons. (A) Slice cultures of heterozygous controls and Mfn2 mutants. Slices were immunostained with TH (red). The first column shows a merged image of TH and mito-Dendra2 fluorescence (green), while the last column is an enlargement of the boxed zone. The asterisks highlight degenerating Mfn2-null neurons that have diminished or absent TH staining. (B) Mito-Dendra2 signal in neuronal projections. Marked depletion of mitochondria in both proximal and distal processes is evident in Mfn2 mutant slices. (C) Quantification of mitochondrial mass normalized to the number of dopaminergic neurons. For each sample, the total mito-Dendra2-positive area in a 5 mm × 5 mm region was measured and normalized to the number of Dendra2-positive neurons. Mitochondrial mass is reported as the percentage area of heterozygous control ± SEM. The Student's t-test was used to evaluate statistical significance (**P < 0.001; n = 5 for mutant slices; n = 7 for control). Scale bar is 10 μm for all images.
Decreased mitochondrial transport along nerve processes in Mfn2 mutants
To monitor mitochondrial transport along dopaminergic axons and dendrites, we performed live imaging of mito-Dendra2 in the slice cultures. For accurate monitoring of mitochondrial dynamics in the dense milieu of dopaminergic projections, we photo-converted mitochondria in a nerve process and tracked the movement of this labeled subpopulation (Fig. 6A, top). From the time-lapse movies, we generated kymograph representations that resolved the complex trajectories of the photo-converted mitochondria (Fig. 6A, bottom). With a vertical time axis, mobile mitochondria create diagonal tracks, whereas stationary mitochondria project as vertical streaks. Consistent with previous studies describing the mobility of mitochondria in neuronal processes (33–35), we found that a subpopulation of mitochondria is highly mobile in control dopaminergic neurons (Fig. 6A and Supplementary Material, Movie S2). Mfn1-null neurons similarly contained a subpopulation of highly mobile mitochondria (Supplementary Material, Fig. S3C and D). In contrast, mitochondrial transport is minimal in Mfn2-null neurons (Fig. 6B and Supplementary Material, Movie S3). In heterozygous controls, 50% of photo-conversion experiments resulted in at least one transport event, defined as directed movement for 5 μm. In Mfn2 mutants, only 14% of photo-conversion experiments showed a transport event (Fig. 6C). Additionally, mobile mitochondria in Mfn2 mutant dopaminergic neurons exhibit more intermittent movements and longer immobile periods. As a result, the average velocity for mitochondria in Mfn2-null slices is also slower relative to controls, 0.05 versus 0.15 μm/s (Fig. 6D).
Figure 6.

Decreased mitochondrial transport in Mfn2 mutant cultures. (A) A representative tracking experiment of an Mfn2 heterozygous control. A subset of mitochondria in the nerve process was photo-converted to red for time-lapse imaging. The images of the photo-converted signal were processed into a kymograph to visualize mitochondrial movement (binary image). Velocity measurements were calculated from the red tracks that overlay mitochondrial trajectories. (B) Representative tracking experiment in an Mfn2 mutant showing the loss of mitochondrial transport. (C) Quantification of mitochondrial transport in neuronal processes. For each photo-conversion experiment, a positive event was defined as the directed movement of more than 5 μm during the 15 min recording session. The graph shows the frequency of photo-conversion experiments that resulted in at least one positive event. For Mfn2 heterozygous slices, 150 experiments were scored; for Mfn2 mutant slices, 138 experiments were scored. The Student's t-test was used to calculate statistical significance (**P < 0.001). (D) A scatter plot of the average velocity of moving mitochondria. The red dots indicate the population averages ± SD. Statistical significance was calculated as in (C) (n = 138 mitochondria in heterozygous slices, n = 38 mitochondria in mutant slices). Scale bar is 5 μm for all images.
Rescue of Mfn2 mutants with L-DOPA treatment
The reduced level of TH-positive terminals in Mfn2 mutant mice implies a deficiency of dopamine in the striatum. We therefore tested whether the administration of L-3,4 dihydroxyphenylalanine (L-DOPA) by peritoneal injection could alleviate the motor defects of Mfn2 mutant mice. With 4–5- and 8–9-week-old mice, we found that L-DOPA administration causes a substantial increase in both the travel distance and the rearing frequency of Mfn2 mutant mice (Fig. 7). Collectively, these results suggest that dopamine deficiency, attributed to axonal loss, underlies the motor deficits in Mfn2 mutants.
