

Abstract
Huntington's disease (HD) is a progressive neurodegenerative disorder caused by trinucleotide repeat (TNR) expansions. We show here that somatic TNR expansions are significantly reduced in several organs of R6/1 mice lacking exon 2 of Nei-like 1 (Neil1) (R6/1/Neil1−/−), when compared with R6/1/Neil1+/+ mice. Somatic TNR expansion is measured by two different methods, namely mean repeat change and instability index. Reduced somatic expansions are more pronounced in male R6/1/Neil1−/− mice, although expansions are also significantly reduced in brain regions of female R6/1/Neil1−/− mice. In addition, we show that the lack of functional Neil1 significantly reduces germline expansion in R6/1 male mice. In vitro, purified human NEIL1 protein binds and excises 5-hydroxycytosine in duplex DNA more efficiently than in hairpin substrates. NEIL1 excision of cytosine-derived oxidative lesions could therefore be involved in initiating the process of TNR expansion, although other DNA modifications might also contribute. Altogether, these results imply that Neil1 contributes to germline and somatic HD CAG repeat expansion.
INTRODUCTION
Huntington's disease (HD) is a progressive neurodegenerative disorder caused by a CAG:CTG repeat expansion in exon 1 of the gene that encodes the polyglutamine-containing protein Huntingtin (1). There is an inverse relationship between trinucleotide repeat (TNR) length and age at disease onset (2). Additionally, after accounting for constitutive repeat length, somatic instability in the human cortex has recently been shown to be a significant predictor of HD onset with larger repeat length gains associated with earlier disease onset (3). TNR length expansion is correlated with HD neuropathology and probably precedes the onset of symptoms (4).
The mechanisms underlying selective neurodegeneration in nearly all disorders remain poorly understood. We recently have shown that the striatum and cortex display a dramatic and periodic expansion that is mechanistically different from the slow expansion observed in most other somatic tissues of the HD transgenic R6/1 mouse model (5). The base excision repair (BER) enzyme 7,8-dihydroxy-8-oxoguanine-DNA glycosylase (Ogg1), which repairs oxidized guanine bases, has been shown to initiate age-dependent TNR expansion in R6/1 mice (6), and deletion of the mismatch repair proteins MutS homolog 2 (Msh2) and Msh3 has been shown to abolish somatic expansion in several HD transgenic mouse models (7–9). Moreover, the stoichiometry of several key BER proteins correlates well with increased somatic TNR instability in the striatum compared with the cerebellum in HD transgenic mice (10).
Nei-like 1 (NEIL1) is one of four mammalian DNA glycosylases involved in removing oxidized lesions as part of BER. The multifunctional DNA glycosylases OGG1 and NTH1 process the abasic (AP) site product following base removal via a single lytic β-elimination reaction, while NEIL1 and NEIL2 nick the DNA strand 3′ to the AP site through an associated β and δ AP lyase activity, leaving phosphates at both ends of the resulting single strand break. The β-elimination product of OGG1 and NTH1 is further processed by apurinic/apyrimidinic endonuclease (APE1), whereas polynucleotide kinase removes the 3′-phosphate to generate a 3′-OH group for ligation after incision by the NEILs [reviewed by Hazra et al. (11)]. If the AP site is reduced or oxidized, flap endonuclease 1 is required for removing a 2–6 nucleotide single-stranded (ss) DNA-flap generated by 5′ AP site incision and strand-displacement synthesis during long patch-BER (12).
Recombinant NEIL1 has been shown to remove pyrimidine-derived lesions like thymine glycol (Tg), 5-hydroxycytosine (5-OHC) and 5-hydroxyuracil (5-OHU) in double- and ssDNA (13–15). 5-OHU is efficiently excised with similar activities in bubble-, duplex- and ssDNA by NEIL1 (16). NEIL1 also excises 7,8-dihydroxy-8-oxoguanine (8-oxoG) in duplex- and ssDNA, although less efficiently than oxidized pyrimidines (13–16). Mouse Neil1 also excises ionizing radiation-induced ring-opened purine-derived lesions 2,6-diamino-4-hydroxy-5-formamidopyrimidine (faPyG) and 4,6-diamono-5-formamidopyrimidine (faPyA), while thymine-derived lesions are excised at a much lower rate (17). NEIL1 and NEIL2 also excise oxidation products of 8-oxoG, such as guanidinhydantoin and spiroiminohydantoin lesions (18). Human NEIL1 excises these hydantoin lesions much more efficiently (>100-fold faster) than the previously reported pyrimidine-derived substrates (19).
Since Ogg1 has been shown to initiate somatic TNR expansion (6), we wanted to examine the role of Neil1 as a possible modifier of repeat stability. Neil1 is ubiquitously expressed (13–15), and the expression increases with age in the brain (20). Neil1 and Ogg1 have structural distinctions, but some overlapping substrate specificities including 8-oxoG and faPyG, although Neil1 prefers cytosine-derived lesions. Furthermore, Neil1 repairs oxidative lesions in ssDNA and bubble structures, which might be important DNA structures in the context of TNR instability (16). Neil1 also repairs lesions in close proximity of DNA breaks (21). These latter activities are absent or significantly weaker in Ogg1 (16,21,22).
