

Abstract
The objective of the study is to quantify the causal effect of β-cell function on type 2 diabetes by minimizing residual confounding and reverse causation. We employed a Mendelian randomization (MR) approach using TCF7L2 variant rs7903146 as an instrument for lifelong levels of β-cell function. We first conducted two sets of meta-analyses to quantify the association of the TCF7L2 variant with the risk of type 2 diabetes among 55 436 cases and 106 020 controls from 66 studies by calculating pooled odds ratio (OR) and to quantify the associations with multiple direct or indirect measures of β-cell function among 35 052 non-diabetic individuals from 31 studies by calculating pooled mean difference. We further applied the method of MR to obtain the causal estimates for the effect of β-cell function on type 2 diabetes risk based on findings from the meta-analyses. The OR [95% confidence interval (CI)] was 0.87 (0.81–0.93) for each five unit increment in homeostasis model assessment of insulin secretion (HOMA-%B) (P = 3.0 × 10−5). In addition, for measures based on intravenous glucose tolerance test, ORs (95% CI) associated with type 2 diabetes risk were 0.24 (0.08–0.74) (P = 0.01) and 0.14 (0.04–0.48) (P = 0.002) for per 1 standard deviation increment in insulin sensitivity index and disposition index, respectively. Findings from the present study lend support to a causal role of pancreatic β-cell function itself in the etiology of type 2 diabetes.
INTRODUCTION
Type 2 diabetes is characterized by insulin resistance in peripheral tissues coupled with a progressive impairment of pancreatic β-cell function. The recent demonstration that most type 2 diabetes-associated variants impact β-cell function provided support of the theory that a background of β-cell insufficiency is more likely to predate and set the stage for type 2 diabetes (1–4). However, the magnitude of the potential causal effect of β-cell function itself on the development of type 2 diabetes remains uncertain (5,6). The majority of previous epidemiological studies on the role of β-cell function in the development of type 2 diabetes were conducted among adults whose β-cell function may have been affected by impaired glucose tolerance from pre-diabetes or undiagnosed diabetes. Therefore, reverse causation is a plausible explanation for previous findings. As a result, findings from the conventional population studies cannot tease apart the impact of β-cell function from that of peripheral insulin/glucose action on the pathogenesis of type 2 diabetes. Moreover, residual confounding from poorly measured or unmeasured factors is always a major concern on the associations observed in observational studies (7).
The ‘Mendelian randomization’ (MR) approach (8–10), using genetic variants that are causally and specifically related to pancreatic β-cell function and type 2 diabetes as an instrument variable, offers the potential to assess and quantify causation using observational data. Studies exploiting the MR approach build upon the rationale that genotypes formed at conception could be indicators of lifetime exposure that are generally independent of confounders such as lifestyle and environmental factors and also free of reverse causation due to temporal preservation using pre-clinical or undiagnosed disease status (8–10). Genetic association studies have identified and confirmed a number of genetic loci significantly associated with type 2 diabetes, of which the TCF7L2 gene has shown the strongest and most widely replicated association (11–13). Importantly, function data from in vitro and in vivo studies demonstrated that TCF7L2 specifically affects pancreatic β-cell function through regulation of insulin production and secretion pathways (14–16), incretin action (15,17,18) and β-cell proliferation and apoptosis (15,19). The risk allele T at rs7903146 variant has recently been shown to be in an open chromatin state in human pancreatic islets accompanied by a change in enhancer activity, indicating that it may be a genuine causal variant in regulating TCF7L2 expression and splice pattern in pancreatic islets (20,21).
In addition, it is worth mentioning that direct and reliable measures of β-cell function require dynamic data obtained from some laborious, costly and invasive ‘gold standard’ methods (22). As a result, these measures are not practically feasible in large population studies and their predictive values for type 2 diabetes remain important yet understudied. MR approach based on the totality of evidence provides a unique opportunity to assess an unbiased association between these ‘gold standard’ measures of β-cell function and type 2 diabetes.
In the present study, based on an MR approach using one TCF7L2 gene variant (i.e. rs7903146) as an instrumental variable, we aimed to provide an alternative quantitative assessment of the impact of various surrogate measures of pancreatic β-cell function on the risk of type 2 diabetes by minimizing residual confounding and reverse causation. To maximize statistical power to obtain robust MR estimates, meta-analysis approach was employed to synthesize the evidence from the available genotype-β-cell function phenotype and genotype-type 2 diabetes studies, respectively.
RESULTS
Characteristics of association studies
Of the 66 studies involving a total of 55 436 cases and 106 020 controls, rs7903146 was the most commonly studied TCF7L2 SNP (Supplementary Material, Table S1). Overall, there were significant differences in the minor allele frequency of rs7903146 in TCF7L2 across different populations; the minor allele T in rs7903146 was quite common (0.16–0.48) in all Caucasians, Africans and Hispanics except for Pima Indians but less frequent (0.02–0.04) in all East Asian populations, including Japanese, Chinese and Korean.
TCF7L2 genotype and type 2 diabetes risk
The T allele of the rs7903146 variant appeared to confer a greater risk for type 2 diabetes (Table 1). The results showed no evidence of significant between-study heterogeneity for the allelic associations. Overall, the random-effect pooled OR of the allelic association for the T allele at rs7903146 was 1.41 (95% CI, 1.37–1.46; P = 2.49 × 10−121), while there was no strong statistical evidence to suggest between-study heterogeneity for all included studies [P-value for Q statistics = 0.11, H = 1.1(1.0–1.3) and I2 = 20 (0–45)].
