

Abstract
Androgen receptor (AR) signaling is essential for prostate cancer (PCa) development in humans. The initiation of prostate malignancy and progression to a castration-resistant stage are largely contributed by the modulation of AR activity through its coregulatory proteins. We and others previously reported that p14 alternative reading frame (ARF) expression is positively correlated with the disease progression and severity of PCa. Here, we provide evidence that p14ARF physically interacts with AR and functions as an AR corespressor in both an androgen-dependent and androgen-independent manner. Endogenous ARF (p14ARF in human and p19ARF in mouse) and AR colocalize in both human PCa cells in vitro and PCa tissues of mouse and human in vivo. Overexpression of p14ARF in PCa cells significantly attenuates the activities of androgen response region (ARR2)-probasin and prostate-specific antigen (PSA) promoters. The forced expression of p14ARF in cells resulted in a suppression of PSA and NK transcription factor locus 1 (NKX3.1) expression. Conversely, knockdown of endogenous p14ARF in human PCa cells with short hairpin RNA enhanced AR transactivation activities in a dose-dependent and p53-independent manner. Furthermore, we demonstrated that p14ARF binds to both the N-terminal domain and the ligand-binding domain of AR, and the human double minute 2 (HDM2)-binding motif of p14ARF is required for the interaction of p14ARF and AR proteins. p14ARF perturbs the androgen-induced interaction between the N terminus and C terminus of AR. Most importantly, we observed that the expression of PSA is reversely correlated with p14ARF in human prostate tissues. Taken together, our results reveal a novel function of ARF in modulation of AR transactivation in PCa.
Prostate cancer (PCa) is the second leading cause of cancer-related deaths in American men (1). Patients with advanced PCa are usually treated by androgen-depletion therapy to block the function of androgen receptor (AR). Despite the initial effectiveness, a majority of patients still died of the recurrence of castration-resistant PCa (CRPC) (2, 3). The aberrant AR signaling, caused by altered AR expression or AR coregulators, is thought to drive the development and progression of PCa including CRPC (4–6).
AR, a member of the nuclear steroid receptor family, is a hormone-dependent transcription factor that regulates multiple genes for male development as well as the initiation and progression of PCa (5, 6). In the literature, there are reports that increased AR levels by gene amplification occur in about 20% of CRPC cases (3, 7, 8). Mutations and variants of AR also contribute to CRPC by their increased sensitivity to low levels of androgen, decreased sensitivity to antiandrogens (9, 10), or loss of the ligand-binding ability (4, 11). A number of AR coregulators, including coactivators and corepressors, have been identified, and their alterations have been postulated to contribute to CRPC (4–6, 12). Unlike coactivators that enhance AR-mediated gene transcription, AR corepressors function to attenuate AR transactivation through remodeling chromatin structure, posttranslational modifications of AR including acetylation, sumoylation, ubiquitylation, or other undefined mechanisms (13–16). Emerging evidence shows that AR signaling is down-regulated in metastasis compared with primary PCa, and cancer cells are likely surviving with minimal androgen signaling upon deprivation therapy (17, 18). Recent results in mouse models revealed that epithelial cells with a decreased AR show a higher proliferation than those with an elevated AR (19–21).
ARF (alternative reading frame) is an alternative transcript of the INK4α/ARF gene locus that concomitantly encodes another product, p16Ink4a, an inhibitor of cyclin-dependent kinases (22). Despite the same gene locus, ARF (p14ARF in human and p19ARF in mouse) is transcribed independently from p16Ink4a due to the unique characteristic of the first exon (23). ARF acts as a tumor suppressor because p19ARF-deficient mice display susceptibility to sarcomas, lymphomas, and pulmonary and mammary adenocarcinomas (24). ARF stabilizes p53 protein by antagonizing the function of mouse double minute 2 (MDM2), the E3 ubiquitin ligase of p53, and sequestering MDM2 in the nucleolus in proliferating cells (24, 25). Elevated p53 results in cell cycle arrest, cell death, or senescence in response to DNA damage and oncogenic stress. ARF displays its tumor suppressor role both in a p53-dependent and p53-independent manner (26–28), which may partly be attributed to its repression of ribosome biogenesis (29).
