

Abstract
It has been reported that inflammation is involved in brain injury after subarachnoid hemorrhage (SAH). Nuclear factor-κB (NF-κB) is a key transcriptional regulator of inflammatory genes. Here, we used pyrrolidine dithiocarbamate(PDTC), an inhibitor of NF-κB, through intracisternal injection to study the role of NF-κB in delayed brain injury after SAH. A total of 55 rabbits were randomly divided into five groups: the control group; the SAH groups including Day-3, 5, and 7 SAH groups (the rabbits in these groups were sacrificed at 3, 5, 7 days after SAH, respectively); and the PDTC group (n = 11 for each group). Electrophoretic mobility shift assay (EMSA) was performed to detect NF-κB DNA-binding activity. The mRNA levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and intercellular adhesion molecule (ICAM)-1 were evaluated by RT-PCR analysis. Deoxyribonucleic acid fragmentation was detected by TUNEL and p65 immunoactivity was assessed by immunohistochemistry. Our results showed the activation of NF-κB after SAH, especially at day 3 and 5. The activated p65 was detected in neurons. NF-κB DNA-binding activity was suppressed by intracisternal administration of PDTC. Increased levels of the TNF-α, IL-1β, and ICAM-1 mRNA were found in the brain at day 5 after SAH, and which were suppressed in the PDTC group. The number of TUNEL-positive cells also decreased significantly in the PDTC group compared with that in the Day-5 SAH group. These results demonstrated that the activated NF-κB in neurons after SAH plays an important role in regulating the expressions of inflammatory genes in the brain, and ultimately contributes to delayed brain injury.
Introduction
In most developed countries, stroke is the third leading cause of death, 20% of which are due to aneurysmal subarachnoid hemorrhage (SAH)[1]. There are far more studies focusing on the contraction of the cerebral arteries than on the pathophysiological changes in the brain after SAH, which also could lead to disastrous outcomes.
Early and delayed brain injury after SAH have been well documented, but the underlying mechanisms still remain unclear. It has been reported that cell death, especially apoptosis, occurs in the brain after exposure to subarachnoid hemolysate or hemorrhage. A previous study demonstrated that SAH could trigger an inflammatory reaction in the brain, which might contribute to cell death[2]. Additionally, increasing evidence indicates that inflammatory cytokines and adhesion molecules, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-8, IL-10, intercellular adhesion molecule (ICAM)-1, and vascular cell adhesion molecule (VCAM)-1, are increased in the cerebrospinal fluid (CSF) after SAH[3]. These increased cytokines and adhesion molecules in CSF are correlated with neurological injury, so it is likely that they contribute to brain injury after SAH[4], [5].
Transcription factor nuclear factor-κB (NF-κB) plays a crucial role in regulating the immunity and inflammation in the central nervous system (CNS)[6]. NF-κB has been reported to be involved in traumatic brain trauma[7], spinal cord injury[8], and ischemia[9], which all share many common pathological events with early and delayed brain injury after SAH. Thus, it seems likely that NF-κB may contribute to brain injury after SAH as well.
Among all chemicals or drugs which could inhibit NF-κB, including glucocorticoids[10], [11], vitamin C [12], vitamin E[13], flavonoids [14], leflunomide [15], cyclosporin A and tacrolimus (FK-506) [16], pyrrolidine dithiocarbamate (PDTC) [17] has been used more widely in the experimental study as the inhibitor of NF-κB for the treatment of inflammatory diseases [18]. Moreover, it has been reported that PDTC could protect against brain ischemia with a wide therapeutic time window via inhibiting the activation of NF-κB in neurons [19]. The κB site decoy oligodeoxynucleotides (ODNs), which blocks the binding of NF-kB to DNA, are also used in a number of animal studies[20], [21]. However, ODNs are degraded rapidly in vivo, therefore high doses of ODNs have to be delivered frequently which prevents their clinical use. NF-κB could be suppressed by means of RNA silencing, but this method need to be evaluated more precisely in vivo.
Thus, the present study was designed to assess the change of NF-kB DNA-binding activity in the brain after SAH and then to determine the potential role of NF-kB in delayed brain injury after SAH. In addition, the influence of PDTC on delayed brain injury after SAH was also investigated.
Materials and Methods
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Care and Use Committee of Nanjing University. All surgeries were performed under anesthesia, and all efforts were made to minimize suffering. Fifty-five male rabbits were randomly assigned to 5 groups:
The animals in group 1 served as controls and intracisternal saline injection was performed (n = 11).