Table 1.
A meta-analysis involving 66 independent studies for rs7903146 for reliably assessing the overall genetic association between the TCF7L2 common polymorphism and type 2 diabetes risk
| TCF7L2 Genotypes | na | Meta-analysis |
Test of heterogeneity |
|||
|---|---|---|---|---|---|---|
| Random-effects OR (95%CI) | P-value | Q-test | H | I2 (%) | ||
| rs7903146 | ||||||
| T/T versus C/C | 53 | 1.75 (1.61–1.91) | 3.85 × 10−40 | <0.0001 | 1.4 (1.2–1.6) | 48 (29–63) |
| C/T versus C/C | 53 | 1.37 (1.31–1.43) | 7.50 × 10−43 | 0.0003 | 1.4 (1.1–1.6) | 45 (24–60) |
| Per T allele (versus C allele) | 49 | 1.41 (1.37–1.46) | 2.49 × 10−121 | 0.11 | 1.1 (1.0–1.3) | 20 (0–45) |
aTotal number of independent studies with the data available (a total of studies included: n = 66 with 55 436 cases and 106 020 controls).
Despite significant race/ethnicity-specific frequencies of the TCF7L2 rs7903146 SNP, we found no evidence of any divergent results for increased diabetes risk associated with the TCF7L2 rs7903146 SNP across diverse ethnic groups (Table 2 and Supplementary Material, Fig. S1). Similarly, the pooled OR estimate did not differ substantially by the adjustment of body mass index (BMI) (yes versus no), statistical models (crude versus adjusted) or study designs (prospective study versus retrospective study) (Table 2).
Table 2.
Major subgroup analyses for the genetic association between rs7903146 TCF7L2 polymorphism and type 2 diabetes
| Subgroups | na | Cases/Controls | rs7903146 |
||
|---|---|---|---|---|---|
| Random effects OR (95% CI) | P-value | Q-test | |||
| Ethnicity | |||||
| European whites | 20 | 15 765/42 408 | 1.41 (1.37–1.45) | 2.44 × 10−98 | 0.74 |
| American whites | 4 | 2033/4320 | 1.51 (1.35–1.69) | 1.19 × 10−12 | 0.27 |
| American African | 3 | 1939/1835 | 1.34 (1.13–1.59) | 7.65 × 10−4 | 0.08 |
| African | 3 | 1432/1165 | 1.41 (1.22–1.64) | 4.01 × 10−6 | 0.23 |
| East Asians | 8 | 16 412/17 055 | 1.47 (1.33–1.63) | 1.22 × 10−13 | 0.22 |
| South Asians | 4 | 3654/2174 | 1.34 (1.24–1.46) | 4.61 × 10−12 | 0.73 |
| BMI adjustment | |||||
| No | 37 | 35 910/55 085 | 1.42 (1.38–1.46) | 5.01 × 10−133 | 0.35 |
| Yes | 16 | 12 641/26 810 | 1.36 (1.26–1.47) | 6.58 × 10−16 | 0.07 |
| Statistical model | |||||
| Crude | 31 | 33 419/48 534 | 1.43 (1.39–1.48) | 4.65 × 10−120 | 0.35 |
| Adjustedb | 19 | 14 585/29 501 | 1.36 (1.28–1.45) | 9.09 × 10−24 | 0.13 |
| Study design | |||||
| Prospective | 5 | 2879/15 910 | 1.37 (1.28–1.47) | 1.20 × 10−17 | 0.89 |
| Retrospective | 44 | 42 084/58 447 | 1.42 (1.37–1.47) | 5.15 × 10−100 | 0.06 |
aTotal number of studies with the data available (A total of studies included: n = 47 with 41 400 cases and 70 794 controls).
bAdjustment for at least two diabetes risk factors.
In addition, neither a funnel plot analysis nor formal tests (Begg's and Egger's tests) showed evidence for the presence of substantial publication bias for the TCF7L2-type 2 diabetes association in different inheritance models (data not shown).
TCF7L2 and quantitative measures of β-cell function among non-diabetic individuals
Overall, carriers of the T allele at rs7903146 (TT and CT) consistently had lower levels of β-cell function measures than non-carriers (Table 3). Compared with those homozygous for the C allele (CC), carriers of the T allele at rs7903146 (TT and C/T) had significantly lower levels of fasting insulin and homeostasis model assessment of insulin secretion (HOMA-%B) and higher fasting glucose and 2 h post-load glucose levels (Table 3). For insulin function parameters assessed by IVGTT including AIR, SI and DI levels (expressed as standardized effect sizes due to substantial heterogeneity in the protocols implemented and units reported from different studies), the TT genotype was associated with their decreased levels when compared with the homozygotes of the C allele at rs7903146. The results did not explicitly indicate any specific heritable models for the associations of TCF7L2 variants with insulin function measures, probably due in part to low statistical power.
Table 3.