More than 30 ARF interacting partners have been identified, but roles of p14ARF in cancers remain poorly understood (22). Loss of ARF is frequently found in various cancers such as sarcoma, lymphoma, and melanoma; nonetheless, ARF up-regulation is reported in human PCa, and its increased level is associated with advanced and metastatic stages (28, 30). Recent studies revealed that ARF loss suppresses the oncogenesis of tumors, at least in some tissues including prostate (28, 31). In this study, we hypothesized that ARF regulates AR activity through a direct interaction that may contribute to the development and progression of PCa. Indeed, we demonstrate that ARF physically interacts with AR, and overexpression of ARF inhibits AR transactivation as well as the expression of AR target genes PSA and NKX3.1 in PCa cells. Conversely, knockdown of ARF enhances AR transactivation of reporter genes and revokes the inhibition of ARF on the N-C interaction of AR in PCa cells. The expression of ARF is reversely correlated with AR activity in human PCa tissue.
Materials and Methods
Constructs of AR mutants and p14ARF short hairpin RNA (shRNA)
Five constructs for deletion mutants of AR were generated to include the N-terminal domain (NTD), NTD plus DNA-binding domain (DBD), DBD plus hinge (H), DBD H plus ligand-binding domain (LBD), and LBD functional domains, respectively. cDNAs encoding AR mutants were amplified by PCR with pCMV3.1–3xFlag-hAR plasmid containing a full-length cDNA of human AR (32). Primer sequences are listed in Supplemental Table 1 (published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). cDNAs of AR mutants were ligated into p3xFlag-CMV10 vector at BglII and BamHI or NotI and BamHI sites using Quick Ligase kit (NEB, Ipswich, Massachusetts). AR mutants were verified by bidirectional sequencing with primers. To generate p14ARF shRNA plasmids, forward and reverse oligonucleotides (Supplemental Table 1) were suspended separately in ddH2O to a concentration of 3 mg/mL, and then 1 μl of each oligonucleotide was added to a volume of 48 μl annealing buffer containing 10mM Tris-HCl (pH 7.8), 50mM NaCl, and 1mM EDTA. The mixture was incubated at 94°C for 4 minutes, 85°C for 4 minutes, 75°C for 4 minutes, 70°C for 4 minutes, and room temperature for 1 hour. Annealed inserts were ligated into pSuper-retro-puro expression vectors at BglII and HindIII sites.
Cell culture, transfection, and short hairpin RNA and RNA interference knockdown
LNCaP, C4-2B (M.D. Anderson, Houston, Texas), CWR22Rv1, and PC3 (American Type Culture Collection, Manassas, Virginia) human PCa cells were grown in RPMI 1640 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C with 5% CO2. HEK293T and Phoenix-Ampho cells were maintained in DMEM complemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. For transient transfection, cells at 70% confluence were transfected with plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, California). To investigate AR-mediated responses, cells were first grown in medium containing 10% charcoal-dextran–stripped FBS (Invitrogen) for 24 hours and then treated with 10 nM dihydrotestosterone (DHT) for 24 hours. Expression of target genes was determined 48 to 72 hours after transfection using quantitative real-time PCR or immunoblotting.
To knock down p14ARF in cells, retroviruses containing p14ARF short hairpin RNA (shRNA) were prepared with Phoenix-Ampho packing cells (Orbigen, San Diego, California; RVC-10001) using p14ARF shRNA constructs. Briefly, Phoenix cells in 10-cm lysine-coated culture plates were transfected with 10 μg p14ARF scrambled or p14ARF shRNA plasmids using Lipofectamine 2000. Forty-eight hours after transfection, retroviral supernatants were collected and filtered through a 0.45-μm filter. Fresh shRNA retroviruses were applied to PC3 cells containing 5 μg/mL Polybrene. Infected cells were grown for an additional 48 hours and selected with medium containing 2 μg/mL puromycin for 3 days, and selected cells were then pooled for analysis.
To ablate ubiquitin ligase for ARF (ULF) in C4-2B cells, cells were transfected with small interfering RNA duplex oligonucleotides ULF-RNA interference (RNAi) 5′-GGUAGUGACUCCACCCAUUUU-3′ (33) synthesized by Dharmacon (Lafayette, Colorado), and control RNAi (Control-siRNA-A; Santa Cruz Biotechnology, Santa Cruz, California) with Lipofectamine 2000 according to manufacturer's instructions. RNAi transfections were performed two times at 24-hour intervals, and cell lysates were collected for protein analysis 48 hours after the second transfection.