The animals in groups 2, 3, 4 were subjected to experimental SAH on day 0 and 2 twice and killed on days 3, 5, and 7, respectively (n = 11 for each group).
The animals in group 5 were subjected to experimental SAH on day 0 and 2 twice, intracisternal administration of PDTC (3 mg/kg) from day 0 to day 5, and then sacrificed on day 5 (n = 11).
Five rabbits in each group were killed for detection the localization of activated NF-κB by immunohistochemical assay of p65 and for detection of deoxyribonucleic acid (DNA) fragmentation by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL). Three rabbits were sacrificed for detection of NF-κB DNA-binding activity by electrophoretic mobility shift assay (EMSA). Others were used for detecting the mRNA of TNF-α, IL-1β, and ICAM-1 by semi-quantitative reverse transcriptase polymerase chain reaction (RT-PCR) analysis.
Induction of Experimental SAH
In groups 2, 3, 4, and 5, SAH was produced according to the two-hemorrhage method [22]. The rabbits were anaesthetized with an intramuscular injection of a mixture of ketamine (25 mg/kg) and droperidol (1.0 mg/kg) on day 0. Under spontaneous breathing, a 21-gauge butterfly needle was percutaneously inserted into the cisterna magna. After withdrawal of 1.5 ml of the CSF, the same amount of non-heparinized fresh autologous auricular arterial blood was injected into the cisterna magna by a 1-min slow injection under aseptic technique. The animals were then kept in a 30° head-down position for 30 min. Once being recovered from anesthesia, they were returned to the feeding room. Forty-eight hours after the first SAH, the second injection was accomplished using the same procedure as described above.
In the animals of the control group, the same technique was applied with injection of sterile saline instead of blood. In the PDTC group, PDTC (3 mg/kg) was injected into cisterna magna in the blood injection manner every 24 hours from 1 min before first blood injection on day 0 to day 5.
On days 3, 5 and 7, the rabbits scheduled for sacrifice were anesthetized again with an intramuscular injection of a mixture of ketamine (40 mg/kg) and droperidol (2.5 mg/kg). The rabbits were exsanguinated and decollated. The brain was removed and certain areas were taken for assay. The tissue for EMSA and RT-PCR was frozen in liquid nitrogen immediately, while the tissue for TUNEL and immunohistochemistry was fixed with 10% neutral buffered formalin.
Nuclear protein extracts and EMSA
Nuclear protein of the tissue was extracted and quantified as described [7]. EMSA was performed using a commercial kit (Gel Shift Assay System; Promega, Madison, WI). Consensus oligonucleotide probe (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was end-labeled with T4-polynucleotide kinase. Nuclear protein (30 µg) was preincubated in a total volume of 9 µl in a binding buffer for 10 min at room temperature. After adding of the 32P-labled oligonucleotide probe, the incubation was continued for 20 min. Reaction was stopped by adding 1 µl of gel loading buffer and the mixture was subjected to nondenaturing 4% polyacrylamide gel electrophoresis in 0.5×TBE(Tris-borate-EDTA) buffer. After electrophoresis was conducted at 390 V for 1 h, the gel was vacuum-dried and exposed to X-ray film (Fuji Hyperfilm) at −70°C.
Immunohistochemical staining
The brain tissue was fixed with the 10% neutral buffered formalin and embedded in paraffin. The tissue sections were used for immunohistochemical staining, which was performed with an anti-p65 polyclonal antibody (diluted 1⊗200, Santa Cruz Biotechnology, Santa Cruz, CA). The sections were incubated with the diluted antibody overnight at 4°C, and then washed, and blocked with 1.6% H2O2 in phosphate-buffered saline (PBS) for 10 min. After washing with PBS, each section was incubated with horseradish peroxidase (HRP)-conjugated IgG (diluted 1⊗500, Santa Cruz Biotechnology, Santa Cruz, CA) for 60 min at room temperature. Diaminobenzidine (DAB) was used as chromogen and counterstaining was done with hematoxylin.