RESULTS
Ogg1 is a DNA glycosylase that repairs 8-oxoG and faPy lesions in DNA and has been shown to initiate CAG repeat expansion in R6/1 mice (6). Neil1 repairs a much broader spectra of oxidative lesions than Ogg1, and thus, we investigated whether Neil1 could be involved in TNR instability by using R6/1 mice lacking exon 2 of Neil1 (R6/1/Neil1−/−). The generation and full characterization of the Neil1−/− mice will be described elsewhere (Rolseth et al., manuscript in preparation).
Upon establishment of the Neil1-deficient HD mouse model, we examined whether germline expansion was affected, since CAG repeats tend to expand when transmitted from a male parent to offspring in both mice and humans (2,23–25). Germline expansion was defined as the increase in repeat length between generations in ear samples taken at 3 weeks of age. We analysed germline expansion from R6/1 male parents with or without functional Neil1 and the corresponding litters (Fig. 1A). It is evident that the repeat tract expands from male R6/1/Neil1+/+ parents to offspring, whereas in male R6/1/Neil1−/− parents, the repeat tract is more stable, with a tendency towards contraction when transferred to offspring (Fig. 1B). The germline transmission in R6/1/Neil1−/− mice was from three successive generations. These results indicate that Neil1 contributes to germline TNR expansion in male R6/1 mice.
Figure 1.
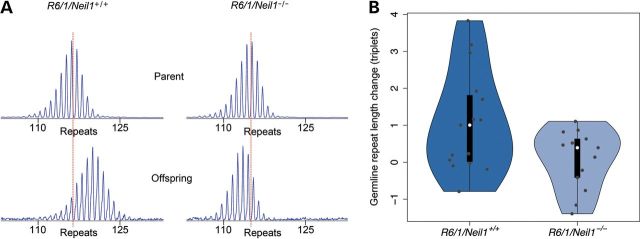
Germline TNR instability was calculated in R6/1/Neil1+/+ and R6/1/Neil1−/− mice by measuring the number of CAG repeats present in ear samples taken at 3 weeks of age from male parent mice and offspring. (A) GeneMapper plots showing germline TNR expansion in offspring from a male R6/1/Neil1+/+ mouse and TNR contraction in offspring from a male R6/1/Neil1−/− mouse. (B) A vioplot showing germline expansion from two male R6/1/Neil1+/+ (n = 15 offspring) and three male R6/1/Neil1−/− mice (n = 13 offspring). Germline expansion for individual R6/1/Neil1+/+ and R6/1/Neil1−/− mice is shown as violin plots to illustrate the distributions present. Individual points are overlaid in grey for completeness. The data show a tendency towards germline contraction in R6/1/Neil1−/− mice.
Next, we investigated somatic expansion in several tissues at 21 weeks of age, including ear (EA), spleen (SP), kidney (KI), liver (LI), olfactory bulb (OB), cortex (CO), striatum (ST) and cerebellum (CE). These samples cover a representative spectrum of tissues with varying degrees of TNR instability (5,23). Somatic expansion was calculated by estimating the change in mean repeat length in the 21-week-old sample in comparison to the 3-week-old ear sample from the same mouse (see Materials and Methods). Notably, the fragment lengths containing the repeat tracts were visibly shorter in several organs of R6/1/Neil1−/− mice compared with R6/1/Neil1+/+ mice (Fig. 2A). Moreover, when both genders were combined, the change in mean repeat length was significantly shorter in all organs tested, except for the cortex, of R6/1/Neil1−/− animals compared with the R6/1/Neil1+/+ counterparts (Fig. 2B). Breaking the data down by gender, the change in mean fragment length was significantly shorter in several organs of male R6/1/Neil1−/− mice, such as the ear, kidney, olfactory bulb, striatum and cerebellum, while the change in TNR length in the brain regions (olfactory bulb, striatum and cerebellum) was significantly shorter in R6/1/Neil1−/− female mice, when compared with their respective R6/1/Neil1+/+ male and female groups. These results suggest that Neil1 modifies TNR instability in both genders, although the effects seem to be somewhat more pronounced in male mice.
Figure 2.

Somatic TNR expansion in R6/1/Neil1+/+ and R6/1/Neil1−/− mice determined by calculation of mean repeat change in each mouse from ear at 3 weeks to tissues at 21 weeks of age. (A) GeneMapper plots showing the distribution of CAG repeat fragment lengths present in each organ from one representative R6/1/Neil1+/+ and one representative R6/1/Neil1−/− mouse. Black arrows and white arrows represent 111 and 138 repeats, respectively. (B) Mean repeat change measured in several organs of male and female R6/1/Neil1+/+ and R6/1/Neil1−/− mice from 3 to 21 weeks of age. N = 19 R6/1/Neil1+/+ (12 males, 7 females) and 11 R6/1/Neil1−/− (6 males, 5 females) mice. Error bars represent ±SEM. *P > 0.05; **P > 0.01; ***P < 0.001; one-tailed unpaired t-test. EA, ear; EA3, ear at 3 weeks; EA21, ear at 21 weeks; SP, spleen; KI, kidney; LI, liver; OB, olfactory bulb; CO, cortex; CE, cerebellum.