Pooled estimates for effects of TCF7L2 polymorphisms on phenotype traits of insulin homeostasis in healthy non-diabetic controls
| Traits | rs7903146: T/T versus C/C |
rs7903146: C/T versus C/C |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n (studies) | n (TT/CC) | Pooled mean difference (95% CI) | P-valuea | Q-test for heterogeneityb | n (studies) | n (CT/CC) | Pooled mean difference (95% CI) | P-valuea | Q-test for heterogeneityb | |
| Fasting measuresc | ||||||||||
| Fasting insulin, pmol/l | 25 | 3421/22 473 | −6.12 (−8.76, −3.48) | 6.0 × 10−6 | <0.001 | 26 | 17 236/26 117 | −2.94 (−4.39, −1.49) | 8.2 × 10−5 | 0.009 |
| Fasting glucose, mmol/l | 31 | 4039/31 013 | 0.084 (0.052, 0.12) | 2.5 × 10−7 | <0.001 | 30 | 18 209/27 446 | 0.02 (0.001, 0.04) | 0.04 | <0.001 |
| HOMA-IR | 17 | 2179/16 717 | −0.06 (−0.14, 0.02) | 0.17 | 0.33 | 18 | 11 415/17 603 | −0.05 (−0.12, 0.01) | 0.12 | 0.03 |
| HOMA-%B | 9 | 463/2479 | −20.11 (−29.4, −10.9) | 2.0 × 10−5 | 0.01 | 10 | 1975/3365 | −21.0 (−39.7, −2.35) | 0.03 | <0.001 |
| OGTT measuresc | ||||||||||
| 2 h post-load glucose, mmol/l | 17 | 1095/10 075 | 0.28 (0.15, 0.42) | 6.0 × 10−5 | <0.001 | 18 | 9189/15 063 | 0.10 (0.05, 0.16) | 0.0003 | 0.02 |
| 2 h post-load insulin, pmol/l | 8 | 587/7398 | −19.3 (−51.1, 12.5) | 0.10 | 0.24 | 9 | 3906/7693 | −15.6 (−29.2, −2.02) | 0.02 | 0.10 |
| IVGTT measuresd | ||||||||||
| AIR | 4 | 160/1045 | −0.12 (−0.29, 0.05) | 0.15 | 0.39 | 5 | 825/1268 | −0.09 (−0.18, 0.01) | 0.05 | 0.42 |
| SI | 4 | 160/1045 | −0.38 (−0.68, −0.09) | 0.01 | 0.03 | 4 | 787/1045 | 0.024 (−0.07, 0.12) | 0.61 | 0.35 |
| DI (SI*AIR) | 4 | 160/1045 | −0.28 (−0.45, −0.11) | 0.001 | 0.22 | 4 | 787/1045 | −0.05 (−0.14, 0.04) | 0.29 | 0.30 |
OGTT, 75 g oral glucose tolerance test; HOMA-IR and HOMA-%B, homeostasis model assessment of insulin resistance and insulin secretion; IVGTT, intravenous glucose tolerance test; AIR, a measure of the first-phase insulin secretion; SI, insulin resistance index; DI, disposition index.
aP-values for pooled mean differences.
bP-values for heterogeneity between studies.
cData are the weighted mean differences for those parameters labeled with units.
dData are the standardized effect sizes used.
Causal association of β-cell function with risk of type 2 diabetes by MR
Table 4 shows the predicted OR of type 2 diabetes associated with per unit increase in direct or indirect measures of β-cell function using rs7903146 variant as an instrumental variable for β-cell function. Increased fasting glucose and post-load glucose levels were strongly associated with increased risk of type 2 diabetes, whereas fasting insulin and 2 h post-load insulin levels were modestly associated with lower diabetes risk. Moreover, HOMA-%B was significantly and inversely associated with risk of type 2 diabetes; the estimated causal OR associated with per 5% increase in HOMA-%B was 0.87 (95% CI, 0.81–0.93). More importantly, MR analysis results indicated that the gold standard measures of β-cell function (i.e. AIR, SI and DI based on IVGTT), were also significantly and inversely associated with diabetes risk, although the estimates may be relatively less precise due to small number and low statistical power. The causal ORs were significant per 1-SD increment in SI (0.24; 95%, 0.08–0.74) and DI (0.14; 95% CI, 0.04–0.48).
Table 4.
MR analyses for the causal association between insulin secretion measures and risk of developing type 2 diabetes
| Variables | rs7903146: T/T versus C/C |
rs7903146: T/C versus C/C |
||
|---|---|---|---|---|
| Pooled OR (95% CI) | P-valuea | Pooled OR (95% CI) | P-valuea | |
| Fasting measures | ||||
| Fasting glucose, 0.1 mmol/l | 2.09 (1.61, 2.71) | 3.7 × 10−8 | 5.63 (2.59, 12.24) | 1.0 × 10−5 |
| Fasting insulin, pmol/l | 0.92 (0.90, 0.95) | 9.6 × 10−9 | 0.92 (0.90, 0.95) | 9.6 × 10−9 |
| HOMA-%B, per 5 units | 0.87 (0.81, 0.93) | 3.0 × 10−5 | 0.96 (0.95, 0.98) | 2.7 × 10−9 |
| OGTT | ||||
| 2 h post-load glucose, 0.1 mmol/l | 1.48 (1.21, 1.80) | 1.0 × 10−4 | 1.54 (1.22, 1.93) | 2.0 × 10−4 |
| 2 h post-load insulin, pmol/l | 0.98 (0.95, 1.00) | 0.05 | 0.98 (0.97, 1.00) | 0.013 |
| IVGTT | ||||
| AIR, per SD | 0.01 (0.00003, 5.23) | 0.15 | 0.03 (0.0005, 1.39) | 0.07 |
| SI, per SD | 0.24 (0.08, 0.74) | 0.01 | —b | —b |
| DI (SI*AIR), per SD | 0.14 (0.04, 0.48) | 0.002 | —b | —b |
OGTT, 75 g oral glucose tolerance test; HOMA-%B, homeostasis model assessment of insulin resistance and insulin secretion; IVGTT, intravenous glucose tolerance test; AIR, a measure of the first-phase insulin secretion; SI, insulin resistance index; DI, disposition index.