RNA extraction and RT-PCR
The levels of TNF-α, IL-1β, and ICAM-1 mRNA expression were determined by RT-PCR. Total RNA was extracted with TriPure Reagent (Roche Diagnostics Corp., Indianapolis, IN, USA) according to the manufacture's instruments. The cDNA synthesis from the isolated RNA was performed using a reverse transcriptional system. Briefly, 4 µg of total RNA was subjected to the first strand cDNA synthesis for 1 h at 42°C in a 20 µl reaction mixture containing 20 U RNase inhibitor, 0.04 µmol of each dNTP, 0.5 µg oligo(dT)15 and 15 U AMV reverse transcriptase (Promega). The reaction was terminated by incubation at 95°C for 5 min. The cDNA was stored at –20°C or used for PCR immediately. PCR amplification was performed in a total volume of 25 µl containing 4 µl cDNA, 0.05 µmol MgCl2, 2.5 U Taq polymerase, 0.02 µmol of each dNTP, specific oligonucleotide primers(Table 1) for TNF-α, IL-1β, ICAM-1 or GAPDH genes, and 2.5 µl 10×Taq polymerase reaction buffer. Each PCR cycle included a denaturation step at 94°C for 30 s, a primer annealing step for 30 s (the temperatures were listed on Table 1), an extension step at 72°C for 50 s, and a final extension step at 72 °C for 7 min. The number of cycles was also shown in the Table 1. Thereafter, the amplified fragments were detected by agarose gel electrophoresis and visualized by ethidium bromide staining. The gel was captured as a digital image and analyzed using Scion Image software (Scion Corp, Maryland, USA). Values for each sample were normalized by the GAPDH as the control.
Table 1. Sequences of PCR primers.
| Target gene | Sense primer (5′ to 3′) | Antisense primer (5′ to 3′) | Annealing temperature (°C) | Number of cycles | Size (bp) |
| TNF-α | GCTGCACTTCAGGCTGATC | CTTGTTCGGGTAGGAGACG | 57 | 34 | 352 |
| IL-1β | ATCTCCTGCCAACCCTACA | CTTTCAGCTCATA CGTGCC | 54 | 34 | 274 |
| ICAM-1 | GCGGCTCAGTGTCTCATTCC | CACGCAGTCCTCGGCTTCT | 58 | 34 | 184 |
| GAPDH | GGAGCCAAAAGGGTCATC | CCAGTGAGTTTCCCGTTC | 57 | 30 | 347 |
Assessment of brain injury
The DNA fragmentation of the brain was detected by TUNEL. The coronal sections were embedded in paraffin and sectioned at 4- µm thickness. In situ cell death detection Kit POD (ISCDD, Boehringer Mannheim, Germany) was used according to the manufacturer's instruction. Briefly, paraffin-embedded sections were mounted on positively charged slides, deparaffinized, rehydrated, and washed thoroughly with distilled water. The tissues were digested with 20 g/ml proteinase K (Boehringer Mannheim, Mannheim, Germany) at room temperature for 15 min to retrieve antigen. Endogenous peroxidase activity was blocked by incubation in 0.3% hydrogen peroxide/methanol in phosphate-buffered saline at 37°C for 30 min. The sections were then incubated with terminal deoxynucleotidyl transferase at 37°C for 60 min, which would add the dioxigenin-conjugated dUTP to the 3′-OH ends of fragmented DNA. Anti-dioxigenin antibody peroxidase was applied to the sections to detect the labeled nucleotides. The sections were stained with DAB and counterstained slightly with hematoxylin. The positive cells were identified, counted, and analyzed under a light microscope by another investigator in a blinded fashion. The extent of brain injury was evaluated by calculating the average positive neuronal cells per 100 neurons, and at least 1000 neurons were counted.
Statistical analysis
All data were presented as mean ± SEM. Software SPSS 11.0 was used for the statistical analysis. Comparison between different groups was made by one-way analysis of variance (ANOVA), followed by Tukey's multiple comparisons test if a significant difference had been determined by ANOVA. A probability value of P<0.05 was considered statistically significant.
Results
Time course of NF-κB DNA-binding activity in the brain after SAH
The temporal activation NF-κB in the brain after SAH was examined by EMSA. The NF-κB DNA-binding activity was up-regulated significantly after SAH compared with that in the control group (P<0.05 vs. control group), especially on day 3 and 5 (P<0.01 vs. control group)(Fig.1A and B). The increased NF-κB DNA-binding activity on day 7 after SAH was not more than that on the day 3 and day 5 SAH groups.
Figure 1. The time course of NF-κB DNA-binding activity detected by EMSA after SAH and the effects of PDTC on NF-κB activation.