Figure 7.
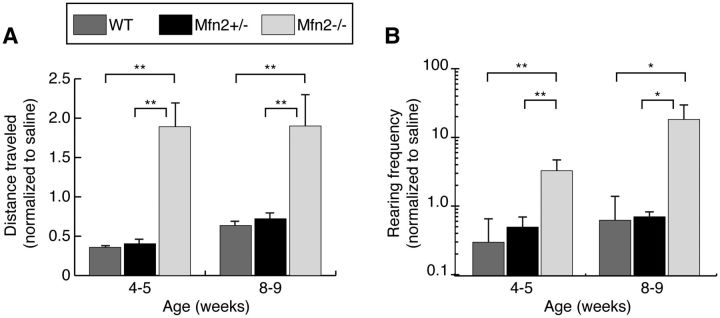
Increased motor activity after L-DOPA injection. Animals were analyzed by an open field test after a control saline injection to obtain baseline activity. They were tested a second time after an L-DOPA injection. Quantification of open field data for total distance traveled (A) and rearing frequency (B). For each animal, the activity after L-DOPA treatment was normalized to the activity after the saline injection. The Student's t-test was used to obtain P-values (*P < 0.05; **P < 0.001; n = 10–15 per genotype), and error bars represent the propagated error.
DISCUSSION
Several genes associated with familial PD—including PINK1, Parkin, DJ-1 and LRRK2—have been linked to mitochondrial dynamics. As a result, we sought to understand the role of mitochondrial dynamics in the nigrostriatal pathway. We deleted the mitofusins Mfn1 and Mfn2 from a subset of dopaminergic neurons, including those of the SNc. Mfn2 mutants exhibit severe locomotive defects, which are preceded by the loss of dopaminergic efferents to the striatum. Importantly, these mice show dysfunction in the striatum and motor deficits weeks earlier than the loss of nigral neurons. This sequence of pathological findings is consistent with retrograde degeneration, in which neuronal deficits initiate distally in the axon terminals and progress backward to the cell bodies. Interestingly, pathological studies of PD brains have suggested a similar ‘dying back’ mode of neurodegeneration based on the disproportionate loss of striatal dopamine relative to the neuronal loss in the SNc (36,37). A recent mouse model of PD with deletions in mitochondrial DNA (38) and administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine to rhesus monkeys (39) also produces this differential pattern of neuronal damage.
We found that dopaminergic neurons lacking Mfn2 have a prominent defect in mitochondrial content and transport in neuronal processes. This latter observation supports growing evidence that the mitofusins are important for mitochondrial movement (40,41). Mechanistically, this link may be related to the observation that Mfn2 can co-immunoprecipitate with exogenously expressed Miro and Milton, which are important components for kinesin-mediated transport of mitochondria along microtubules (41). In contrast to the severe defects found in Mfn2-null animals, we found that Mfn1-null mice show no apparent movement disorder, and correspondingly, no defects in the nigrostriatal circuit or in mitochondrial transport. At present, it is unknown whether this distinction is simply due to differential levels of these proteins in dopaminergic cells or to functional differences between Mfn1 and Mfn2 (42).
The identification of PD-related genes, such as α-synuclein, PINK1, Parkin, DJ-1 and LRRK2, has led to the development of numerous mouse models of PD. Although some of these models show modest decreases in striatal dopamine and associated motor impairments, they generally fail to recapitulate the progressive loss of dopaminergic neurons that is the pathological hallmark of PD (43,44). In contrast, disruption of mitochondrial function by the loss of mitochondrial transcription factor A (Tfam) results in dopamine cell death that is associated with reduced mitochondrial DNA content and severe respiratory chain deficiency (45,46). In our study, we find that dopaminergic neurons lacking the mitochondrial fusion gene Mfn2 exhibit fragmented mitochondria that fail to be transported within the axon. An important feature of our mouse model is the clear demonstration of progressive degeneration at the cellular level. Early deficits are regionally restricted to the nerve terminals, and it takes 2 months to progress to neuronal cell loss. The time span between distal defects and cell loss presents a window for investigating the cellular mechanisms leading to the degeneration of dopaminergic neurons.