Coimmunoprecipitation and Western blotting
To precipitate Flag-tagged AR and GFP/Myc-tagged p14ARF, HEK293T cells were transfected with the empty vector, pCMV3.1-3xFlag-AR, GFP-p14ARF-pcDNA4/TO, or pcDNA3-Myc3-p14ARF expressing plasmids. Two days after transfection, cells were harvested in RIPA buffer (1× PBS [pH 7.4], 2 mM EDTA, 1% Triton X-100, 0.5% or 0.25% sodium deoxycholate, and protease inhibitor). Cell lysates were sonicated briefly and centrifuged at 13 200 rpm for 30 minutes. The supernatant was precleared with 100 μl mouse IgG or 100 μl protein A/G plus agarose beads and then immunoprecipitated with the following antibodies: 1 μg mouse anti-GFP (Invitrogen), 3 μg mouse anti-C-Myc (9E10; Santa Cruz), and 1 or 3 μg normal mouse IgG (Sigma Chemical Co, St Louis, Missouri) for a negative control, all in combination with 30 μl protein A/G plus agarose or 30 μl mouse anti-Flag M2 agarose gel (Sigma) at 4°C for 16 hours, respectively. Antibodies used for Western blotting were mouse anti-Flag M2 (Sigma), living color mouse anti-GFP (Clontech, Palo Alto, California), rabbit anti-AR (N-20; Santa Cruz), and mouse anti-p14ARF (MS850P1; Thermo Scientific, Pittsburgh, Pennsylvania), and horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Piscataway, New Jersey). Immunoblot signals were detected by ECL plus Western blotting reagents (GE Healthcare).
To map AR-binding sites to ARF, HEK293T cells were cotransfected with Myc-p14ARF plasmids and individual AR mutants, p3xFlag-CMV10-hAR-NTD, p3xFlag-CMV10-hAR-NTD-DBD, p3xFlag-CMV10-hAR-DBD-H, p3xFlag-CMV10-hAR-DBD-H-LBD, and p3xFlag-CMV10-hAR-LBD, respectively. Similarly, to locate p14ARF-binding sites to AR, cells were cotransfected with pCMV3.1-3xFlag-AR and pcDNA3-Myc3-p14ARF (full-length ARF), or individual ARF mutants, pcDNA3-Myc3-p14ARF-ΔN64, pcDNA3-Myc3-p14ARF-ΔN45, pcDNA3-Myc3-p14ARF-ΔN30, and pcDNA3-Myc3-p14ARF-ΔN15, respectively. Two days after transfection, cells were harvested in modified immunoprecipitation buffer (1× PBS [pH 7.4], 2 mM EDTA, 0.5% Triton X-100, 0.25% sodium deoxycholate, and protease inhibitor). Antibodies for coimmunoprecipitations are mouse anti-C-Myc (9E10; Santa Cruz) and mouse anti-Flag M2 agarose.
Immunofluorescence and immunohistochemistry staining
LNCaP cells grown on coverslips were cultured in starvation medium for 24 hours. Cells were stimulated for 5 hours with 200 ng/mL IGF-I in the presence or absence of 10 nM DHT, fixed with methanol at −20°C for 15 minutes, and then incubated with rabbit anti-AR (N-20, 1:200; Santa Cruz) or rabbit anti-Fibrillarin (C13C3, 1:400; Cell Signaling Technology, Danvers, Massachusetts) and mouse anti-p14ARF (4C6/4, 1:200; Cell Signaling) for 1 hour at room temperature. After washing, cells were incubated with donkey anti-rabbit Alexa Fluor 488 (Invitrogen; 1:1000) and goat anti-mouse IgG Alexa Fluor 568 (Invitrogen; 1:1000) for 30 minutes. Nuclei were counterstained with diamidino-2-phenylindole (DAPI) (Invitrogen) at a final concentration of 1 μg/mL for 10 minutes. Stained slides were mounted with Vectashield antifade aqueous medium (Vector Laboratories, Burlingame, California) and sealed with nail polish. Images were recorded with Nikon A1R confocal microscopy.