Recently, a novel method to define tissue-specific TNR instability has been published (26). This method calculates an instability index from GeneMapper traces by including peak heights within a threshold. The peak heights are normalized to one and multiplied with the change from the highest peak in the tail. We used the same approach with some minor adaptations (see Materials and Methods) to confirm our conclusions described above. In Figure 3A, the normalized and weighted peak heights for the striatum of male R6/1/Neil1+/+ and R6/1/Neil1−/− mice are presented. It is therefore visualized that male R6/1/Neil1−/− mice can be divided into two populations that display different distributions in the striatum (Fig. 3A). Population 1 shows a large reduction in expansion compared with the male R6/1/Neil1+/+ mice, while population 2 shows a small reduction in expansion (evident in inset of Fig. 3A). The instability index is shown in Figure 3B for several organs of male and female mice. It is apparent that the instability index is significantly reduced in the striatum of both male and female R6/1/Neil1−/− mice compared with the respective R6/1/Neil1+/+ mice. In addition, the instability index was significantly reduced in the ear, spleen, cortex and cerebellum of male R6/1/Neil1−/− mice. These results show a consistent expansion-reducing effect of Neil1 deficiency in nearly all tissue types, with the most pronounced effect in the brain tissues of male mice.
Figure 3.

The instability index in R6/1/Neil1+/+ and R6/1/Neil1−/− mice. (A) The normalized and weighted peak heights in the striatum of R6/1/Neil1+/+ and R6/1/Neil1−/− male mice. The two sub-populations of R6/1/Neil1−/− mice are easily identified, with one population showing very strong retardation of expansion rate, while the other population displays a reduction in the expansion rate which is shown inset for clarity. (B) The instability index divided by gender, genotype and organ, with the difference between the means for each genotype shown on the right. N = 19 R6/1/Neil1+/+ (12 males, 7 females) and 11 R6/1/Neil1−/− (6 males, 5 females) mice. EA, ear; SP, spleen; KI, kidney; LI, liver; OB, olfactory bulb; CO, cortex; CE, cerebellum. •P > 0.1; *P > 0.05; one-tailed unpaired t-test. Error bars represent ±SD. (C) Neil1 mRNA expression in WT and HD R6/1 mice. The bars represent the average of the calculated and normalized mRNA from two cDNA synthesis reactions for each mouse. N = 7 WT (4 males, 3 females) and 8 HD (4 males, 4 females) R6/1 mice. Error bars represent ±SEM. *P > 0.05; **P > 0.01; *** P < 0.001; two-tailed unpaired t-test.
In order to obtain further insights into how Neil1 could influence TNR instability, we analysed the mRNA expression of Neil1 by quantitative PCR (qPCR) in wild-type (WT) and HD R6/1 mice. As shown in Figure 3C, the expression of Neil1 was moderately down-regulated in brain regions of HD mice, particularly in the cerebellum, relative to the WT samples. To explore for any gender differences, Neil1 mRNA expression was determined in male and female R6/1 WT and HD mice, but no statistically significant differences were observed between genders (Fig. 3C).
Based on these data, we decided to test the incision activity of human NEIL1 on different 5-OHC or 8-oxoG containing oligonucleotide substrates in vitro. In a subset of these substrates, the base lesion was located at different positions within the loop of a hairpin, and in the other substrates the base damage was located within either a double-stranded or ssDNA molecule (Fig. 4A). NEIL1 exhibited the highest activity on the single-stranded and duplex 5-OHC-containing control substrates (Fig. 4B). NEIL1 base excision and strand cleavage activity for each of the lesion-containing hairpin substrates were ∼2.5–6.4-fold lower than the ssOHC oligonucleotide. We point out that we observed incision activity on the CAG hairpin substrates only at the higher concentration of NEIL1 (100 ng; data reported in Fig. 4C), whereas for the cognate substrates, ssOHC and 34OHC-34G, we saw incision product even at the lower NEIL1 concentration (10 ng; Fig. 4B). As would be expected, NEIL1 appears to process the duplex DNA substrates via a combined β, δ-elimination reaction, whereas OGG1 processes the abasic site product via β-elimination only (Fig. 4B). We note that NEIL1 excised 8-oxoG only in the duplex substrate, albeit with poor efficiency, but did not significantly remove this base lesion when placed in the hairpin loop at either 10 or 100 ng of enzyme (data not shown).
Figure 4.