MATERIALS AND METHODS
Generation of Mfn mutant mice
Conditional mouse lines of Mfn1 and Mfn2 have been described previously (47). The Slc6a3-Cre driver was obtained from the Jackson Laboratory (B6.SJL-Slc6a3tm1.1(cre)Bkmn/J). All experiments were approved by the Caltech Institute Animal Care and Use Committee.
Open field test
Animals were placed in a 50 cm × 50 cm white Plexiglass box and allowed an adaptation period of 30–60 min prior to being analyzed. Activity was recorded for two consecutive sessions, each lasting 15 min, by a ceiling-mounted video camera. The Ethovision software (Noldus, Leesburg, VA, USA) was used to measure the distance, velocity, rearing frequency and immobility of the mice.
In the L-DOPA experiments, animals were first injected intraperitoneally with saline and monitored by open field analysis to obtain baseline activity. Subsequently, animals were administered a cocktail of l-3,4-dihydroxyphenylalanine methyl ester (25 mg/kg, Sigma) and the DOPA decarboxylase inhibitor benserazide hydrochloride (5 mg/kg, Sigma). After 60 min, animals were followed by open field analysis.
Histological analysis
Animals were sacrificed after anesthesia with halothane. Brains were dissected and fixed overnight at 4°C in 10% neutral buffered formalin. The caudal portions of the brains were trimmed in an acrylic matrix (2 mm from the end) before specimens were mounted and sectioned with the Leica VT1200S vibratome. Brains were sliced into consecutive sections of 50 µm for the striatum or 35 µm for the midbrain. For counting, every fourth midbrain slice was processed for TH (1:1000, Chemicon) immunohistochemistry following the manufacturer's protocol (Vectastain elite ABC kit, Vector Labs). To enhance antigenicity, slides were boiled for 40 min in 10 mm sodium citrate buffered at pH 6. Sections were developed with 3,3′-diaminobenzidine and subsequently immersed in a 0.1% cresyl violet acetate solution for Nissl counterstain. Each slide contained a set of homozygous, heterozygous and wild-type samples to minimize staining variability between samples. Two reviewers, blinded to the genotypes, counted TH-immunoreactive and Nissl-positive cells at 100× magnification. For each animal, nine sections spanning the midbrain were counted. Total counts from the heterozygotes and homozygotes were normalized to the age-matched wild-type controls. Densitometry of the TH signal in the striatum has been described (39). Briefly, the Nikon Elements software was used for computer-assisted measurement of TH intensity in the striatal area. The same threshold was maintained across all samples on the slide. For each animal, three sections, spanning the rostral-caudal extent of the striatum, were measured, summed and normalized to wild-type measurements.
For fluorescent Nissl staining, sections were stained with Nissl conjugated Alexa 633 (1:500, Molecular Probes) for 1 h at room temperature. Sections were rinsed with several times with phosphate buffered saline prior to mounting with Cytoseal.
Imaging and microscopy analysis
Images were acquired on a Zeiss LSM 710 confocal microscope using EC-Plan-Neofluar 40×/1.3 oil or Plan-Apochromat 63×/1.4 oil objectives. Z-stack acquisitions oversampled twice the thickness of the optical slice, and Zen 2009 analysis software was used for maximum z-projections. To photo-convert Dendra2, a small region was irradiated with the 405 nm laser (4% laser power) for 60 iterations at a scan speed of 6.3–12.61 µs/pixel. For live imaging, slices were submerged in Tyrode's buffer (Sigma) supplemented with 25 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and 6.5 mg/ml of glucose and stabilized with a slice anchor (Warner Instruments). Slices were imaged on a stage-top heated platform maintained at 35°C. Four fields were imaged in each slice and time-lapse movies were acquired at 20 s intervals for 15 min. Custom macros were written for ImageJ software to produce kymographs and to measure velocity traces. In quantifying mitochondrial area, noise reduction in maximally z-projected images utilized the median and Liptschitz top hat filters. Subsequently, the dynamic thresholding plugin was applied to segment mitochondria, and the ‘Analyze particles’ algorithm in ImageJ provided the quantitation of mitochondrial signal. Manual counts of mito-Dendra2-positive cells in the z-stacks used the ‘Cell counter’ algorithm.
