

Abstract
BACKGROUND
Mounting evidence from animal studies shows that anesthetic exposure in early life leads to apoptosis in the developing nervous system. This loss of neurons has functional consequences in adulthood. Clinical retrospective reviews have suggested that multiple anesthetic exposures in early childhood are associated with learning disabilities later in life as well. Despite much concern about this phenomenon, little is known about the mechanism by which anesthetics initiate neuronal cell death. Caenorhabditis elegans, a powerful genetic animal model, with precisely characterized neural development and cell death pathways, affords an excellent opportunity to study anesthetic-induced neurotoxicity. We hypothesized that exposing the nematode to volatile anesthetics early in life would induce neuron cell death, producing a behavioral defect that would be manifested in adulthood.
METHODS
After synchronization and hatching, larval worms were exposed to volatile anesthetics at their 95% effective concentration for 4 hours. On day 4 of life, exposed and control worms were tested for their ability to sense and move to an attractant (i.e., to chemotax). We determined the rate of successful chemotaxis using a standardized chemotaxis index.
RESULTS
Wild-type nematodes demonstrated striking deficits in chemotaxis indices after exposure to isoflurane (ISO) or sevoflurane (SEVO) in the first larval stage (chemotaxis index: untreated, 85 ± 2; ISO, 52 ± 2; SEVO, 47 ± 2; P < 0.05 for both exposures). The mitochondrial mutant gas-1 had a heightened effect from the anesthetic exposure (chemotaxis index: untreated, 71 ± 2; ISO, 29 ± 12; SEVO, 24 ± 13; P < 0.05 for both exposures). In contrast, animals unable to undergo apoptosis because of a mutation in the pathway that mediates programmed cell death (ced-3) retained their ability to sense and move toward an attractant (chemotaxis index: untreated, 76 ± 10; ISO, 73 ± 9; SEVO, 76 ± 10). Furthermore, we discovered that the window of greatest susceptibility to anesthetic neurotoxicity in nematodes occurs in the first larval stage after hatching (L1). This coincides with a period of neurogenesis in this model. All values are means ± SD.
CONCLUSION
These data indicate that anesthetics affect neurobehavior in nematodes, extending the range of phyla in which early exposure to volatile anesthetics has been shown to cause functional neurological deficits. This implies that anesthetic-induced neurotoxicity occurs via an ancient underlying mechanism. C elegans is a tractable model organism with which to survey an entire genome for molecules that mediate the toxic effects of volatile anesthetics on the developing nervous system.
Multiple animal studies have demonstrated that anesthetic exposure during a critical period of synaptogenesis causes cell death in the brains of young rodents and nonhuman primates; these animals demonstrate deficits in learning and memory later in life.1–6 How these findings translate to humans is still unclear. Retrospective analyses of select pediatric populations have suggested that children with multiple anesthetic exposures have an increased likelihood of developing learning disabilities and behavioral problems.7–10 However, the retrospective nature of these studies, which contain many confounding factors, makes it impossible to assign causality. (For a review, see Sun11 2010.)
A fundamental question about anesthetic-induced neurotoxicity remains unanswered: by what mechanism do anesthetics trigger widespread neuronal apoptosis in the developing brain? Studies in rodents have suggested that anesthetics alter neurotrophin signaling, e.g., via brain-derived neurotrophic factor, which in turn can lead to cell death.12,13 Other studies have implicated mitochondrial function as a contributing factor to anesthetic-induced neurotoxicity.14,15 However, these effects may not be the sole mechanisms by which anesthesia leads to neurotoxicity. Likewise, the links between anesthetic exposure and initiators of this signaling are not easily identified in complex animals like mammals.
Apoptosis is a crucial process that occurs during normal development of vertebrates and invertebrates. Much of the current understanding of apoptosis comes from research initially performed in Caenorhabditis elegans, in which it was termed programmed cell death. Genetic studies in the nematode have led to the identification of critical highly conserved genes involved in this process, such as ced-3 and ced-916–18 (the gene names ced are meant to reflect the phenotype of cell death). CED-3 and CED-9 are homologous to mammalian Caspase-9 and Bcl-2, respectively.19 Because C elegans has been an indispensable organism in the study of apoptotic mechanisms in the past, we expect it to be invaluable in the elucidation of factors involved in apoptosis that follows anesthetic exposure.