NEIL1 incision activity and binding to oligonucleotide substrates containing a 5-OHC lesion. (A) Design of hairpin and control oligonucleotides. Hairpin oligonucleotides contain a base lesion in the 3-nucleotide (CAG1) or 4-nucleotide (CAG2) loop. The hairpin structured oligonucleotides contained BamHI, SacII and EcoRI restriction sites on the linear double-stranded portion of the hairpin to verify hairpin formation (10). (B) Purified NEIL1 had reduced incision activity on the hairpin substrates compared with the duplex control oligonucleotides and the single-stranded 34OHC substrate. (C) Incision and binding activities of NEIL1. Left column: hairpin, single-stranded (ss) and 5-hydroxyC (OHC)-containing substrates used. Centre column: % incision = incised product/(incised product + substrate). Right column: % binding = protein-DNA complex/(complex + unbound substrate). Data shown are the average and standard deviation of three independent experimental values derived from assays performed at 100 ng NEIL1. See Materials and Methods for further details.
Subsequent electrophoretic mobility shift assays (EMSAs) revealed that NEIL1 bound all oligonucleotide substrates, with the possible exception of ssOHC (Fig. 4C); the low complex formation seen with ssOHC likely stems from the inability of NEIL1 to bind the efficiently produced incision product. As seen with the enzymatic assays above, the duplex 5-OHC-containing DNA was the preferred substrate for stable NEIL1 complex formation. The binding of the hairpin substrates was in general in good agreement with the incision efficiency (summarized in Fig. 4C).
DISCUSSION
In humans, the HD-associated TNR has been shown to be unstable in more than 80% of meiotic transmissions, presenting both expansions and contractions with the largest expansions occurring during paternal transmission (2). The HD CAG repeat in transgenic mice exhibits an expansion bias during paternal transmission and a contraction bias during maternal transmission (23–25). In R6/1/Csb−/− mice, a 5-fold increase in intergenerational expansions was seen compared with the R6/1 control counterparts (27). Conversely, the presence or absence of Ogg1 did not significantly affect the level of intergenerational instability in R6/1 mice (27). We find that Neil1 depletion results in fewer expansions and increased contractions during paternal transmissions, when compared with control mice. Thus, our data are more analogous to the absence of Msh2, which abolished paternal HD TNR expansions and resulted in an increased occurrence of contractions (9,28). We have recently shown that treatment with the antioxidant anthocyanin inhibited mean TNR expansion in the testis of R6/1 mice (Møllersen et al., submitted for publication), suggesting that oxidative damage might promote germline expansion. This observation indicates that reduced germline expansion in male R6/1/Neil1−/− mice could be due to unprocessed oxidative insults on DNA.
In HD patients, all tissues have been shown to display some repeat mosaicism, with the greatest change being detected in the brain and sperm (29). Regions within the human brain showing the most obvious neuropathology, such as the striatum and the cortex, also display the greatest mosaicism, whereas the cerebellum, an apparently unaffected brain region, exhibits the lowest degree of CAG instability (29,30). HD transgenic mice similarly exhibit somatic TNR expansion in several organs, including the cortex, olfactory bulb, striatum, thalamus, kidney and liver (23,30,31). We have recently found that tissues believed to be stable in R6/1 mice exhibit slow expansion, although this expansion was mechanistically different from the dramatic and periodic expansion present in the cortex and striatum (5).
By two different methods, we have shown that the lack of functional Neil1 results in significantly reduced somatic TNR expansion in several organs of R6/1 mice. When female and male groups were combined, the mean somatic TNR expansion was reduced in the ear, spleen, kidney, liver, olfactory bulb, striatum and cerebellum of R6/1/Neil1−/− mice. While there is some discrepancy between the two methods for calculation of somatic expansion, both show the same tendency: expansion is significantly reduced in specific non-brain and brain regions of R6/1/Neil1−/− male mice, and is significantly reduced only in certain brain regions of female R6/1/Neil1−/− mice.
Remarkably, half of the R6/1/Neil1−/− male mice showed an extreme expansion inhibition (Fig. 3A), and therefore, the overall effect on repeat stability was more pronounced in male mice. Only one R6/1/Neil1−/− female mouse had this extreme TNR inhibition in all tested organs. However, the total number of female mice studied was lower, so the reduced presence of R6/1/Neil1−/− female mice displaying an extreme reduction in TNR expansion could be a coincidence. The reason for the two populations that display different TNR expansions within the R6/1/Neil1−/− genotype is not known, but could be due to variable penetrance. The obesity phenotype of Neil1−/− mice also lacks 100% penetrance (32).
Ogg1 has been shown to initiate somatic CAG repeat expansion in R6/1 mice, presumably by commencing an error-prone BER response (6). In particular, expansion was prevented in roughly 70% of the R6/1/Ogg1−/− animals. In the remaining animals, tissue-specific, age-dependent expansion was not observed in either the brain or liver of age- and gender-matched R6/1 littermates (6). It therefore appears that there are two populations of both R6/1/Neil1−/− and R6/1/Ogg1−/− mice that exhibit either a significant effect or no effect on somatic repeat expansion. Notably, we have performed a statistical analysis on mean TNR expansion between R6/1/Neil1−/− mice and R6/1/Neil1+/+ mice of both genders in which we have excluded all mice with extreme expansion inhibition, and we still found statistically significant differences between genotypes in the striatum, olfactory bulb and cerebellum (data not shown).