Immunofluorescence staining of ARF and AR in mouse and human PCa tissues was performed with paraffin or cryosections in 5 μm thickness. Sections were incubated in 3% H2O2, boiled in antigen retrieval citrate buffer [pH 6.0] for 15 minutes, and blocked with 10% FBS in 1× PBS containing 0.5% Triton X-100 for 30 minutes. Primary antibodies were rabbit anti-AR (N-20, 1:50; Santa Cruz) and mouse anti-p14ARF (MS850P1, 1:25; Thermo Scientific) or rat anti-p19ARF (5-C3–1, 1:100; Santa Cruz). Slides were incubated with primary antibodies in 1× PBS containing 1% BSA and 0.5% Triton X-100 overnight at 4°C and then incubated with secondary antibodies Alexa Fluor 568 (1:3000) and Alexa Fluor 488 (1:2000) in 1× PBS containing 1% BSA and 0.5% Triton X-100 for 1 hour at room temperature. Sections were counterstained with TO-PRO-3 (1:1000) or 1 μg/mL DAPI to visualize nuclei.
Human prostate tissue microarray slides were obtained from Biomax (Rockville, Maryland), consisting of 40 cases in 80 cores. Tissue sections were deparaffinized and rehydrated in alcohol and then treated with 3% H2O2 and antigen retrieval buffer in the same procedure of immunofluorescence staining. Anti-p14ARF (4C6/4, 1: 50; Cell Signaling), anti-AR (N-20, 1:200) and anti-PSA (A67-B/E3, 1:100; Santa Cruz) were used for immunohistochemistry (IHC) staining. Sections were incubated with biotinylated secondary antibodies, and the immune complex was visualized with ABC kit using the chromogen diaminobenzidine substrate (Vector). Nuclei were counterstained with Mayer's hematoxylin solution (Sigma). The scores of ARF, AR, and PSA staining were graded as 0 (negative staining), 1 (weak staining), 2 (moderate staining), or 3 (strong staining) according to their intensities. The percentage scores of their staining were graded as 0 (no cells stained), 1 (<10% cells stained), 2 (10%–50% cells stained), and 3 (>50% cells stained) (16).
Dual-luciferase reporter assay
LNCaP, C4-2B, and CWR22Rv1 cells were grown in 24-well plates and transfected with 2.5 ng pRL-CMV Renilla reporter, 200 ng ARR2-PB-Luc or pGL3-PSA-Luc reporter, and pcDNA3-Myc3-p14ARF or pCI-neo-p14ARF plasmids. For PC3-scramble and PC3-shARF cells, cells in 24-well plates were transfected with 2.5 ng pRL-CMV Renilla reporter, 200 ng ARR2-PB-Luc reporter, 200 ng pCMV3.1-3xFlag-AR, and GFP-p53 or p53 mutant plasmids. For each transfection, the total amounts of DNA were equalized with pcDNA3.1 or pCI-neo vector plasmid. Twenty-four hours after transfection, the medium was replaced with the androgen starvation medium and treated with 10 nM DHT for 24 hours. To test the effects of ARF on the interaction between the N and C termini of AR, PC3-scramble, and PC3-shARF cells were transfected with Renilla and ARR2-PB-Luc reporters, and 200 ng of the N and/or C terminus of AR mutant plasmids. For HEK293T cells, 200 ng p14ARF plasmid were cotransfected with the same set of reporter plasmids for PC3-shARF cells. Luciferase activities in cell lysates were measured with a standard dual-luciferase reporter assay system (Promega, Madison, Wisconsin) and normalized by the Renilla activities. The luciferase activity relative to the untreated control was determined, and results are presented as means (± SD) of triplicates.