A. The representative autoradiogram showed the NF-κB DNA-binding activity in each group. B. The time course of NF-κB DNA-binding activity after SAH. Quantification of the DNA-binding activity of NF-κB was performed by densitometric analysis. The NF-κB DNA-binding activity was up-regulated significantly after SAH, especially on day 3 and 5, while restored on day 7. C. the effects of intracisternal administration of PDTC on NF-κB DNA-binding activity. It was shown that NF-κB DNA-binding activity was suppressed after treatment with PDTC. 1 stands for the control group. 2, 3 and 4 stand for the D-3, 5 and 7 SAH groups. 5 stands for the PDTC groups. Results are represented as means ± SEM from five independent experiments in each group. *P<0.05 vs. control group, **P<0.01 vs. control group, #P<0.05 vs. D-5 SAH group.
Localization of activated NF-κB
We examined the localization of activated NF-κB in the brain by immunohistochemical staining. It was shown that NF-κB p65 immunoactivity was mainly presented in neurons, especially in the nucleus, which implied that NF-κB was activated after SAH (Fig.2).
Figure 2. Localization of activated NF-κB detected by immunohistochemistry.
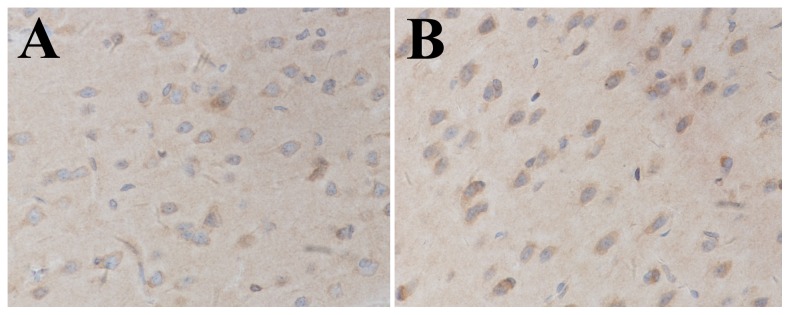
NF-κB p65 immunoactivity was mainly presented in neurons in the SAH group. Furthermore, NF-κB p65 immunoactivity mainly located in the nuclei of neurons.
Effect of PDTC on cell injury in the brain
Cell injury in the brain was assessed by detecting the DNA fragmentation using TUNEL staining (Fig. 3A, B, C, and D). The TUNEL-positive cells mainly distribute on the dorsal temporal cortex and the temporal poles. The number of TUNEL-positive cells was much higher in the day-5 SAH group (group 3) compared with the control group (P<0.01). We intracisternally administrated the inhibitor of NF-κB, PDTC, in the SAH model in group 5. It was found that NF-κB DNA-binding activity in the group 5 decreased significantly compared with group 3 (P<0.01), which indicates that intracisternal administration of PDTC could effectively inhibit the DNA-binding activity of NF-κB in the brain. The number of TUNEL-positive cells in the group 5 with PDTC treatment also decreased compared with the group 3 (P<0.05). In addition, the levels of the TNF-α, IL-1β, and ICAM-1 mRNA in the brain were detected by RT-PCR (Fig. 4A and B), which were all significantly increased in the SAH group (P<0.05 vs. control group). There are statistical differences between the levels of TNF-α, IL-1β, and ICAM-1 mRNA in the group 3 and the group 5 (P<0.05).
Figure 3. The cell injury in the brain was assessed by detecting the DNA fragmentation using TUNEL staining.

Panel 1. It was shown that the cells have a normal structure and are light blue-stained. In the SAH group, the dystrophic and brown-stained TUNEL-positive cells are observed in the cortex. And the TUNEL-positive cells in the PDTC group are less than that in the SAH group. Panel 2. Quantification of the TUNEL staining showed that the TUNEL-positive cells are significantly increased in the SAH group compared with that in the control group. In the PDTC group, the TUNEL-positive cells are significantly decreased compared with that in the SAH group. **P<0.01 vs. control group, #P<0.05 vs. SAH group.
Figure 4. The gene expressions of TNF-α, IL-1β, and ICAM-1 in the brain.

The representative autoradiograms of the RT-PCR results of TNF-α, IL-1β, ICAM-1. B) Relative amount of TNF-α, IL-1β and ICAM-1 mRNA. The levels of TNF-α, IL-1β and ICAM-1 mRNA increased after SAH and was suppressed in the PDTC group. **P<0.01 vs. control group, ##P<0.01 vs. SAH group.
Discussion
The main findings of the present study are: 1) NF-κB DNA-binding activity increased after SAH, and peaked on day 3 and 5, which could all be suppressed by PDTC, an inhibitor of NF-κB; 2) cell death in the brain after SAH could also be alleviated by intracisternal administration of PDTC; 3) NF-κB may contribute to delayed brain injury via regulating the gene expressions of TNF-α, IL-1β, and ICAM-1 in the brain.