aP-values for pooled estimates.
bPower was limited to yield stable estimates from available data.
DISCUSSION
Findings from the present study provide some evidence for a causal role of pancreatic β-cell function in the development of type 2 diabetes. First, our systematic analysis of up to 55 436 diabetes cases and 106 020 controls demonstrated robust associations of the genetic variants of TCF7L2 gene with both measures of β-cell function and the risk of type 2 diabetes. Moreover, our MR causal estimates by leveraging available genetic association evidence showed that lifelong averaged levels of various β-cell function measures were significantly and inversely associated with risk of type 2 diabetes. Such significant MR estimates indicate a causal role of β-cell function alone in the etiology of type 2 diabetes free of reverse causation and confounding from many factors after birth, especially during adulthood.
Our MR approach using TCF7L2 genotypes as an instrument represents an important and unique quantitative approach to assess the causal impact of genetically determined β-cell function on risk of type 2 diabetes. In agreement with recent evidence suggesting the role of TCF7L2 polymorphisms in the insulin function pathway (14–16,21), our meta-analysis showed that the TCF7L2 genotypes were robustly associated with measures of β-cell function, supporting the hypothesis that the genetic effects on type 2 diabetes are likely mediated through β-cell function as the intermediate phenotypic product. The strength of the instrument is determined by the absolute magnitude of its association with the intermediate phenotype. Although we found evidence of an association between TCF7L2 variants and surrogates of insulin resistance such as fasting glucose, fasting insulin and 2 h glucose responses, such associations were expected because these biomarkers are not very specific and may to some extent reflect the concomitant β-cell function. Functional and genetic data supported that rs7903146 is the causal variant at this locus (17,18,21,23). Although the precise molecular mechanisms remain to be determined, there is no clear evidence that the TCF7L2 alleles associated with diabetes risk have pleiotropic effects on metabolic or physiological systems other than pancreatic β-cell function (15,21).
The robustness of associations between TCF7L2 variants and type 2 diabetes in genetically heterogeneous populations based on our systematic analyses of up to 55 436 diabetes cases and 106 020 controls further strengthens the precision and certainty of our inferences. In previous studies, the heterogeneous nature of observational studies and limited sample sizes make the results from individual studies less reliable. Furthermore, ethnic/racial differences in the frequencies of these known TCF7L2 variants are pronounced, with the allele frequency of the minor T allele at rs7903146 being low in East Asians but common in other populations. Such genetic heterogeneity of TCF7L2 in populations of various ethnic origins not only affects the power of individual studies but also indicates different patterns and magnitudes of linkage disequilibrium (LD) between variants in this gene among different ethnic groups (i.e. population-specific LD). Despite substantial ethnic difference in the frequency of TCF7L2 risk allele, we observed a robust association between TCF7L2 genotypes and type 2 diabetes risk in a strong genotype–dose manner with similar magnitude across subgroups. In addition, the assumption of no LD with causal variants or population stratification required for genetic instruments seemed not to be violated in our MR approach. Our meta-analysis synthesizing the final result from each ethnically homogenous population minimized the potential of population stratification.
Meeting all three basic assumptions (8,9), in the present study, the MR analysis based on the evidence from the genotype–intermediate phenotype and the genotype–disease relations provided strong evidence supporting and quantifying the causal role of β-cell function in the etiology of type 2 diabetes. It has been hypothesized that impairments in early β-cell development can lead to fetal malnutrition and predisposes individuals to the development of type 2 diabetes later in life (24–26). Beyond obesity and insulin resistance, β-cell function has been increasingly recognized as a central cause of type 2 diabetes. Because of measurement error, however, a single measure of β-cell function is not sufficient in capturing lifelong β-cell function levels. Dynamic and longitudinal measures of β-cell function are usually not practically feasible in large population studies. In addition, clinical assessment of insulin function is usually conducted during adulthood and has a hyperbolic relation with insulin action (22). As such, studies based on these assessments cannot separate the impact of β-cell function from that of insulin/glucose action in peripheral tissues. As genetic variants in the TCF7L2 gene exert their effects across a lifetime, our estimates of genotype–phenotype relation may reflect the vast majority of variation of β-cell function measures attributable to TCF7L2 polymorphisms. Our MR estimates of the impact of β-cell function on type 2 diabetes risk may therefore largely represent a lifelong effect —a measure of average lifelong exposure, which incorporates various mitigating, compensating and attenuating patterns and lifestyle-related changes over a lifetime. In other words, our MR analysis provided precise estimates of the causal link from genetically determined lifetime levels of β-cell function to adult-onset type 2 diabetes, which were free of reverse causation or confounding by insulin resistance or impaired glucose metabolisms or other diabetes risk factors in adulthood.