Real-time RT-PCR
Total RNAs were extracted from transfected LNCaP, C4-2B, and CWR22Rv1 cells using TRIZOL reagent (Invitrogen). Five micrograms of total RNA were used to prepare cDNAs by reverse transcription using SuperScript III first-strand synthesis kit (Invitrogen). Real-time quantitative PCR was performed with a Bio-Rad (Hercules, California) CFX96 real-time system in triplicates in a 20 μl reaction volume consisting of 10 μl 2× SYBR Green PCR Master Mix (Applied Biosystems, Foster City, California), 2 μl cDNAs (for comparative cycle threshold [CT] method) or 8 μl of a 1:20 dilution of cDNAs (for standard curve method), and 0.5μM of each primer for each sample, using the forward and reverse primers listed in Supplemental Table 1. Standard curves were generated using pooled cDNAs and a 4-fold dilution series (diluted from 1:1 to 1:256) for each pair of primers. An aliquot of each sample was used as PCR templates to obtain the CT. The level of target mRNA was determined either by a standard curve and then normalized with β-actin or by comparative CT method (ΔΔCT). The relative fold change of target mRNA expression was presented as a ratio of the level of target mRNA in treated cells to that in untreated control cells.
Statistical analysis
Statistical analyses were conducted using either Student's t test or χ2 test, except that correlation analysis was performed using Pearson correlation test, and the values of P < .05 were considered as statistically significant.
Results
ARF physically interacts with AR
AR has substantial coregulators contributing to molecular mechanisms of PCa (5, 6), and ARF physically interacts with more than 30 partners (22). Most importantly, both ARF and AR are remarkably up-regulated in PCa (8, 28, 30). Therefore, we reasoned that ARF may impact the development and progression of PCa by regulating AR activity through a direct interaction. To test this hypothesis, HEK293T cells were cotransfected with Flag-tagged human AR and GFP-p14ARF or Myc-p14ARF plasmids, and coimmunoprecipitation was performed using anti-Flag, anti-C-Myc, or anti-GFP antibody. As a result, ARF protein was indeed detected in the immunoprecipitates with anti-Flag when cells were cotransfected with both AR and ARF plasmids, whereas no coimmunoprecipitation between AR and ARF was found in cells cotransfected with vectors, Myc-ARF (Figure 1A), or GFP-ARF plasmid alone (Supplemental Figure 1A). To verify this interaction, reciprocal immunoprecipitations were performed using anti-Myc or anti-GFP antibody, respectively. As expected, AR protein was detected in the immunoprecipitates with anti-Myc or anti-GFP (Figure 1B and Supplemental Figure 1B). These results provided the biochemical evidence of a physical interaction between ARF and AR proteins in cells.
Figure 1.

ARF physically interacts with AR. A, Coimmunoprecipitation of ARF and AR. HEK293T cells were cotransfected with Flag-AR and/or Myc-p14ARF plasmids. Cell lysates were immunoprecipitated with anti-Flag and blotted with anti-p14ARF or anti-C-Myc. B, A reciprocal immunoprecipitation with anti-C-Myc antibody. Lysates of cells transfected with empty vector, Flag-AR, or Myc-p14ARF plasmids were used as negative controls for A and B. Normal mouse IgG was used as an antibody control. C, Constructs of AR deletion mutants. D, ARF interacts with AR at specific domains and motifs. p14ARF is recruited to N and C termini of AR. The overexpressed AR mutants are indicated as arrowheads. The heavy chain (top) and light chain (bottom) of the antibody are indicated with arrows. AR mutant plasmids were cotransfected with pcDNA3-Myc-p14ARF into HEK293T cells, respectively. After 48 hours, cell lysates were immunoprecipitated using anti-Myc or normal mouse IgG antibodies (Supplemental Figure 1C). E, Constructs of N-terminal deletion mutants of ARF. F, The N-terminal motif of p14ARF binds to AR. The full-length ARF and mutants of ARF were individually coexpressed with Flag-AR in HEK293T cells. After 48 hours, cell lysates were immunoprecipitated with anti-Flag and anti-C-Myc antibody, and proteins were analyzed by Western blotting.