Chemotaxis, a fundamental behavior of C elegans, is mediated by a set of sensory neurons with well-characterized morphologies and genetic controls.20 An animal’s ability to sense and move toward an attractant can be assessed with a simple chemotaxis assay.21 We hypothesized that anesthetic exposure in larval nematodes would cause neuronal apoptosis, and that the neuronal loss would be manifest in dysfunctional chemotaxis. We then hypothesized that such induction of chemotactic dysfunction could be prevented by blocking apoptosis, which we accomplished by means of a targeted mutation to the apoptotic pathway. C elegans is an ideal organism in which to investigate anesthetic-associated neuroapoptosis because of its short life cycle, straightforward genetics, and low cost. Studies in the nematode may reveal the causes and mechanisms of anesthetic-induced neurotoxicity.
METHODS
C elegans Strains
Wild-type N2 and ced-3 were obtained from the Caenorhabditis Genetics Center (Minneapolis, MN). The gas-1 was isolated and first characterized in our laboratory. 22 Animals are grown on agar plates spread with Escherichia coli OP50 and maintained at 20°C as previously described.22
Synchronizing and Anesthetic Protocol
Adult nematodes were placed on plates for 8 hours to lay eggs. Worms were then anesthetized at different larval stages. Nematodes progress through 4 larval stages (L1, L2, L3, and L4) and reach adulthood on day 4 of life (at 20°). Anesthetic exposure was performed for 4 hours at the 95% effective concentration for isoflurane (ISO) or sevoflurane (SEVO) in glass chambers at 20°C as previously described.23 Anesthetic concentrations were confirmed by gas chromatography.
Chemotaxis Assay
On day 4 of life, nematodes were subjected to a chemotaxis assay, described by Bargmann and Horvitz.21 All experiments were performed at 20°C. Each assay involved 50 to 100 adult worms.
Large (9-cm) nematode growth media22 agar plates were prepared for the chemotaxis assay in the following manner (Fig. 1): the corners of an equilateral triangle were marked with dots on the bottom of the plate, with the points 4 cm apart. The triangle’s apex marked the start point. An attractant (1 drop of E coli OP50) was placed on the agar at the bottom left point, and allowed to dry. Immediately before the assay, 1 μL of 1 M sodium azide in S. Basal was placed on the OP50 spot, as well as on the bottom right spot.
Figure 1.

Chemotaxis assay plate setup. A large NGM plate is prepared by marking the corners of an equilateral triangle. One drop of OP50 Escherichia coli is placed at the “attractant” spot and allowed to dry. Immediately before the assay, 1 μL of 1 M sodium azide is placed on both the attractant and the “trap” spots. After washing, worms are placed at the start point and allowed to move around the plate for 45 minutes, at which point the chemotaxis index is calculated. Chemotaxis index = (no. of worms at attractant − no. of worms at trap)/(no. of total) × 100.
When the nematodes reached adulthood early on day 4 of life, they were washed with cold S. Basal into 1.6 mL Eppendorf tubes. Washing proceeded with centrifugation at 600g and resuspension in S. Basal 3 times. After the final wash, the supernatant was removed and the worm pellet was gently transferred to the start point of the chemotaxis plate. Residual liquid was dispersed with a platinum wire and evaporation occurred over 5 to 10 minutes. The nematodes were allowed to move on the plate for 45 minutes, at which point the chemotaxis index was determined. Worms immobilized by sodium azide within a 1.5-cm diameter of the attractant or the blank trap were considered to be at these respective sites; those outside these borders were included in the total number of worms on the plate. The chemotaxis index is calculated as (no. of worms at attractant − no. of worms at trap)/(no. of total) × 100 as described by Bargmann and Horvitz.21
Statistical Analysis
This protocol was performed 5 times and the results pooled and averaged. Analysis of variance (ANOVA) was used for determining the significance of the difference among the means. Errors and error bars represent the SD of the mean. Post hoc analysis was performed using a t test to compare matched samples followed by a Bonferroni correction to control for multiple comparisons. Because we compared 3 groups for the effects of anesthetic on each of the different strains (2 degrees of freedom), we defined significance as P < 0.05/2 (0.025). For the comparison of N2 strains at different ages, there were 5 measurements with each anesthetic giving 4 degrees of freedom. Thus, significance was defined as P < 0.05/4 (0.01).