Organotypic slice cultures
Preparations of sagittal organotypic slices have been described (36). We made modifications to the angle of sectioning to improve the preservation of nigrostriatal projections (32). The rotating magnetic stage from the Leica VT1200S vibratome was tilted so that sections could be acquired between 10° and 15° from the midline. Pups were sacrificed at postnatal days 10–12. Typically, only two slices (one per hemisphere) of 330 μm thickness contained the nigrostriatal pathway. Slices acquired at 1–1.2 mm lateral from the midline were retained for culturing. Cultures were fed three times a week using the Stoppini media (37). Brain slices were equilibrated in culturing conditions for at least 2 weeks prior to experimentation. For immunofluorescence, membranes around the slices were trimmed and fixed in 4% paraformaldehyde–lysine–periodate overnight at 4°C. Slices were permeabilized with 1% Triton X-100 for 30 min and incubated with blocking buffer (2% goat serum, 1% bovine serum albumin and 0.1% Triton X-100) for 4–6 h at room temperature. Samples were incubated with anti-TH antibody overnight at 4°C, followed by secondary antibody (goat anti-rabbit IgG Alexa 568, Molecular Probes) for 2 h.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (GM062967 to D.C.C.), Howard Hughes Medical Instituteand the Thomas Hartman Foundation. Funding to pay the Open Access publication charges for this article was provided by the Howard Hughes Medical Institute.
ACKNOWLEDGEMENTS
We thank Andrew D. Steele and Paul H. Patterson for use of equipment and software, Sally A. Kim for advice on organotypic slice cultures and Hsiuchen Chen for advice on mouse crosses. We are grateful to the Chan lab for discussion and comments on the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. doi:10.1016/S0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 2.Parker W.D., Jr, Swerdlow R.H. Mitochondrial dysfunction in idiopathic Parkinson disease. Am. J. Hum. Genet. 1998;62:758–762. doi: 10.1086/301812. doi:10.1086/301812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langston J.W., Ballard P., Tetrud J.W., Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. doi:10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 4.Cannon J.R., Greenamyre J.T. Neurotoxic in vivo models of Parkinson's disease recent advances. Prog. Brain Res. 2010;184:17–33. doi: 10.1016/S0079-6123(10)84002-6. doi:10.1016/S0079-6123(10)84002-6. [DOI] [PubMed] [Google Scholar]
- 5.Irrcher I., Aleyasin H., Seifert E.L., Hewitt S.J., Chhabra S., Phillips M., Lutz A.K., Rousseaux M.W., Bevilacqua L., Jahani-Asl A., et al. Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010;19:3734–3746. doi: 10.1093/hmg/ddq288. doi:10.1093/hmg/ddq288. [DOI] [PubMed] [Google Scholar]
- 6.Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. doi:10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 7.Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. doi:10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. doi:10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 9.Krebiehl G., Ruckerbauer S., Burbulla L.F., Kieper N., Maurer B., Waak J., Wolburg H., Gizatullina Z., Gellerich F.N., Woitalla D., et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One. 2010;5:e9367. doi: 10.1371/journal.pone.0009367. doi:10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X., Yan M.H., Fujioka H., Liu J., Wilson-Delfosse A., Chen S.G., Perry G., Casadesus G., Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Detmer S.A., Chan D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. doi:10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 12.Chen H., Chan D.C. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum. Mol. Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. doi:10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J.W., Yang L., Beal M.F., Vogel H., Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl Acad. Sci. USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. doi:10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. doi:10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y., Ouyang Y., Yang L., Beal M.F., McQuibban A., Vogel H., Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl Acad. Sci. USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. doi:10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng H., Dodson M.W., Huang H., Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl Acad. Sci. USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. doi:10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka A., Cleland M.M., Xu S., Narendra D.P., Suen D.F., Karbowski M., Youle R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. doi:10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G., et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. doi:10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Petrie T.G., Liu Y., Liu J., Fujioka H., Zhu X. Parkinson's disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012;121:830–839. doi: 10.1111/j.1471-4159.2012.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas K.J., McCoy M.K., Blackinton J., Beilina A., van der Brug M., Sandebring A., Miller D., Maric D., Cedazo-Minguez A., Cookson M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011;20:40–50. doi: 10.1093/hmg/ddq430. doi:10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bäckman C.M., Malik N., Zhang Y., Shan L., Grinberg A., Hoffer B.J., Westphal H., Tomac A.C. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. doi:10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q.Y., Palmiter R.D. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. doi:10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]