Repeat expansion is not altered in R6/1/Aag−/− or R6/1/Nth1−/− mice (6). Alkyladenine glycosylase, Aag, excises a variety of alkylated bases (33), whereas the Nth1 glycosylase processes a variety of oxidized pyrimidine derivatives and thus repairs many of the same lesions as Neil1. The result with the R6/1/Aag−/− model suggests that the base modifications repaired by this DNA glycosylase are not relevant to TNR instability. The difference observed between the Neil1 and Nth1 mice, in terms of somatic repeat expansion, suggests that Neil1 excises lesions not recognized by Nth1, or lesions in bubble or ssDNA.
We tested whether the expression levels of Neil1 in WT and HD R6/1 mice could contribute to TNR expansion and found that Neil1 was moderately down-regulated in several brain regions of R6/1 HD mice, most notably in the cerebellum. The expression level of Ogg1 has also been shown to be lower in the cerebellum, but unchanged in the striatum, of R6/1 HD mice compared with WT, although the biochemical activity of Ogg1 was higher in the striatum relative to the cerebellum in both R6/1 WT and HD mice (10). Since Neil1 was only moderately down-regulated, it is not likely that the expression level of Neil1 dictates TNR expansion. The expression level of Neil1 was also similar in male and female R6/1 mice, and therefore the expression of Neil1 cannot explain the TNR expansion differences between genders.
We found that the incision activity of NEIL1 on 5-OHC and 8-oxoG lesions in duplex DNA was significantly stronger than the activity on hairpin substrates, a pattern similar to what was previously observed for OGG1 (10,34). We speculate that the lower activity of NEIL1 for base lesions in hairpins may lead to the accumulation of damage within CAG repeat sequences. Since our knockout mouse studies indicate a role for NEIL1 in modulating repeat expansion, we propose a model similar to what has been outlined for OGG1, whereby the accumulation of base damage within CAG tracts can ultimately drive an error-prone BER response, which drives expansion of the TNR and is influenced by numerous factors, including protein stoichiometry and damage position (6,10,35). Although, as shown previously by others (13–16), our results indicate that NEIL1 efficiently processes ssDNA substrates, a role for incision of lesions in ssDNA in TNR expansion is presently unclear, although could potentially be linked to transcription.
The activity of NEIL1 on 8-oxoG substrates measured by us and others was weak. Thus, the strong inhibition of TNR expansion in R6/1/Neil1−/− mice is probably due to processing of pyrimidine-derived lesions. NEIL1 has also been shown to have strong activity towards hydantoin lesions, which are further oxidation products of 8-oxoG (19). Such lesions might well be relevant for CAG stability. Additionally, Neil−/− mice seem to accumulate faPyA and faPyG in the liver, kidney and brain, although the increase in faPyG in the brain was modest (36). FaPy lesions are also substrates for Neil1 and Ogg1 and one could hypothesize that repair of faPys contribute to TNR expansion in R6/1 mice. To summarize, our results indicate that Neil1 excises lesions in duplex DNA, and that lack of this activity suppresses TNR expansion in R6/1/Neil1−/− mice.
Since both Neil1 and Ogg1 influence TNR expansion in R6/1 mice, it is reasonable to assume that antioxidants, which might reduce oxidative base damage formation, could inhibit the expansion process. Coenzyme Q10, which is an antioxidant and cofactor in the electron transport chain, reduced the 8-oxoG levels in the brain and urine of R6/2 mice (37). We have recently showed that treatment with anthocyanin antioxidants reduced TNR expansion in brain and male gonads of R6/1 mice (Møllersen et al., submitted for publication). However, the overall effects of these antioxidants on somatic TNR expansion were modest in comparison to the contribution of Neil1 in the R6/1/Neil−/− mice.
In conclusion, Neil1 is an important modifier of both germline and somatic TNR expansion in R6/1 mice. In future studies, it would be interesting to examine whether HD/Neil−/−/Ogg1−/− double knockout mice would have a combined effect on somatic repeat expansion and whether TNR expansion is correlated to behavioural performance.
MATERIALS AND METHODS
Animals
B6CBA-Tg(HDexon1)61pb/J mice of the R6/1 line with exon 1 of the human HD gene (38) were crossed for two to three generations with C57BL/6J mice with or without deletion of exon 2 in mouse Neil1 containing the active site (Rolseth et al., manuscript in preparation) and the offspring were analysed for TNR instability. The R6/1/Neil1−/− mice were observed regularly. They appeared as R6/1 mice and developed Huntingtin aggregates in the brain at 21 weeks of age (data not shown). The mice were housed in transparent polycarbonate cages with controlled temperature and humidity, and fed Rat and Mouse No. 3 maintenance diet (Special Diet Services) and drinking water ad libitum. All experimental procedures were approved by the section for comparative medicine at Oslo University Hospital and the Norwegian animal research authority, and complied with national laws, institutional regulations and EU Directive 86/609/EEC governing the use of animals in research.