RESULTS
Wild-type nematodes (N2) demonstrated striking deficits in chemotaxis indices after exposure to ISO or SEVO as first-stage larvae (L1), age 0 to 8 hours (chemotaxis indices: untreated ISO control, 84 ± 2; L1 exposed, 52 ± 2, P = 0.0002; untreated SEVO control, 91 ± 3; L1 exposed, 47 ± 2, P = 0.0001). Anesthetic exposure at later stages (L2–4) did not produce significant abnormalities in chemotaxis (Fig. 2). Specifically, animals exposed within the first 4 hours of hatching were the most severely affected by anesthetic exposure (data not shown). All values for chemotaxis indices are means ± SD.
Figure 2.
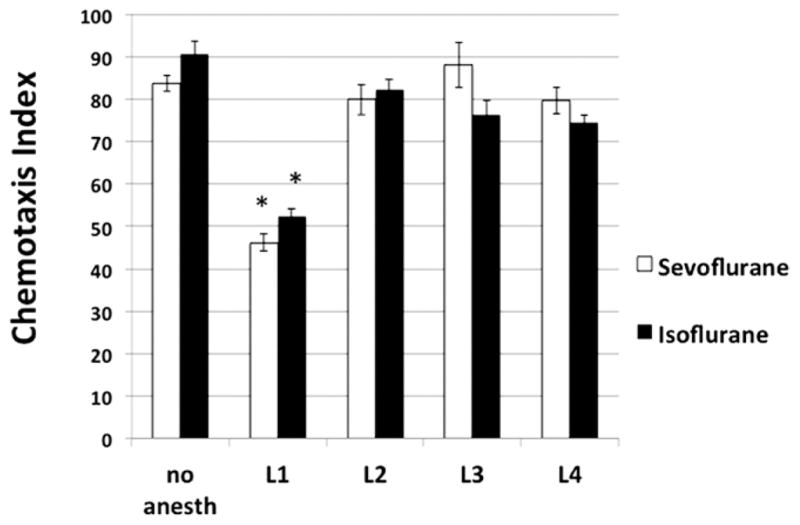
Chemotaxis indices for young adult Caenorhabditis elegans reaching chemoattractant (bacterial food). Animals were exposed to anesthetic for 4 hours during the larval stage shown on the x-axis. The chemotaxis index is decreased after early exposure to a volatile anesthetic but not after an exposure later in life. Error bars are standard deviations of the mean. *Different from control of same anesthetic exposure, P < 0.01.
Current research on anesthetic neurotoxicity in mammals suggests the involvement of mitochondria,14,15 and unquestionably identifies apoptosis as the common pathway for this phenomenon.5 We therefore compared the effects of mutations in mitochondria (gas-1) and the apoptotic pathway (ced-3) on the behavioral effects of early anesthetic exposure. The gas-1 mutants are anesthetic sensitive; ced-3 mutants cannot undergo apoptosis. Synchronized N2, gas-1, and ced-3 worms were exposed to volatile anesthetics during the early L1 stage of development, and then allowed to grow to adulthood. Each worm strain was assayed for chemotaxis on day 1 of adulthood. Untreated animals in all 3 groups had similar chemotaxis indices (N2, 85 ± 2; ced-3, 76 ± 10; gas-1, 71 ± 2). Using new control data for N2 (N2: untreated, 85 ± 2; ISO, 52 ± 10, P = 0.004; SEVO, 47 ± 10, P = 0.003), an even larger decrease in the chemotaxis index was seen in gas-1 animals exposed to volatile anesthetic (gas-1: untreated, 71 ± 2; ISO, 29 ± 12, P = 0.0002; SEVO, 24 ± 13, P = 0.001). In contrast, ced-3 mutants retained their ability to sense and move toward an attractant (ced-3: untreated, 76 ± 10; ISO, 73 ± 9; SEVO, 76 ± 10) (Fig. 3). Consistent with our hypothesis, both wild-type and anesthetic-sensitive (i.e., gas-1) nematodes demonstrated neurological deficits after larval exposure to anesthesia; animals unable to undergo apoptosis did not exhibit impaired chemotaxis after an identical exposure. (Note that the ISO-and SEVO-treated N2 data in these experiments had the same average values as in the larval experiments described above. However, the measurements were done independently. The variation in the individual tests for wild-type controls in the mutant comparisons was greater than in the larval experiments leading to larger standard deviations.)