Like all studies adopting MR approach, some limitations inherent in MR analysis merit consideration (8–10). First, hypothetically, MR analysis is analogous to a randomized controlled trial of a drug treatment or intervention—where there is always a degree of non-adherence to the treatment drug, non-compliance to protocol, drop-out, drop-in and necessary compensatory/rescue drugs used in placebo/control groups that mimic canalization and incomplete penetrance. While MR analysis does require several important assumptions that need to be carefully evaluated prior to making causal inference, some of these assumptions cannot be empirically verified and requires subject-matter knowledge (e.g. absence of pleiotropy/heterogeneity of effects and canalization/the buffering effects of germline variants by developmental compensation via other biologic pathways) (8–10). For example, it remains difficult to evaluate the assumptions of no canalization and no gene–environment interactions, which would require further experimental and intervention work. In the present study, however, the robustness and the strength of associations of multiple measures of β-cell function with type 2 diabetes risk using both conventional multivariable method and the MR analysis provide further some assurance of precision and certainty of our inferences. Secondly, β-cell function measures derived from IVGTT are usually regarded as the gold standard measures. Due to the limited number of available studies with IVGTT measures and the heterogeneity in IVGTT test protocols, confidence intervals (CI) of odds ratio (OR) or relative risk (RR) estimates of the genotype–phenotype association and the MR estimates of causal effect based on the IVGTT measures were somewhat wider and may be relatively less precise.
In conclusion, findings from the present study based on MR approach and systematic analyses of available population data lend support to the causal role of pancreatic β-cell function in the pathogenesis of type 2 diabetes. These findings not only further highlight the compelling need for the development and application of validated β-cell function measures for population studies, but also further support using preservation of pancreatic β-cell function as a target for the prevention or treatment of type 2 diabetes in general population.
MATERIALS AND METHODS
Study selection
Relevant studies were identified by searching MEDLINE, PubMed, EMBASE and NCBI OMIM databases for all published genetic association studies up to February 1, 2010, using the search terms ‘TCF7L2’, ‘pancreatic beta-cell function’, ‘insulin secretion’, ‘insulin resistance’, ‘glucose metabolism’, ‘diabetes mellitus’, ‘type 2 diabetes’ and ‘diabetes’. Additional studies were retrieved through a hand-search of references from original reports. Two common variants (i.e. rs7903146 and rs12255372) have been widely studied for TCF7L2 gene. Overall, more studies are available for the rs7903146 variant such that statistical power from available data was much higher for rs7903146 than rs12255372 in terms of both genotype–phenotype and genotype–diabetes associations. Moreover, the rs7903146 variant is generally in strong LD with the rs12255372 variant except in several African groups. In addition, it has been demonstrated that the variant affects β-cell function by altering different genomic regulatory functions in pancreatic islets (27). In the present study, our main analysis focused on the rs7903146 variant only. All studies on this topic were considered eligible if they had data on the associations of the rs7903146 variant with the risk of type 2 diabetes and/or diabetes-related quantitative traits. Only English language articles were identified and included.
Two authors (Y.S. and C.L.) independently reviewed each published paper and extracted relevant information examining the associations of TCF7L2 gene variants with the risk of type 2 diabetes. Two separate authors (E.Y. and L.C.) independently reviewed each published paper and extracted relevant information examining the associations of TCF7L2 variants with intermediate metabolic phenotypes. When a study reported results on different subpopulations, we considered each subpopulation as a separate study in the meta-analysis. When necessary, missing information (such as genotype distributions, standard deviations or 95% CI) was obtained by direct contact with the original authors.
Of the 647 retrieved articles that were evaluated and abstracted, 575 studies were excluded (Fig. 1). These included non-human studies, review/editorials, case only studies, studies with abstract only, meta-analysis, redundant reports of the same population and reports that did not provide relevant data. The final data set of our meta-analyses included 72 published articles that provided 66 independent studies for rs7903146. Data on TCF7L2 genotypes and diabetes-related metabolic traits were also available depending on the phenotype (e.g. for rs7903146 genotypes, the sample sizes were 160 and 1045 for the IVGTT measures comparing TT with CC and 18209 and 27446 for the comparison of CT and CC on fasting glucose).
Figure 1.

Flow chart of study selection for meta-analysis of the association of TCF7L2 polymorphism (rs7903146) and risk of type 2 diabetes and multiple β-cell function measures.
Data extraction
For each of the articles reporting the TCF7L2-type 2 diabetes association, we included studies that provided estimates of RRs or ORs of type 2 diabetes or data that permitted estimation of these parameters. We developed a database that included the first author's name, year of publication, study population, sample size, mean age for cases and controls, mean age at diagnosis, mean BMI comparisons of allele frequencies between cases and controls, genotype distribution in cases and controls and the estimates of RRs or ORs of type 2 diabetes associated with the genotypes of TCF7L2. For studies reporting the relation of TCF7L2 with diabetes-related phenotypes, we extracted the number of subjects, the means and standard deviations of quantitative metabolic phenotypes that may indirectly or directly reflect β-cell function, including fasting plasma glucose and insulin levels, HOMA-B, plasma levels of glucose and insulin at 2 h after a 75 g oral glucose tolerance test (OGTT) and acute insulin response (AIR), insulin sensitivity index (SI) and disposition index (DI) derived from intravenous glucose tolerance test (IVGTT). Information on homeostasis model assessment index of insulin resistance (HOMA-IR) was also extracted to evaluate the specificity of the association with insulin sensitivity when compared with the β-cell function parameters discussed earlier. When results were not presented in the published papers, attempts were made to obtain additional data by directly contacting 24 corresponding authors.