The time course of NF-κB activation in the brain after SAH
The up-regulation of NF-κB DNA-binding activity occurred early after SAH. NF-κB DNA-binding activity was increased and almost reached the peak on day 3, the first day after the second blood injection, which might result in the early brain injury after SAH. Many pathological factors can induce the activation of NF-κB in the brain or in the neurons in vitro, such as elevation of glutamate[23], focal or global cerebral ischemia[9], [24], [25], and free radical generation[26]. Those events also occur in the brain immediately after SAH, which may result from the raised intracranial pressure (ICP), reduced cerebral blood flow (CBF) and cerebral perfusion pressure (CPP), and dysfunction of cerebral auto-regulation[27], [28].
Nevertheless, the up-regulation of NF-κB DNA-binding activity on day 5 and day 7 might contribute to delayed brain injury. In another study, we demonstrated that the delayed cerebral vasospasm in this two-hemorrhage SAH model began on day 5 [29]. Thus, the pathological alteration in the brain caused by cerebral vasospasm-induced brain ischemia may result in the molecular alteration in the brain, which might include the up-regulation of NF-κB DNA-binding activity since abundant data suggest the activation of NF-κB induced by cerebral ischemia [30], [31], [32], [33]. On day 7, NF-κB DNA-binding activity decreased profoundly compared with that on day 3 and day 5, which accords with the recovery of cerebral vasospasm in this two-hemorrhage SAH model (in this animal model, the time course of cerebral vasospasm after SAH was similar but not the same as that in humans)[29]. These findings implied that cerebral vasospasm could induce the pathological alteration in the brain including activation of NF-κB. Further studies were encouraged to clarity the details between the delayed cerebral vasospasm and the activation of NF-κB in the brain after SAH.
Moreover, NF-κB was demonstrated to be activated in the brain and to play a crucial role in brain injury after cerebral ischemia or traumatic brain injury via regulating the expressions of inflammatory genes [6], [7], [9]. Thus, the NF-κB pathway is very critical for the pathophysiology after brain injury. In the present study, the gene expressions of the cytokines and adhesion molecules were also up-regulated in the brain after SAH, which is consistent with one previous study from other group[2]. Because of the key role of the activated NF-κB in inflammation, it would be reasonable to assume that inhibiting the NF-κB can ameliorate the inflammation in the brain post injury and then attenuate the entire damage process after SAH. Therefore, in this study, we used the PDTC, a confirmed effective inhibitor of NF-κB. It was demonstrated to inhibit the NF-κB DNA-binding activity in the brain and to reduce the levels of TNF-α, IL-1β, and ICAM-1 gene expressions.
Taken together, all of these findings indicated that NF-κB plays a potential role in pathophysiology of the brain after SAH in rabbits. Its long-lasting action pattern makes it to be a possible target for pharmacological intervention in brain injury after SAH.
NF-κB and delayed brain injury
Some studies have reported that DNA fragmentation or cell death occurred in the brain in different SAH models[34], [35], [36], [37]. But the underlying mechanisms have not been fully identified yet. Also, it has been reported that heat shock proteins were induced by experimental SAH in vivo or by the blood products in the subarachnoid space in vitro. They were regarded as the sensitive markers of neuronal injury and were believed to be related to the cell death after SAH[35], [37], [38]. Additionally, Prunell and his colleagues' study indicated that inflammatory reaction in the brain might have significant contributions to neuronal injury after SAH[2]. In the present study, the gene expressions of the cytokines and adhesion molecular showed a strong correspondence with the degree of DNA fragmentation.
Although NF-κB plays a key role in regulating the inflammatory reaction, the role of NF-κB in cell death and cell survival after brain injury remains controversial. It appears that NF-κB is involved in both processes, not only in vitro experiments but also in vivo studies[39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49]. Thus, we speculated that a balance could be present in the role of NF-κB in inducing and reducing cell death in the brain. Recently, several studies showed that NF-κB contributed to the cell death in cerebral ischemia [25], [39], [43], [50]. Our results clearly showed that inhibiting the DNA binding activity of NF-κB could attenuate the cell death in the brain after experimental SAH.
The mechanism by which NF-κB contributes to cell death might be the genes regulated by NF-κB. NF-κB activation can lead to the increase of gene expression of some factors such as TNF-α, IL-1β, ICAM-1 and nitric oxide synthetase (NOS)[46], [50], which are all believed to aggravate the cell death in the brain. However, the exact mechanisms need to be clarified in further studies. Characterization of the relationship between NF-κB activation and DNA fragmentation or cell death in the brain after SAH may result in the identification of target molecules to develop new therapeutic interventions.