ARF interacts with the N and C termini of AR
To define the interacting regions of ARF and AR, we first mapped the interaction domains of AR protein required for its binding to ARF protein using AR mutants. Flag-AR deletion mutants (Figure 1C), including ARNTD (1–551), ARNTD+DBD (1–640), ARDBD+H (552–667), ARDBD+H+LBD (552–917), and ARLBD (668–917), were generated from human AR (32). Each AR mutant was cotransfected with Myc-p14ARF in HEK293T cells, and then coimmunoprecipitations were performed with anti-Myc antibody. All AR mutants expressed truncated proteins in predicted molecular weights (Figure 1D, arrowheads). It should be noted that ARNTD and ARNTD+DBD displayed some bands with reduced molecular sizes, which were probably degraded AR mutants. The protein level of p14ARF was unexpectedly lower when coexpressed with ARNTD+DBD as compared with other AR mutants. We also found that all AR fragments including ARNTD, ARNTD+DBD, ARDBD+H+LBD, and ARLBD, but ARDBD+H, displayed the interaction with ARF (Figure 1D), in the same manner as the full-length AR (Figure 1A). As shown in controls, AR mutants were not immunoprecipitated by normal mouse IgG in the presence of ARF overexpression or by mouse anti-C-Myc antibody in the absence of Myc3-p14ARF overexpression (Supplemental Figure 1, C and D). These data suggest that both NTD and C-terminal LBD of AR are responsible for AR binding with ARF.
N-terminal human double minute 2 (HDM2)-binding motif of ARF is required to interact with AR
Literatures report that two N-terminal motifs of ARF, A1 (3RRFLVTLR10) and A2 (21RVFVVHIPR28), interact with oncoprotein HDM2 (34). A LXXLL motif-like sequence, 45VLMLL50, is found in the primary amino acids of ARF. The LXXLL has been identified as the interactive motif of some AR coregulators such as cAMP-response element-binding protein steroid receptor coactivator 1 and receptor interacting protein 140 (35). Therefore, we investigated whether these ARF motifs (or motif-like sequence) also played an important role in ARF-AR binding. We coexpressed the full-length ARF (Myc-ARF) or Myc-ARF deletion mutants (Figure 1E) with AR in HEK293T cells and performed coimmunoprecipitation using anti-AR and anti-Myc antibodies, respectively. As shown in Figure 1F, the expression of full-length ARF and ARF mutants was detected, but ARFΔN64 unexpectedly showed an increased molecular mass relative to ARFΔ45 as previously reported, which is likely attributed to protein modifications (36). The reciprocal immunoprecipitation results revealed that only ARFΔN15, not other deletion fragments ARFΔN30, ARFΔN45, or ARFΔN64 of ARF, retains the binding capacity to AR and mimics the activity of the full-length ARF (Figure 1F). Based on the location of the A2 motif (21RVFVVHIPR28) of ARF within the N-terminal region of ARFΔN15 and the observation that fragments deficient of A2 motif lose the ability to bind AR, we concluded that the HDM2-binding motif of ARF is essential for the interaction of ARF and AR proteins. Taken together, these results indicate that ARF interacts with the N and C termini of AR, and the N-terminal HDM2-binding motif of ARF is required to interact with AR.
ARF colocalizes with AR in PCa cells in vitro and prostate tumors in vivo
AR activation requires several steps from the agonist stimulation, translocation to nucleus, and binding to androgen-responsive elements in the promoter regions of target genes. We asked ourselves whether the ARF-AR interaction occurs intrinsically in PCa cells in vitro and prostate tumors in vivo. To elucidate the relevance of ARF and AR interaction in cancer cells, we explored the colocalization of ARF and AR in LNCaP cells. Because LNCaP cells do not show a detectable level of endogenous ARF, we stimulated them with IGF-I after starvation in the presence or absence of DHT (37). The immunofluorescence results demonstrated that the endogenous ARF protein was excluded from nucleoli upon IGF-I stimuli as indicated by Fibrillarin, a nucleolar marker (Figure 2A, bottom panel). This notion suggests that ARF and AR predominantly colocalize in the nucleoplasm of LNCaP cells after IGF-I induction (Figure 2A, top panel), which is not dependent upon the addition of DHT to the culture (Supplemental Figure 5).
Figure 2.

Endogenous ARF and AR colocalize in PCa cells in vitro and PCa tissues in vivo. A, Immunofluorescence images of AR (green, anti-AR), Fibrillarin (green, anti-Fibrillarin), and ARF (red, anti-p14ARF) staining in LNCaP cells. Cells were starved, induced by IGF-I, and then treated with DHT. B, Prostate tissue sections of Pten/p53 mice were immunostained with anti-AR (red) and anti-p19ARF (5-C3-1) (green). C, Sections of human PCa tissues were immunostained with anti-AR and anti-p14ARF, and nuclei were counterstained with DAPI. The colocalization (yellow) of ARF and AR was observed in prostate tissues from mouse and human. Scale bars, 5 μm (A, top panel), 10 μm (A, lower panel), 20 μm (B), and 10 μm (C).