Statistical analyses
Association of TCF7L2 variants with intermediate phenotypes and type 2 diabetes
To maximize statistical power, a meta-analysis was performed to provide robust effect estimates of the associations of TCF7L2 variant with measures of β-cell function and risk of type 2 diabetes, which are required for the MR algorithm. We calculated the ORs and 95% CI for the associations between TCF7L2 genotypes and risk of type 2 diabetes in three different inheritance models. When the adjusted ORs were not available, we used the crude ORs reported or calculated from the raw data. We performed pre-defined stratified meta-analyses to explore potential heterogeneity (effect modification) of the TCF7L2 genotype-diabetes association by ethnicity group (European whites, American whites, American African, African, East Asian and South Asian), BMI adjustment (yes or no), statistical model (unadjusted crude model or adjusted model) and study design (prospective versus retrospective).
For all continuous measures relevant to β-cell function, we converted all data to the same units (for better clinical interpretation of results than using standardized units) and estimated mean values from medians and log-transformed data when only those values were available. We then calculated the mean difference for the comparisons of the minor allele carriers (TT or CT for rs7903146) versus homozygotes of the common allele (CC for rs7903146). To incorporate both within-study and between-study variability, we used DerSimonian and Laird's (27) random-effects model with inverse-variance (standard error) weighting of individual trial results. Each mean difference was weighted according to the inverse of its pooled variance. Then we calculated the weighted mean difference and 95% CI to pool the results for each continuous outcome using random-effects models. For those measures based on IVGTT on different scales according to different protocols, we decided to calculate standardized mean differences, which is the difference in means divided by a pooled standard deviation.
For a meta-analysis of k studies, the Cochran's Q statistic was used to examine the extent of heterogeneity (28–30). A P-value < 0.1 is suggestive of the presence of study heterogeneity (31–33). Because the Cochran's Q statistic has low statistical power27, the I2 statistic was also calculated (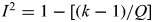 , which describes the percentage of total variation due to the study heterogeneity across the k studies) (32). Thus, percentages of around 25% (I2 = 25), 50% (I2 = 50) and 75% (I2 = 75) would mean low, medium and high heterogeneity, respectively (32). The H statistic was also used as a complement to the I2 statistic in assessing study heterogeneity; an H < 1.2 indicates little heterogeneity and an H > 1.5 raises caution regarding notable heterogeneity (32).
, which describes the percentage of total variation due to the study heterogeneity across the k studies) (32). Thus, percentages of around 25% (I2 = 25), 50% (I2 = 50) and 75% (I2 = 75) would mean low, medium and high heterogeneity, respectively (32). The H statistic was also used as a complement to the I2 statistic in assessing study heterogeneity; an H < 1.2 indicates little heterogeneity and an H > 1.5 raises caution regarding notable heterogeneity (32).
We assessed publication bias using Begg's modified funnel plots, in which the OR was plotted on a logarithmic scale (log[OR]) against its standard error from each study. Publication bias was also assessed by two formal tests: Begg's adjusted rank correlation test and Egger's regression asymmetry test (34,35). The former was used to examine if there is significant correlation between the effect estimates and their variances and the latter uses an inverse-variance weighted regression of the effect sizes on their precision (the inverse of standard error) to test whether the intercept deviates significantly from zero. Meta-analyses including tests of publication bias were performed using the STATA statistical software (Version 8.0, STATA Corp, College Station, TX, USA). Statistical significance was defined as two-tailed α < 0.05.
Causal inference using MR analysis
The MR approach incorporating information on both the genotype–intermediate phenotype association and genotype–disease association into one analytical framework may allow for an unbiased estimate of the intermediate phenotype–disease association (Figs 2 and 3). In the MR paradigm, an instrumental variable has to satisfy the following three criteria: (i) the genotype should be robustly associated with intermediate phenotype (e.g. TCF7L2 gene – β-cell function measures); (ii) the genotype should not be associated with confounding factors that bias the association between intermediate phenotype and disease outcome and (iii) absence of pleiotropy, as the genotype should exert its effect on the clinical outcome only through the specific intermediate phenotype (e.g. effect of TCF7L2 gene on type 2 diabetes risk via β-cell function alone). Based on available evidence (1–3,10,14–16,23,36), TCF7L2 genotypes seemed to meet these assumptions very well. Thus, the MR coefficient estimates using TCF7L2 genotypes as instruments would provide the causal association between β-cell function and type 2 diabetes risk free of bias due to reverse causation and residual confounding.
Figure 2.

Directed acyclic graph (DAG) encoding the causal relationship between TCF7L2 genetic instrumental variables G, intermediate β-cell function phenotype X and type 2 diabetes outcome Y that satisfies three key assumptions.
Figure 3.

Scheme for calculation of an unconfounded estimate of the effect of an unit increase in each of β-cell function measures on the risk of type 2 diabetes, based on the concept of MR.