PDTC and delayed brain injury
PDTC has been characterized as a relatively selective inhibitor of NF-κB activation mainly by preventing the degradation of I-κB[19], [50]. In addition, PDTC has metal-chelating and antioxidant properties, both of which can mediate NF-κB inhibition [19], [50]. PDTC has been reported to stimulate cell death by suppressing activation of NF-κB in various cancer cells[49], [51]. And some previous studies also showed the neuroprotective effects of PDTC in brain injury, which was also mediated by suppressing activation of NF-κB[19], [50]. In the present study, PDTC was demonstrated to suppress the activation of NF-κB and to ameliorate the cell death in the brain after SAH. Hence, it is reasonable to draw a conclusion that the neuroprotective effects of PDTC were also mediated by suppressing the activation of NF-κB.
A previous study has reported that PDTC analogous could inhibit HIV progression in patients, which suggests the broad prospects for the clinical application of PDTC[52]. PDTC and its structurally modified derivatives with possible greater efficacy may represent potential therapies against SAH.
Limitations
Some limitations of the present study should be noted. First, PDTC has the antioxidant capacity, and was regarded as an antioxidant agent in many studies [53], [54]. Therefore, it is possible that the beneficial effect of PDTC is at least partially based on its antioxidative functions. Second, although we used this two-hemorrhage rabbit model of SAH aiming to produce severe vasospasm and brain injury similar to that in humans[22], the effect of vasospasm on the delayed brain injury should be examined in this model. In addition, another study of our group has showed that cerebral vasospasm could be attenuated partially by PDTC in this animal model[29], so studies exploring the detailed mechanisms of delayed brain injury have been initiated in our lab. Third, because cell death caused by early brain injury was failed to be excluded from that detected on day 5 post SAH in this study, it is essential to validate our findings in dog SAH models, in which the interval between early brain injury and delayed brain injury would be longer.
Conclusion
Activated NF-κB in the neurons after SAH contributes to delayed brain injury by regulation of the gene expressions of the cytokines and ICAM-1 in the brain. The NF-κB inhibitor, PDTC, could attenuate the delayed brain injury after SAH.
Funding Statement
The National Natural Science Foundation of China (NSFC): No. 81000503(MZ), No. 81070921, and No. 81271297 (JS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Phillips LH, 2nd Whisnant JP, O′Fallon WM, Sundt TM, Jr The unchanging pattern of subarachnoid hemorrhage in a community. Neurology 30: 1034–1040. [DOI] [PubMed] [Google Scholar]
- 2. Prunell GF, Svendgaard NA, Alkass K, Mathiesen T (2005) Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery 56: 1082– 1092 discussion 1082–1092. [PubMed] [Google Scholar]
- 3. Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, et al. (2003) Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery 53: 123–133 discussion 133–125. [DOI] [PubMed] [Google Scholar]
- 4. Aihara Y, Kasuya H, Onda H, Hori T, Takeda J (2001) Quantitative analysis of gene expressions related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke 32: 212–217. [DOI] [PubMed] [Google Scholar]
- 5. Mathiesen T, Andersson B, Loftenius A, von Holst H (1993) Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J Neurosurg 78: 562–567. [DOI] [PubMed] [Google Scholar]
- 6. Kaltschmidt B, Widera D, Kaltschmidt C (2005) Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta 1745: 287–299. [DOI] [PubMed] [Google Scholar]
- 7. Hang CH, Shi JX, Tian J, Li JS, Wu W, et al. (2004) Effect of systemic LPS injection on cortical NF-kappaB activity and inflammatory response following traumatic brain injury in rats. Brain Res 1026: 23–32. [DOI] [PubMed] [Google Scholar]
- 8. Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, et al. (1998) Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci 18: 3251–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salminen A, Liu PK, Hsu CY (1995) Alteration of transcription factor binding activities in the ischemic rat brain. Biochem Biophys Res Commun 212: 939–944. [DOI] [PubMed] [Google Scholar]