Figure 3.
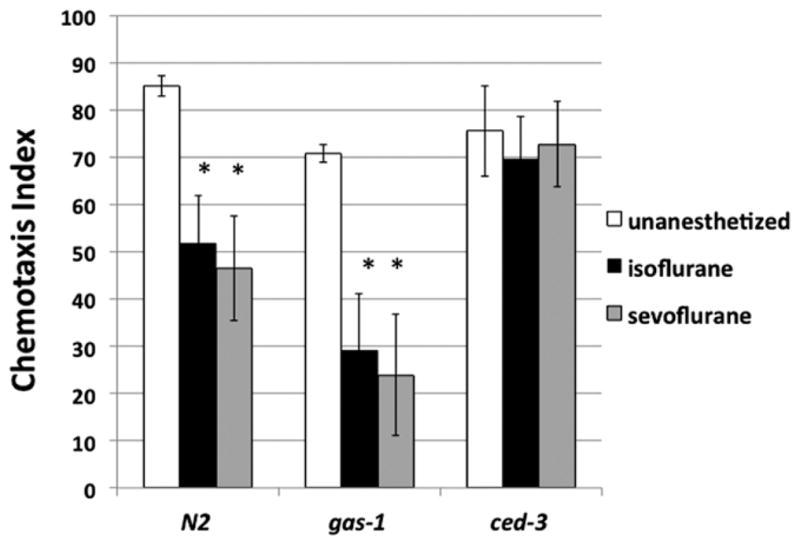
Chemotaxis indices for young adult N2 (wild-type), gas-1, and ced-3 mutants, with and without anesthetic exposure in the first larval stage. Unexposed wild-type animals located the attractant with a chemotaxis index of 85 ± 2 (see Methods section). When N2 animals exposed to anesthetics as larvae reached adulthood, they were defective in this ability. The gas-1 mutant has mitochondrial dysfunction and is exquisitely sensitive to volatile anesthetics. The gas-1 animals exposed to anesthetics had profoundly impaired chemotactic abilities. The ced-3 mutant is incapable of apoptosis, and did not demonstrate changes in chemotaxis after anesthetic exposure. Error bars are standard deviations of the mean. n mt 250 for each value. *Different from control of same genotype, P < 0.025.
DISCUSSION
We have demonstrated that nematodes experience anesthetic-induced neurological damage as evidenced by abnormalities in chemotaxis after a 4-hour larval volatile anesthetic exposure. The period of susceptibility is in the first larval stage, in the first few hours after hatching. Anesthetic-sensitive gas-1 worms exhibited the most marked impairment in chemotaxis after a larval anesthetic exposure. Intriguingly, those worms with a mutation that prevents apoptosis from progressing (ced-3) were apparently protected from the neurotoxic effect of volatile anesthetics. These findings are important for several reasons. That a simple organism, such as the nematode, is susceptible to anesthetic-induced neurotoxicity suggests that the mechanism underlying this phenomenon is ancient. C elegans thus becomes a potential model organism for the further study of anesthetic-related neuronal damage. This prospect is exciting because intrinsic to the nematode are the benefits of relatively simple genetics, a quick and inexpensive method of inducing changes in gene expression via RNA interference, and the rapid production of results due to the animal’s short generation time.