Tissues, genotyping and sizing of TNR
At 3 weeks of age, an ear biopsy was taken from each mouse for genotyping and to measure the number of repeats present at this age. At 21 weeks of age, the mice were sacrificed by cervical dislocation, the organs were harvested, frozen on dry ice and stored at −70°C. DNA from all tissues was isolated according to the DNeasy® Blood & Tissue kit (Qiagen GmbH, Germany). CAG repeats were sized by PCR with primers 5′-FAM-atgaaggccttcgagtccctcaagtccttc-3′ and 5′-ggcggctgaggaagctgagga-3′ according to ref. (38) with slight modifications. Approximately 75 ng of genomic DNA was amplified with 0.15 U AmpliTaq® Gold DNA polymerase with PCR Buffer II, 1.25 mm MgCl2 (Applied Biosystems, Carlsbad, CA, USA), 5% Dimethyl Sulfoxide (Sigma-Aldrich Co. LLC) and 2.5 mm dNTPs (Applied Biosystems). The cycling conditions were 94°C for 10 min, 35 cycles of 94°C for 30 s, 64°C for 30 s, 72°C for 2 min and a final extension at 72°C for 10 min. The FAM-labelled PCR products were mixed with GeneScan™ - 600 LIZ® Size Standard and HiDi™ Formamide (Applied Biosystems) and run on an ABI 3730 Genetic Analyzer (Applied Biosystems). Sizing of the PCR fragments was done by using the GeneMapper® Software Version 3.7 (Applied Biosystems).
Calculation of mean TNR expansion and instability index
The weighted mean repeat value was determined by including all the peaks in the GeneMapper sample plots that were within 10% of the highest peak, and calculated using the following formula: Mean number of repeats = (∑(peak heights × basepair lengths)/∑peak heights) − 86)/3, with subtraction of 86 base pairs without repeat sequences in the fragment. The mean somatic expansion in every mouse was finally determined by subtracting the mean repeat value in the ear sample taken at 3 weeks of age from the mean repeat value in each organ harvested at 21 weeks of age. At 3 weeks of age, the CAG repeat length is similar in all organs (5).
Calculation of the instability index was performed as described (26), but with the use of a 10% peak threshold value instead of 20%, and ear samples taken at 3 weeks of age instead of tail. At 3 weeks of age, ear and tail samples harbour the same CAG repeat length (Møllersen et al., unpublished data). In short: the highest peak in the GeneMapper sample plots for each analysis was identified and peaks with heights less than 10% of the highest peak were excluded (10% threshold). Normalized peak heights were calculated by dividing the peak height of each peak by the sum of the heights of all peaks. The change in repeat length of each peak was deduced from the highest peak in the ear sample determined in the mouse at 3 weeks of age (main allele). The normalized peak heights were multiplied by the changes from the main allele, and the instability index was the sum of the normalized and weighted peak heights.
RNA extraction, cDNA synthesis and qPCR
Mouse tissues were dissected, immediately put in liquid nitrogen and stored at −80°C. The tissues were lysed using Lysing Matrix D tubes (MP Biomedicals LLC, CA, USA) and placed in a Mini-Beadbeater™ for homogenization. Total RNA isolation was performed using the PureLink™ RNA Mini Kit (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol.
After RNA isolation, the samples were treated with recombinant RNase-free DNase I with Protector RNase inhibitor (Roche Applied Science, Penzberg, Upper Bavaria, Germany) according to the manufacturer's instructions to digest any remaining DNA. After DNase I treatment and cleanup of RNA, the samples were analyzed by agarose gel electrophoresis to visually check the RNA quality. The RNA yields were measured using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA).
The High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to reversely transcribe the RNA samples, using 200 ng RNA in each reaction to make cDNA.
The qPCR reaction comprised of 2 µl cDNA, 10 pmol Neil1 forward 5′-GAG ATC CTG TAC CGG CTG AAG A-3′ and reverse 5′-GGT TCT GTA GTT TGG CCT TGA TCT-3′ primers and 5 µL 2x POWER SYBR® Green PCR Master Mix (Applied Biosystems) in a total reaction volume of 10 µl and was performed on a OneStepPlus™ Real-Time PCR System (Applied Biosystems). GAPDH with sense 5′-TCG TCC CGT AGA CAA AAT GGT-3′ and antisense 5′-CGC CCA ATA CGG CCA AA-3′ primers was used as endogenous control. Both GAPDH and β-actin were tested and proven to be suitable as endogenous controls. Each cDNA sample was run in duplicates and the qPCR reactions were repeated with new cDNA synthesis. Relative amounts were calculated using the relative standard curve method and normalized to GAPDH.