- 10. Roman-Blas JA, Jimenez SA (2006) NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage 14: 839–848. [DOI] [PubMed] [Google Scholar]
- 11. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M (1995) Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270: 286–290. [DOI] [PubMed] [Google Scholar]
- 12. Carcamo JM, Pedraza A, Borquez-Ojeda O, Golde DW (2002) Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 41: 12995–13002. [DOI] [PubMed] [Google Scholar]
- 13. Calfee-Mason KG, Spear BT, Glauert HP (2004) Effects of vitamin E on the NF-kappaB pathway in rats treated with the peroxisome proliferator, ciprofibrate. Toxicol Appl Pharmacol 199: 1–9. [DOI] [PubMed] [Google Scholar]
- 14. Bremner P, Heinrich M (2002) Natural products as targeted modulators of the nuclear factor-kappaB pathway. J Pharm Pharmacol 54: 453–472. [DOI] [PubMed] [Google Scholar]
- 15. Manna SK, Mukhopadhyay A, Aggarwal BB (2000) Leflunomide suppresses TNF-induced cellular responses: effects on NF-kappa B, activator protein-1, c-Jun N-terminal protein kinase, and apoptosis. J Immunol 165: 5962–5969. [DOI] [PubMed] [Google Scholar]
- 16. Meyer S, Kohler NG, Joly A (1997) Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-kappaB activation. FEBS Lett 413: 354–358. [DOI] [PubMed] [Google Scholar]
- 17. Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA (1992) Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med 175: 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chabicovsky M, Prieschl-Grassauer E, Seipelt J, Muster T, Szolar OH, et al. (2010) Pre-clinical safety evaluation of pyrrolidine dithiocarbamate. Basic Clin Pharmacol Toxicol 107: 758–767. [DOI] [PubMed] [Google Scholar]
- 19. Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjanheikki J, et al. (2004) Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem 91: 755–765. [DOI] [PubMed] [Google Scholar]
- 20. Isomura I, Morita A (2006) Regulation of NF-kappaB signaling by decoy oligodeoxynucleotides. Microbiol Immunol 50: 559–563. [DOI] [PubMed] [Google Scholar]
- 21.Aoki T, Kataoka H, Nishimura M, Ishibashi R, Morishita R, et al. (2012) Regression of intracranial aneurysms by simultaneous inhibition of nuclear factor-kappaB and Ets with chimeric decoy oligodeoxynucleotide treatment. Neurosurgery 70: 1534–1543; discussion 1543. [DOI] [PubMed]
- 22. Zhou ML, Shi JX, Zhu JQ, Hang CH, Mao L, et al. (2007) Comparison between one- and two-hemorrhage models of cerebral vasospasm in rabbits. J Neurosci Methods 159: 318–324. [DOI] [PubMed] [Google Scholar]
- 23. Guerrini L, Blasi F, Denis-Donini S (1995) Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci U S A 92: 9077–9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP (1997) Global ischemia activates nuclear factor-kappa B in forebrain neurons of rats. Stroke 28: 1073–1080 discussion 1080–1071. [DOI] [PubMed] [Google Scholar]
- 25. Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, et al. (1999) NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med 5: 554–559. [DOI] [PubMed] [Google Scholar]
- 26. Crack PJ, Taylor JM (2005) Reactive oxygen species and the modulation of stroke. Free Radic Biol Med 38: 1433–1444. [DOI] [PubMed] [Google Scholar]
- 27. Sehba FA, Bederson JB (2006) Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res 28: 381–398. [DOI] [PubMed] [Google Scholar]
- 28. Cahill J, Calvert JW, Zhang JH (2006) Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 26: 1341–1353. [DOI] [PubMed] [Google Scholar]
- 29. Zhou ML, Shi JX, Hang CH, Cheng HL, Qi XP, et al. (2007) Potential contribution of nuclear factor-kappaB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab 27: 1583–1592. [DOI] [PubMed] [Google Scholar]
- 30. Gabriel C, Justicia C, Camins A, Planas AM (1999) Activation of nuclear factor-kappaB in the rat brain after transient focal ischemia. Brain Res Mol Brain Res 65: 61–69. [DOI] [PubMed] [Google Scholar]
- 31. Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, et al. (2000) Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab 20: 592–603. [DOI] [PubMed] [Google Scholar]
- 32. Seegers H, Grillon E, Trioullier Y, Vath A, Verna JM, et al. (2000) Nuclear factor-kappa B activation in permanent intraluminal focal cerebral ischemia in the rat. Neurosci Lett 288: 241–245. [DOI] [PubMed] [Google Scholar]