We then investigated ARF-AR interaction in vivo by examining their colocalization in PCa tissues from mouse and human. Because both p19ARF and AR are up-regulated in prostates of Pten/p53 conditional null mice, we performed the staining of ARF and AR in prostate cryosections of Pten/p53 mice. The results showed that p19ARF and AR proteins in mouse prostate tumors were predominantly localized in the nucleus of cells. Notably, p19ARF and AR displayed their colocalization in nucleus of tumor cells (Figure 2B). This finding encouraged us to examine the ARF-AR interaction in human PCa specimens. Consistent with the literature and results from mouse tissues, p14ARF was found in the nucleus (28), and AR was predominantly detected in nucleus of epithelial cells (8). Most importantly, the intensive colocalization of ARF and AR proteins was observed in the most nuclei of cancerous epithelial cells (Figure 2C and Supplemental Figure 2). These results provide essential evidence in vivo to support that ARF and AR proteins are making physical contacts in malignant cells, implicating a vital role in PCa progression.
Endogenous ARF inhibits the expression of AR downstream target PSA
It is reported that ULF is an E3 ligase for p14ARF, and ULF suppression results in p14ARF accumulation in cells (33). To further examine the physiological relevance of the endogenous interaction of ARF and AR, we tested the regulatory effects of ARF alteration on the expression of AR downstream target PSA in C4-2B cells upon ULF knockdown. As shown in Supplemental Figure 2, ULF knockdown led to a significant increase of the protein level of p14ARF in both a DHT-dependent and -independent manner. Strikingly, the elevation of PSA protein was dramatically suppressed upon p14ARF induction after ULF knockdown as compared with the control when cells were treated with DHT, whereas their AR levels remain unchanged. This result further suggests that AR activity is inhibited by ARF at the endogenous level in PCa cells.
Overexpressed ARF inhibits AR transactivation
We next evaluated the biological effects of ARF-AR binding on AR transactivation. To do so, LNCaP, C4-2B, and CWR22Rv1 cells were cotransfected with ARF expression plasmids or empty vectors, along with ARR2-probasin promoter-luciferase (ARR2-PB-Luc) reporter or PSA promoter-luciferase (PSA-Luc) reporter, two reporters commonly used to study AR action. After transfection, cells were treated with 10 nM DHT, and cell lysates were collected to determine luciferase activities. Both reporters indicated that ARF overexpression results in about 2- to 6-fold attenuation of AR transcriptional activities in both androgen-sensitive LNCaP cells and androgen-insensitive C4-2B and CWR22Rv1 cells (Figure 3). In addition, this attenuation caused by ARF was regulated in a dose-dependent manner, independent of AR promoter type and DHT induction (Figure 3, A–F, and Supplemental Figures 3 and 4, A–F). To understand whether the protein level of AR was also regulated by ARF, cell lysates were subjected to Western blotting. As shown, ARF overexpression did not result in significant changes of AR protein level as compared with controls (Supplemental Figure 4, G–L). Results indicate that the decrease of AR transactivation by ARF is not caused by a reduced level of AR protein. Taken together, these results suggest that ARF functions as a repressor in regulating AR transactivation in PCa cells.
Figure 3.