- 23.Palmiter R.D. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann. N.Y. Acad. Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. doi:10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kish S.J., Shannak K., Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. New Eng. J. Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. doi:10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- 25.Stoessl A.J. Neuroimaging in Parkinson's disease. Neurotherapeutics. 2011;8:72–81. doi: 10.1007/s13311-010-0007-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirsch E., Graybiel A.M., Agid Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. doi:10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 27.Damier P., Hirsch E.C., Agid Y., Graybiel A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999;122(Pt 8):1437–1448. doi: 10.1093/brain/122.8.1437. doi:10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 28.Pham A.H., McCaffery J.M., Chan D.C. Mouse lines with photo-activatable mitochondria (PhAM) to study mitochondrial dynamics. Genesis. 2012 doi: 10.1002/dvg.22050. doi:10.1002/dvg.22050. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gahwiler B.H., Capogna M., Debanne D., McKinney R.A., Thompson S.M. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. doi:10.1016/S0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- 30.Cho S., Wood A., Bowlby M.R. Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr. Neuropharmacol. 2007;5:19–33. doi: 10.2174/157015907780077105. doi:10.2174/157015907780077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ammari R., Lopez C., Fiorentino H., Gonon F., Hammond C. A mouse juvenile or adult slice with preserved functional nigro-striatal dopaminergic neurons. Neuroscience. 2009;159:3–6. doi: 10.1016/j.neuroscience.2008.10.051. doi:10.1016/j.neuroscience.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 32.Beurrier C., Ben-Ari Y., Hammond C. Preservation of the direct and indirect pathways in an in vitro preparation of the mouse basal ganglia. Neuroscience. 2006;140:77–86. doi: 10.1016/j.neuroscience.2006.02.029. doi:10.1016/j.neuroscience.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Misgeld T., Kerschensteiner M., Bareyre F.M., Burgess R.W., Lichtman J.W. Imaging axonal transport of mitochondria in vivo. Nat. Meth. 2007;4:559–561. doi: 10.1038/nmeth1055. doi:10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- 34.Kang J.S., Tian J.H., Pan P.Y., Zald P., Li C., Deng C., Sheng Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. doi:10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheng Z.H., Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. doi:10.1038/nrg3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hornykiewicz O. Biochemical aspects of Parkinson's disease. Neurology. 1998;51:S2–S9. doi: 10.1212/wnl.51.2_suppl_2.s2. doi:10.1212/WNL.51.2_Suppl_2.S2. [DOI] [PubMed] [Google Scholar]
- 37.Cheng H.C., Ulane C.M., Burke R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. doi:10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickrell A.M., Pinto M., Hida A., Moraes C.T. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci. 2011;31:17649–17658. doi: 10.1523/JNEUROSCI.4871-11.2011. doi:10.1016/0306-4522(91)90080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pifl C., Schingnitz G., Hornykiewicz O. Effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on the regional distribution of brain monoamines in the rhesus monkey. Neuroscience. 1991;44:591–605. doi: 10.1016/0306-4522(91)90080-8. doi:10.1083/jcb.200211046. [DOI] [PubMed] [Google Scholar]
- 40.Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. doi:10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misko A., Jiang S., Wegorzewska I., Milbrandt J., Baloh R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. doi:10.1038/nature07534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. doi:10.1016/j.neuron.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 43.Dawson T.M., Ko H.S., Dawson V.L. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. doi:10.1016/S1474-4422(11)70227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesselet M.F., Richter F. Modelling of Parkinson's disease in mice. Lancet Neurol. 2011;10:1108–1118. doi: 10.1016/S1474-4422(11)70227-7. doi:10.1073/pnas.0605208103. [DOI] [PubMed] [Google Scholar]
- 45.Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A., et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl Acad. Sci. USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. doi:10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterky F.H., Lee S., Wibom R., Olson L., Larsson N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl Acad. Sci. USA. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. doi:10.1016/j.cell.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]