In one of the earliest studies of anesthetic neurodegeneration in rodents, Jevtovic-Todorovic et al.2 demonstrated that exposure to midazolam, nitrous oxide, and ISO, in postnatal day 7 rats, resulted in widespread histological evidence of apoptosis as well as deficits in spatial reference memory when subjected to behavioral testing. In the Morris water maze, exposed young adults had slower rates of memory acquisition than control animals; when retested as adults, exposed animals also demonstrated impaired retention performance.2 Similar results have been obtained in nonhuman primates. Neonatal rhesus monkeys exposed to 24 hours of ketamine demonstrated persistent cognitive defects when tested out to 3 years of age.6 Chemotaxis in C elegans is a highly developed and fundamental neurological process, which requires neuronal complexity and development. The chemosensory organs contain 11 pairs of neurons that express a wide array of chemosensory receptors. Chemosensation facilitates the recognition of food sources, the avoidance of noxious environments, and is relied upon for mating.24 Thus, while the organisms and the assays differ in scale and complexity, we believe that the impaired chemotaxis we have observed in anesthetic-exposed nematodes is analogous to learning and memory deficits seen in rodents and primates exposed to anesthesia at an early age.
Studies in other animal models have shown that apoptosis is the common pathway involved in the neuronal insult incurred by anesthesia, and our results in the ced-3 mutant suggest that this commonality extends to C elegans. We have not yet screened for direct cellular or morphological evidence of apoptosis in nematodes immobilized by volatile anesthetics. However, we have found that animals unable to undergo programmed cell death because of a mutation in ced-3 retain their ability to chemotax after exposure to volatile anesthetics as larvae. This strongly suggests that apoptosis is the critical pathway linking anesthetic exposure to behavioral deficiencies in worms as in mammals. Our results also imply possible therapeutic maneuvers; if apoptosis is blocked, permanent neurological damage can possibly be prevented.
A period of rapid brain growth and synaptogenesis is thought to be a critical factor determining the vulnerability of the nervous system to anesthetic-induced damage. We sought to determine this window of susceptibility for C elegans by anesthetizing N2, ced-3, and gas-1 worms at each developmental stage. Because animals exposed to ISO in the L1 stage had the most striking deficits in chemotaxis, we conclude that L1 is the time when the C elegans nervous system is most vulnerable to the detrimental effects of anesthetics. While not surprising that this vulnerability occurs during a larval stage, it is interesting to put these results in the context of neural development. Neurons comprise approximately one-third of all the somatic cells of the adult animal. The final 302 neurons that comprise the nervous system of the adult hermaphrodite arise from 222 cells that develop during the first half of embryogenesis.25 However, in late L1, most of the ventral nerve cord motor neurons are produced, and cells migrate to assume their place in the cord. Also at this time, existing synapses are modified to accommodate these new neurons.17 When anesthesia is delivered during the L1 phase of development, it may interfere with either neurogenesis or synapse formation.
As noted previously, the mechanisms by which anesthetics initiate apoptosis remain largely unknown. Brain-derived neurotrophic factor has been implicated in the neurotoxicity of nitrous oxide, midazolam, and ISO12; recent work has suggested that another component of the neurotrophin pathway (the p75 neurotrophin receptor) is involved in propofol neurotoxicity.13 Other work focuses on the role of mitochondria: mitochondrial morphology appears to be altered in the neurons and synapses of young rats exposed to anesthetics. The functional consequences of mitochondrial damage might include leakage of mitochondrial proteins such as cytochrome c, increased generation of reactive oxygen species (ROS), and decreased adenosine triphosphate production.14 In a follow-up study of the role of ROS in anesthetic-induced neurotoxicity, Boscolo et al.15 found that coadministration of the ROS scavenger EUK-134 with isoflurane general anesthesia resulted in less mitochondrial damage and neuronal loss compared with the general anesthesia–only group. In our experiments, the marked effect on chemotaxis seen in gas-1 (a mitochondrial mutant already known to be anesthetic sensitive) also lends credence to the hypothesis that mitochondrial damage contributes to anesthetic-induced neurotoxicity.