- 33. Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, et al. (2004) Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 35: 987–991. [DOI] [PubMed] [Google Scholar]
- 34. Matz PG, Copin JC, Chan PH (2000) Cell death after exposure to subarachnoid hemolysate correlates inversely with expression of CuZn-superoxide dismutase. Stroke 31: 2450–2459. [DOI] [PubMed] [Google Scholar]
- 35. Prunell GF, Svendgaard NA, Alkass K, Mathiesen T (2005) Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J Neurosurg 102: 1046–1054. [DOI] [PubMed] [Google Scholar]
- 36. Matz PG, Fujimura M, Chan PH (2000) Subarachnoid hemolysate produces DNA fragmentation in a pattern similar to apoptosis in mouse brain. Brain Res 858: 312–319. [DOI] [PubMed] [Google Scholar]
- 37.Matz P, Weinstein P, States B, Honkaniemi J, Sharp FR (1996) Subarachnoid injections of lysed blood induce the hsp70 stress gene and produce DNA fragmentation in focal areas of the rat brain. Stroke27 :504– 512; discussion 513. [DOI] [PubMed] [Google Scholar]
- 38. Matz PG, Sundaresan S, Sharp FR, Weinstein PR (1996) Induction of HSP70 in rat brain following subarachnoid hemorrhage produced by endovascular perforation. J Neurosurg 85: 138–145. [DOI] [PubMed] [Google Scholar]
- 39. Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, et al. (2005) Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab 25: 30–40. [DOI] [PubMed] [Google Scholar]
- 40. Pizzi M, Goffi F, Boroni F, Benarese M, Perkins SE, et al. (2002) Opposing roles for NF-kappa B/Rel factors p65 and c-Rel in the modulation of neuron survival elicited by glutamate and interleukin-1beta. J Biol Chem 277: 20717–20723. [DOI] [PubMed] [Google Scholar]
- 41. Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, et al. (1999) Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci 19: 4023–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, et al. (2003) Forebrain-specific neuronal inhibition of nuclear factor-kappaB activity leads to loss of neuroprotection. J Neurosci 23: 9403–9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hill WD, Hess DC, Carroll JE, Wakade CG, Howard EF, et al. (2001) The NF-kappaB inhibitor diethyldithiocarbamate (DDTC) increases brain cell death in a transient middle cerebral artery occlusion model of ischemia. Brain Res Bull 55: 375–386. [DOI] [PubMed] [Google Scholar]
- 44. Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, et al. (1998) Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kappaB in rats. Stroke 29: 677–682. [DOI] [PubMed] [Google Scholar]
- 45. Wen Y, Yang S, Liu R, Perez E, Yi KD, et al. (2004) Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res 1008: 147–154. [DOI] [PubMed] [Google Scholar]
- 46. Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269: 4705–4708. [PubMed] [Google Scholar]
- 47. Ziegler-Heitbrock HW, Sternsdorf T, Liese J, Belohradsky B, Weber C, et al. (1993) Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J Immunol 151: 6986–6993. [PubMed] [Google Scholar]
- 48. Malaguarnera L, Pilastro MR, DiMarco R, Scifo C, Renis M, et al. (2003) Cell death in human acute myelogenous leukemic cells induced by pyrrolidinedithiocarbamate. Apoptosis 8: 539–545. [DOI] [PubMed] [Google Scholar]
- 49. Bian X, Opipari AW Jr, Ratanaproeksa AB, Boitano AE, Lucas PC, et al. (2002) Constitutively active NFkappa B is required for the survival of S-type neuroblastoma. J Biol Chem 277: 42144–42150. [DOI] [PubMed] [Google Scholar]
- 50. Crack PJ, Taylor JM, Ali U, Mansell A, Hertzog PJ (2006) Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 37: 1533–1538. [DOI] [PubMed] [Google Scholar]
- 51. Cheng AC, Huang TC, Lai CS, Kuo JM, Huang YT, et al. (2006) Pyrrolidine dithiocarbamate inhibition of luteolin-induced apoptosis through up-regulated phosphorylation of Akt and caspase-9 in human leukemia HL-60 cells. J Agric Food Chem 54: 4215–4221. [DOI] [PubMed] [Google Scholar]
- 52. Reisinger EC, Kern P, Ernst M, Bock P, Flad HD, et al. (1990) Inhibition of HIV progression by dithiocarb. German DTC Study Group. Lancet 335: 679–682. [DOI] [PubMed] [Google Scholar]
- 53. Bowie A, O′Neill LA (2000) Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol 59: 13–23. [DOI] [PubMed] [Google Scholar]
- 54. Verhaegen S, McGowan AJ, Brophy AR, Fernandes RS, Cotter TG (1995) Inhibition of apoptosis by antioxidants in the human HL-60 leukemia cell line. Biochem Pharmacol 50: 1021–1029. [DOI] [PubMed] [Google Scholar]