NEIL1 incision assays and EMSAs
Hairpin DNA substrates with specific modifications (Fig. 4A) were purchased from Midland Certified Reagent Company, Inc. (Midland, TX, USA). [γ32P]ATP 5′ radiolabelled oligonucleotides were generated as described (39). After annealing, the labelled oligonucleotides were purified from unincorporated [γ32P]ATP by using a Bio-Rad Micro Bio Spin P30 column and verified as previously described (10). NEIL1 and OGG1 incision assays were performed by incubating 5′-32P-labeled CAG1oxoG, CAG2oxoG, CAG1OHC, CAG2OHC, ssoxoG and ssOHC (0.2 pmol) or duplex 34oxoG or 34OHC (0.4 pmol) with hNEIL1 (10 and 100 ng) and hOGG1 protein (30 ng; New England Biolabs) in NEB2 buffer at 37°C for 1 h. Reactions were halted by addition of stop buffer [95% formamide, 20 mm ethylenediaminetetraacetic acid (EDTA), 0.5% bromophenol blue and 0.5% xylene cyanol], and then heated at 95°C for 5 min. Reaction products were resolved by 15% polyacrylamide urea denaturing gel electrophoresis and imaged using a Typhoon phosphorimager.
NEIL1 EMSAs were performed by incubating 5′-32P labelled substrates (0.1 pmol for all substrates) with hNEIL1 (100 ng) in 1× reaction buffer (70 mm MOPS, pH 7.5, 1 mm EDTA, 1 mm DTT, 5% glycerol). Samples were incubated for 15 min on ice and subsequently resolved on an 8% non-denaturing polyacrylamide gel by electrophoresis at 100 V at 4°C. Gels were imaged using a Typhoon phosphorimager.
Statistical analysis
Student's t-test was used for normally distributed qPCR data and to compare mean repeat expansion values in all sampled organs between R6/1/Neil1+/+ and R6/1/Neil1−/− mice. Since the a priori hypothesis was that repeat expansion would be less in R6/1/Neil1−/− mice, as observed in R6/1/Ogg1−/− mice (6), the t-test was one-sided.
FUNDING
This work was supported by the Norwegian Research Council (L.M., K.J.G., A.B.), the Norwegian Cancer Society (G.A.H., L.T.), the Centre for Molecular Biology and Neuroscience (A.D.R.), the Health Region South-East of Norway (A.D.R.), Oslo University Hospital (M.B., A.K.) and the Intramural Research Program of the National Institute on Aging, NIH (J.L.I. and D.M.W.). Funding to pay the Open Access publication charges for this article was provided by the Health Region South-East of Norway.
ACKNOWLEDGEMENTS
We thank Dr Mari Kaarbø for valuable qPCR advice and Prof. Vidar Gundersen for helping with dissection of striatum.
Conflict of Interest statement. None declared.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. doi:10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 2.Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M., Abbott M. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat. Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. doi:10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 3.Swami M., Hendricks A.E., Gillis T., Massood T., Mysore J., Myers R.H., Wheeler V.C. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 2009;18:3039–3047. doi: 10.1093/hmg/ddp242. doi:10.1093/hmg/ddp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shelbourne P.F., Keller-McGandy C., Bi W.L., Yoon S.R., Dubeau L., Veitch N.J., Vonsattel J.P., Wexler N.S., Arnheim N., Augood S.J. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet. 2007;16:1133–1142. doi: 10.1093/hmg/ddm054. doi:10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
- 5.Møllersen L., Rowe A.D., Larsen E., Rognes T., Klungland A. Continuous and periodic expansion of CAG repeats in Huntington's disease R6/1 mice. PLoS Genet. 2010;6:e1001242. doi: 10.1371/journal.pgen.1001242. doi:10.1371/journal.pgen.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kovtun I.V., Liu Y., Bjørås M., Klungland A., Wilson S.H., McMurray C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. doi:10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manley K., Shirley T.L., Flaherty L., Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999;23:471–473. doi: 10.1038/70598. doi:10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 8.Owen B.A., Yang Z., Lai M., Gajek M., Badger J.D., Hayes J.J., Edelmann W., Kucherlapati R., Wilson T.M., McMurray C.T. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005;12:663–670. doi: 10.1038/nsmb965. doi:10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler V.C., Lebel L.A., Vrbanac V., Teed A., te Riele R.H., MacDonald M.E. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum. Mol. Genet. 2003;12:273–281. doi: 10.1093/hmg/ddg056. doi:10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- 10.Goula A.V., Berquist B.R., Wilson D.M., III, Wheeler V.C., Trottier Y., Merienne K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum In Huntington's disease transgenic mice. PLoS Genet. 2009;5:e1000749. doi: 10.1371/journal.pgen.1000749. doi:10.1371/journal.pgen.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hazra T.K., Das A., Das S., Choudhury S., Kow Y.W., Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amst.) 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. doi:10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klungland A., Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. doi:10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morland I., Rolseth V., Luna L., Rognes T., Bjørås M., Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. doi:10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hazra T.K., Izumi T., Boldogh I., Imhoff B., Kow Y.W., Jaruga P., Dizdaroglu M., Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl Acad. Sci. USA. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. doi:10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takao M., Kanno S., Kobayashi K., Zhang Q.M., Yonei S., van der Horst G.T., Yasui A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J. Biol. Chem. 2002;277:42205–42213. doi: 10.1074/jbc.M206884200. doi:10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 16.Dou H., Mitra S., Hazra T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. doi:10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 17.Jaruga P., Birincioglu M., Rosenquist T.A., Dizdaroglu M. Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry. 2004;43:15909–15914. doi: 10.1021/bi048162l. doi:10.1021/bi048162l. [DOI] [PubMed] [Google Scholar]