Clearly, the molecules and mechanisms mediating the developing neuron’s response to anesthesia are just beginning to be elucidated. The remarkable fact that anesthetic exposure in larval nematodes produces behavioral deficits in adult animals underscores the ancient origin of the neurotoxic effects of volatile anesthetics seen in other species. In studies by members of our laboratory, every gene that affects anesthetic sensitivity in C elegans has a close homolog expressed in the nervous system of mammals.26–28 Of note, the apoptotic pathway was first discovered and defined in C elegans, and its components are highly conserved across phylogeny. Therefore, the molecules and mechanisms involved in anesthetic neurotoxicity discovered through genetic studies in the nematode are likely to have correlates in mammals and humans.
Anesthetic neurotoxicity is not just a topic of interest to anesthesiologists and researchers; it is now considered a public health concern, having attracted the attention of the U.S. Food and Drug Administration and the population at large.11 The molecular mechanisms linking anesthetic exposure and neuron cell death must be identified for this phenomenon to be prevented. C elegans is an excellent model organism in which to study this critical issue because it lends itself to rapid and inexpensive genetic approaches. Furthermore, because of its relatively small genome, it can be subjected to whole-genome screens that produce unbiased results and may reveal novel participants in the anesthetic-induced apoptosis pathway. We plan to use RNA interference to sequentially alter expression of genes in the nematode genome and look for changes in susceptibility to anesthetic-induced neurotoxicity. There is also the potential to use green fluorescent protein fusion strains to facilitate detection of apoptosis in vivo. Using approaches such as these, studies in C elegans offer many powerful tools to clarify how early exposure to anesthetics causes neuronal defects.
Acknowledgments
Funding: This work was supported in part by National Institutes of Health grant HD068241, and in part by the Department of Anesthesiology of University Hospitals of Cleveland.
The authors acknowledge the research technicians Beatrice Predoi and Elyce Opheim for their assistance with this project.
Footnotes
The authors declare no conflict of interest.
Reprints will not be available from the authors.
This report was previously presented, in part, at the International Anesthesia Research Society Annual Meeting 2010, and the Society for Pediatric Anesthesia Winter Meeting 2012.
DISCLOSURES
Name: Katherine R. Gentry, MD.
Contribution: This author helped conduct the study and write the manuscript.
Attestation: Katherine R. Gentry has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Louise M. Steele, PhD.
Contribution: This author helped conduct the study and write the manuscript.
Attestation: Louise M. Steele has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Margaret M. Sedensky, MD.
Contribution: This author helped design the study and write the manuscript.
Attestation: Margaret M. Sedensky has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Philip G. Morgan, MD.
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: Philip G. Morgan has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files.
This manuscript was handled by: Marcel E. Durieux, MD, PhD.
References
- 1.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 2.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jevtovic-Todorovic V, Carter LB. The anesthetics nitrous oxide and ketamine are more neurotoxic to old than to young rat brain. Neurobiol Aging. 2005;26:947–56. doi: 10.1016/j.neurobiolaging.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi H, Dikkes P, Soriano SG. Repeated administration of ketamine may lead to neuronal degeneration in the developing rat brain. Paediatr Anaesth. 2002;12:770–4. doi: 10.1046/j.1460-9592.2002.00883.x. [DOI] [PubMed] [Google Scholar]