β-cell function is evaluated using multiple indexes on a continuous scale, and the TCF7L2 genotype is considered as binary (either TT versus CC or TC versus CC). Based on the estimates of the log odds ratio (log OR) of type 2 diabetes given TCF7L2 genotype and the mean difference in the phenotype levels between the two TCF7L2 genotypes, we estimated the MR causal effect of the phenotype levels on type 2 diabetes using two models (A and B) described by Thompson et al. (also summarized in one Supplementary Material) (37). The two models take into account the correlations between genotype–phenotype and genotype–disease associations from the same studies under different assumptions. Model B assumes independence between the heterogeneities on the genotype–phenotype association and phenotype–disease association (i.e. the between-studies correlation of heterogeneities equals to zero) while Model A assumes only no within-study correlation, i.e. the study specific OR estimates are independent of the phenotype level differences. We conducted further analyses to justify the use of Model B by first testing the independence assumption and then by comparing Model B and Model A using the observed −2log likelihood principal. Our analyses found no evidence to reject the independence assumption. Furthermore, Model B is either substantially better than or about equivalent to Model A by the −2log likelihood criterion. In addition, under the assumption that the diabetes prevalence is relatively low in the general population, the OR obtained from our MR approach of case–control data can be interpreted as a causal OR (38). All MR estimations were conducted using the R software.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
C.Z., E.Y., C.L. and E.F.S. are supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health. T.J.V. is supported by grants from the National Institutes of Health (ES017876 and HD060696).
REFERENCES
- 1.Dupuis J., Langenberg C., Prokopenko I., Saxena R., Soranzo N., Jackson A.U., Wheeler E., Glazer N.L., Bouatia-Naji N., Gloyn A.L., et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010;42:105–116. doi: 10.1038/ng.520. doi:10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingelsson E., Langenberg C., Hivert M.F., Prokopenko I., Lyssenko V., Dupuis J., Magi R., Sharp S., Jackson A.U., Assimes T.L., et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes. 2010;59:1266–1275. doi: 10.2337/db09-1568. doi:10.2337/db09-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saxena R., Hivert M.F., Langenberg C., Tanaka T., Pankow J.S., Vollenweider P., Lyssenko V., Bouatia-Naji N., Dupuis J., Jackson A.U., et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010;42:142–148. doi: 10.1038/ng.521. doi:10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P., Zeggini E., Huth C., Aulchenko Y.S., Thorleifsson G., et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010;42:579–589. doi: 10.1038/ng.609. doi:10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahlqvist E., Ahluwalia T.S., Groop L. Genetics of type 2 diabetes. Clin. Chem. 2011;57:241–254. doi: 10.1373/clinchem.2010.157016. doi:10.1373/clinchem.2010.157016. [DOI] [PubMed] [Google Scholar]
- 6.Bonnefond A., Froguel P., Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol. Med. 2010;16:407–416. doi: 10.1016/j.molmed.2010.06.004. doi:10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Greenland S., Robins J.M. Identifiability, exchangeability, and epidemiological confounding. Int. J. Epidemiol. 1986;15:413–419. doi: 10.1093/ije/15.3.413. doi:10.1093/ije/15.3.413. [DOI] [PubMed] [Google Scholar]
- 8.Lawlor D.A., Harbord R.M., Sterne J.A., Timpson N., Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. doi:10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 9.Smith G.D., Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int. J. Epidemiol. 2004;33:30–42. doi: 10.1093/ije/dyh132. doi:10.1093/ije/dyh132. [DOI] [PubMed] [Google Scholar]
- 10.Smith G.D., Timpson N., Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann. Med. 2008;40:524–541. doi: 10.1080/07853890802010709. doi:10.1080/07853890802010709. [DOI] [PubMed] [Google Scholar]
- 11.Grant S.F., Thorleifsson G., Reynisdottir I., Benediktsson R., Manolescu A., Sainz J., Helgason A., Stefansson H., Emilsson V., Helgadottir A., et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006;38:320–323. doi: 10.1038/ng1732. doi:10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 12.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S., et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. doi:10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 13.Zeggini E., Scott L.J., Saxena R., Voight B.F., Marchini J.L., Hu T., de Bakker P.I., Abecasis G.R., Almgren P., Andersen G., et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. doi:10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith U. TCF7L2 and type 2 diabetes–we WNT to know. Diabetologia. 2007;50:5–7. doi: 10.1007/s00125-006-0521-z. doi:10.1007/s00125-006-0521-z. [DOI] [PubMed] [Google Scholar]
- 15.van de Bunt M., Gloyn A.L. From genetic association to molecular mechanism. Curr. Diab. Rep. 2010;10:452–466. doi: 10.1007/s11892-010-0150-2. doi:10.1007/s11892-010-0150-2. [DOI] [PubMed] [Google Scholar]