- 18.Hailer M.K., Slade P.G., Martin B.D., Rosenquist T.A., Sugden K.D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst.) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. doi:10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Krishnamurthy N., Zhao X., Burrows C.J., David S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. doi:10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rolseth V., Runden-Pran E., Luna L., McMurray C., Bjørås M., Ottersen O.P. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair (Amst.) 2008;7:1578–1588. doi: 10.1016/j.dnarep.2008.06.007. doi:10.1016/j.dnarep.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsons J.L., Kavli B., Slupphaug G., Dianov G.L. NEIL1 is the major DNA glycosylase that processes 5-hydroxyuracil in the proximity of a DNA single-strand break. Biochemistry. 2007;46:4158–4163. doi: 10.1021/bi0622569. doi:10.1021/bi0622569. [DOI] [PubMed] [Google Scholar]
- 22.Dobbs T.A., Palmer P., Maniou Z., Lomax M.E., O'Neill P. Interplay of two major repair pathways in the processing of complex double-strand DNA breaks. DNA Repair (Amst.) 2008;7:1372–1383. doi: 10.1016/j.dnarep.2008.05.001. doi:10.1016/j.dnarep.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Mangiarini L., Sathasivam K., Mahal A., Mott R., Seller M., Bates G.P. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat. Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. doi:10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- 24.Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F., et al. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. doi:10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 25.Kovtun I.V., Therneau T.M., McMurray C.T. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington's disease gene. Hum. Mol. Genet. 2000;9:2767–2775. doi: 10.1093/hmg/9.18.2767. doi:10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.M., Zhang J., Su A.I., Walker J.R., Wiltshire T., Kang K., Dragileva E., Gillis T., Lopez E.T., Boily M.J., et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC. Syst. Biol. 2010;4 doi: 10.1186/1752-0509-4-29. doi:10.1186/1752-0509-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovtun I.V., Johnson K.O., McMurray C.T. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY) 2011;3:509–514. doi: 10.18632/aging.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovtun I.V., Thornhill A.R., McMurray C.T. Somatic deletion events occur during early embryonic development and modify the extent of CAG expansion in subsequent generations. Hum. Mol. Genet. 2004;13:3057–3068. doi: 10.1093/hmg/ddh325. doi:10.1093/hmg/ddh325. [DOI] [PubMed] [Google Scholar]
- 29.Telenius H., Kremer B., Goldberg Y.P., Theilmann J., Andrew S.E., Zeisler J., Adam S., Greenberg C., Ives E.J., Clarke L.A. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 1994;6:409–414. doi: 10.1038/ng0494-409. doi:10.1038/ng0494-409. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy L., Evans E., Chen C.M., Craven L., Detloff P.J., Ennis M., Shelbourne P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. doi:10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy L., Shelbourne P.F. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum. Mol. Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. doi:10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- 32.Sampath H., Batra A.K., Vartanian V., Carmical J.R., Prusak D., King I.B., Lowell B., Earley L.F., Wood T.G., Marks D.L., et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011;300:E724–E734. doi: 10.1152/ajpendo.00387.2010. doi:10.1152/ajpendo.00387.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Brien P.J., Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. doi:10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 34.Jarem D.A., Wilson N.R., Delaney S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. 2009;48:6655–6663. doi: 10.1021/bi9007403. doi:10.1021/bi9007403. [DOI] [PubMed] [Google Scholar]
- 35.Goula A.V., Pearson C.E., Della M.J., Trottier Y., Tomkinson A.E., Wilson D.M., III, Merienne K. The nucleotide sequence, DNA damage location, and protein stoichiometry influence the base excision repair outcome at CAG/CTG repeats. Biochemistry. 2012;51:3919–3932. doi: 10.1021/bi300410d. doi:10.1021/bi300410d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan M.K., Ocampo-Hafalla M.T., Vartanian V., Jaruga P., Kirkali G., Koenig K.L., Brown S., Lloyd R.S., Dizdaroglu M., Teebor G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair (Amst.) 2009;8:786–794. doi: 10.1016/j.dnarep.2009.03.001. doi:10.1016/j.dnarep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith K.M., Matson S., Matson W.R., Cormier K., Del Signore S.J., Hagerty S.W., Stack E.C., Ryu H., Ferrante R.J. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington's disease mice. Biochim. Biophys. Acta. 2006;1762:616–626. doi: 10.1016/j.bbadis.2006.03.004. doi:10.1016/j.bbadis.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. doi:10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 39.Berquist B.R., McNeill D.R., Wilson D.M., III Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J. Mol. Biol. 2008;379:17–27. doi: 10.1016/j.jmb.2008.03.053. doi:10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]