- 5.Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, Olney JW. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–97. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paule MG, Li M, Allen RR, Liu F, Zou X, Hotchkiss C, Hanig JP, Patterson TA, Slikker W, Wang C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–30. doi: 10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiMaggio C, Sun LS, Kakavouli A, Byrne MW, Li G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J Neurosurg Anesthesiol. 2009;21:286–91. doi: 10.1097/ANA.0b013e3181a71f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sprung J, Flick RP, Wilder RT, Katusic SK, Pike TL, Dingli M, Gleich SJ, Schroeder DR, Barbaresi WJ, Hanson AC, Warner DO. Anesthesia for cesarean delivery and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;111:302–10. doi: 10.1097/ALN.0b013e3181adf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMaggio C, Sun LS, Li G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth Analg. 2011;113:1143–51. doi: 10.1213/ANE.0b013e3182147f42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, Sprung J, Weaver AL, Schroeder DR, Warner DO. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105(suppl 1):i61–8. doi: 10.1093/bja/aeq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–15. doi: 10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 13.Pearn ML, Hu Y, Niesman IR, Patel HH, Drummond JC, Roth DM, Akassoglou K, Patel PM, Head BP. Propofol neurotoxicity is mediated by p75 neurotrophin receptor activation. Anesthesiology. 2012;116:1–10. doi: 10.1097/ALN.0b013e318242a48c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology. 2011;115:992–1002. doi: 10.1097/ALN.0b013e3182303a63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boscolo A, Starr JA, Sanchez V, Lunardi N, Digruccio MR, Ori C, Erisir A, Trimmer P, Bennett J, Jevtovic-Todorovic V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: the importance of free oxygen radicals and mitochondrial integrity. Neurobiol Dis. 2012;45:1031–41. doi: 10.1016/j.nbd.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengartner MO, Horvitz HR. The ins and outs of programmed cell death during C. elegans development. Philos Trans R Soc Lond B Biol Sci. 1994;345:243–6. doi: 10.1098/rstb.1994.0100. [DOI] [PubMed] [Google Scholar]
- 17.Horvitz HR, Shaham S, Hengartner MO. The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol. 1994;59:377–85. doi: 10.1101/sqb.1994.059.01.042. [DOI] [PubMed] [Google Scholar]
- 18.Driscoll M. Molecular genetics of cell death in the nematode Caenorhabditis elegans. J Neurobiol. 1992;23:1327–51. doi: 10.1002/neu.480230919. [DOI] [PubMed] [Google Scholar]
- 19.Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7:97–108. doi: 10.1038/nrm1836. [DOI] [PubMed] [Google Scholar]
- 20.Chalfie M, White J. The nervous system. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. pp. 380–3. [Google Scholar]
- 21.Bargmann CI, Horvitz HR. Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron. 1991;7:729–42. doi: 10.1016/0896-6273(91)90276-6. [DOI] [PubMed] [Google Scholar]
- 22.Kayser EB, Morgan PG, Sedensky MM. GAS-1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology. 1999;90:545–54. doi: 10.1097/00000542-199902000-00031. [DOI] [PubMed] [Google Scholar]
- 23.Morgan PG, Sedensky MM, Meneely PM. Multiple sites of action of volatile anesthetics in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1990;87:2965–9. doi: 10.1073/pnas.87.8.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bargmann CI. WormBook: The Online Review of C elegans Biology. Pasadena, CA: Wormbook; 2005. Chemosensation in C. elegans created October 25, 2006. Available at: http://www.ncbi.nlm.nih.gov/books/NBK19746/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White JG, Horvitz HR, Sulston JE. Neurone differentiation in cell lineage mutants of Caenorhabditis elegans. Nature. 1982;297:584–7. doi: 10.1038/297584a0. [DOI] [PubMed] [Google Scholar]
- 26.Kayser EB, Morgan PG, Sedensky MM. Mitochondrial complex I function affects halothane sensitivity in Caenorhabditis elegans. Anesthesiology. 2004;101:365–72. doi: 10.1097/00000542-200408000-00017. [DOI] [PubMed] [Google Scholar]
- 27.Falk MJ, Kayser EB, Morgan PG, Sedensky MM. Mitochondrial complex I function modulates volatile anesthetic sensitivity in C. elegans. Curr Biol. 2006;16:1641–5. doi: 10.1016/j.cub.2006.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphrey JA, Hamming KS, Thacker CM, Scott RL, Sedensky MM, Snutch TP, Morgan PG, Nash HA. A putative cation channel and its novel regulator: cross-species conservation of effects on general anesthesia. Curr Biol. 2007;17:624–9. doi: 10.1016/j.cub.2007.02.037. [DOI] [PubMed] [Google Scholar]