- 16.Weedon M.N. The importance of TCF7L2. Diabet. Med. 2007;24:1062–1066. doi: 10.1111/j.1464-5491.2007.02258.x. doi:10.1111/j.1464-5491.2007.02258.x. [DOI] [PubMed] [Google Scholar]
- 17.Shu L., Matveyenko A.V., Kerr-Conte J., Cho J.H., McIntosh C.H., Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009;18:2388–2399. doi: 10.1093/hmg/ddp178. doi:10.1093/hmg/ddp178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villareal D.T., Robertson H., Bell G.I., Patterson B.W., Tran H., Wice B., Polonsky K.S. TCF7L2 variant rs7903146 affects the risk of type 2 diabetes by modulating incretin action. Diabetes. 2010;59:479–485. doi: 10.2337/db09-1169. doi:10.2337/db09-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu L., Sauter N.S., Schulthess F.T., Matveyenko A.V., Oberholzer J., Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57:645–653. doi: 10.2337/db07-0847. doi:10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 20.Gaulton K.J., Nammo T., Pasquali L., Simon J.M., Giresi P.G., Fogarty M.P., Panhuis T.M., Mieczkowski P., Secchi A., Bosco D., et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010;42:255–259. doi: 10.1038/ng.530. doi:10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansson O., Zhou Y., Renstrom E., Osmark P. Molecular function of TCF7L2: Consequences of TCF7L2 splicing for molecular function and risk for type 2 diabetes. Curr. Diab. Rep. 2010;10:444–451. doi: 10.1007/s11892-010-0149-8. doi:10.1007/s11892-010-0149-8. [DOI] [PubMed] [Google Scholar]
- 22.Bergman R.N., Ader M., Huecking K., Van Citters G. Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes. 2002;51(Suppl. 1):S212–S220. doi: 10.2337/diabetes.51.2007.s212. doi:10.2337/diabetes.51.2007.S212. [DOI] [PubMed] [Google Scholar]
- 23.Lyssenko V., Lupi R., Marchetti P., Del Guerra S., Orho-Melander M., Almgren P., Sjogren M., Ling C., Eriksson K.F., Lethagen A.L., et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest. 2007;117:2155–2163. doi: 10.1172/JCI30706. doi:10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahn S.E., Zraika S., Utzschneider K.M., Hull R.L. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52:1003–1012. doi: 10.1007/s00125-009-1321-z. doi:10.1007/s00125-009-1321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maedler K. Beta cells in type 2 diabetes - a crucial contribution to pathogenesis. Diabetes Obes. Metab. 2008;10:408–420. doi: 10.1111/j.1463-1326.2007.00718.x. doi:10.1111/j.1463-1326.2007.00718.x. [DOI] [PubMed] [Google Scholar]
- 26.Meier J.J. Linking the genetics of type 2 diabetes with low birth weight: a role for prenatal islet maldevelopment? Diabetes. 2009;58:1255–1256. doi: 10.2337/db09-0225. doi:10.2337/db09-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DerSimonian R., Laird N.M. Meta-analysis in clinical trials. Control Clin. Trials. 1986;7:177–188. doi: 10.1016/0197-2456(86)90046-2. doi:10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 28.Cochran W.G. The combination of estimates from different experiments. Biometrics. 1954;10:101–129. doi:10.2307/3001666. [Google Scholar]
- 29.Laird N.M., Mosteller F. Some statistical methods for combining experimental results. Int. J. Technol. Assess. Health Care. 1990;6:5–30. doi: 10.1017/s0266462300008916. doi:10.1017/S0266462300008916. [DOI] [PubMed] [Google Scholar]
- 30.Whitehead A., Whitehead J. A general parametric approach to the meta-analysis of randomized clinical trials. Stat. Med. 1991;10:1665–1677. doi: 10.1002/sim.4780101105. doi:10.1002/sim.4780101105. [DOI] [PubMed] [Google Scholar]
- 31.Hedges L.V., Pigott T.D. The power of statistical tests in meta-analysis. Psychol. Methods. 2001;6:203–217. doi:10.1037/1082-989X.6.3.203. [PubMed] [Google Scholar]
- 32.Higgins J., Thompson S., Deeks J., Altman D. Statistical heterogeneity in systematic reviews of clinical trials: a critical appraisal of guidelines and practice. J. Health Serv. Res. Policy. 2002;7:51–61. doi: 10.1258/1355819021927674. doi:10.1258/1355819021927674. [DOI] [PubMed] [Google Scholar]
- 33.Sutton A.J., Abrams K.R., Jones D.R., Sheldon T.A., Song F. Systematic reviews of trials and other studies. Health Technol. Assess. 1998;2:1–276. [PubMed] [Google Scholar]
- 34.Begg C.B., Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50:1088–1101. doi:10.2307/2533446. [PubMed] [Google Scholar]
- 35.Egger M., Davey Smith G., Schneider M., Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315:629–634. doi: 10.1136/bmj.315.7109.629. doi:10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Y., Lin Y., Zhang Y., Yang J., Liu H., Zhang B. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: a large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC Med. Genet. 2009;10:15. doi: 10.1186/1471-2350-10-15. doi:10.1186/1471-2350-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson J.R., Minelli C., Abrams K.R., Tobin M.D., Riley R.D. Meta-analysis of genetic studies using Mendelian randomization–a multivariate approach. Stat. Med. 2005;24:2241–2254. doi: 10.1002/sim.2100. doi:10.1002/sim.2100. [DOI] [PubMed] [Google Scholar]
- 38.Bowden J., Vansteelandt S. Mendelian randomization analysis of case-control data using structural mean models. Stat. Med. 2011;30:678–694. doi: 10.1002/sim.4138. doi:10.1002/sim.4138. [DOI] [PubMed] [Google Scholar]