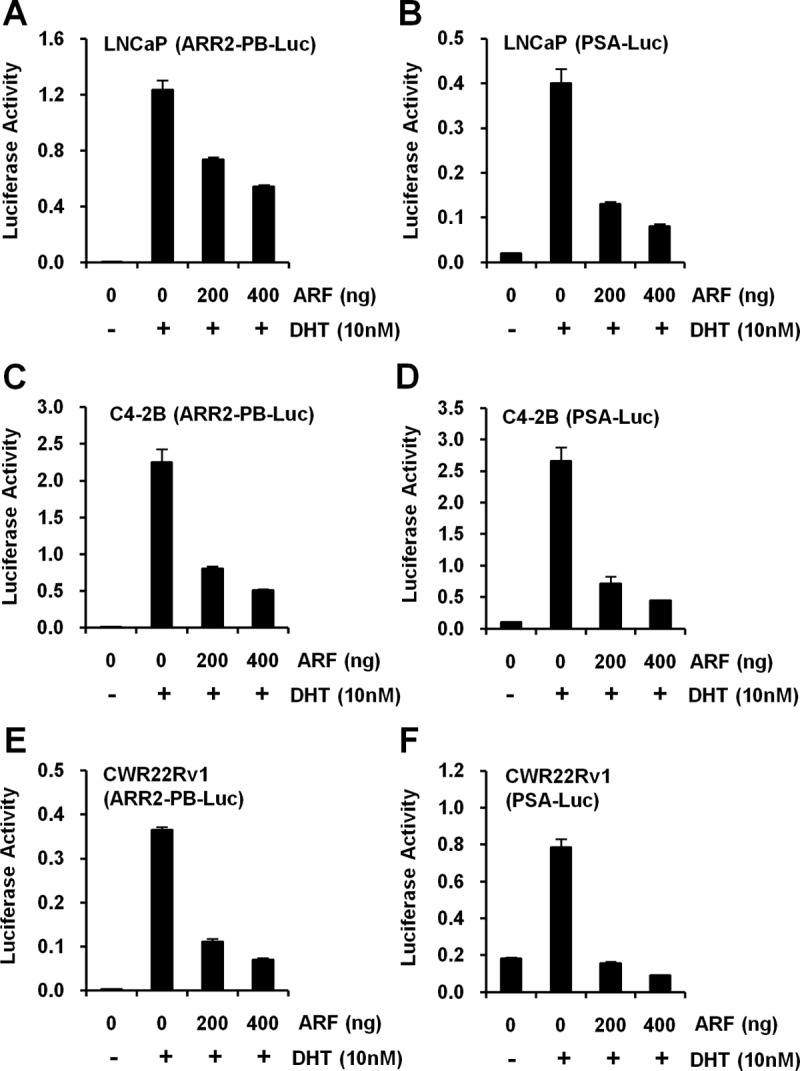
ARF inhibits AR-mediated transactivation. A and B, Effects of p14ARF expression on AR activity in LNCaP cells. C and D, Effects of p14ARF expression on AR activity in C4-2B cells. pcDNA3-Myc3-p14ARF plasmid, ARR2-PB-Luc (A), and pGL3-PSA-Luc (B) reporters were used. E and F, Effects of p14ARF expression on AR activity in CWR22Rv1 cells. pCI-neo-p14ARF plasmid was used. Eighteen hours after transfection, cells were replaced with androgen starvation medium and treated with 10nM DHT or vehicle control (Supplemental Figure 3) for 24 hours before harvesting. The luciferase activities (firefly luciferase light unit/Renilla luciferase) were determined and presented as means (± SD) of triplicates. The levels of AR and p14ARF proteins in lysates were analyzed by Western blotting using anti-AR and anti-C-Myc or anti-p14ARF antibodies (Supplemental Figure 4).
ARF down-regulates PSA and NKX3.1 expression in PCa cells
Next, we investigated the functional consequence of ARF elevation in modulating AR-driven gene expression. First, we transiently overexpressed ARF in LNCaP cells and then examined the level of endogenous PSA and NKX3.1, two AR target genes. Quantitative real-time PCR analysis showed that ARF overexpression resulted in a decrease of PSA and NKX3.1 transcription in response to DHT (Figure 4, A–C, left panel), whereas no PSA reduction was found in the DHT negative control. Similarly, this ARF-induced down-regulation of PSA and NKX3.1 mRNA was also observed in C4-2B and 22Rv1 cells upon DHT treatment (Figure 4, A–C, middle and right panels). We also found that the ectopic expression of p14ARF results in an inhibition of PSA and NKX3.1 at the mRNA level even in the absence of DHT in CWR22Rv1 cells but not in C4-2B cells. This is likely caused by the altered biological features of cancer cell lines that may exhibit different sensitivities to ARF. The relatively small reduction of PSA and Nkx3.1 expression after ectopic ARF expression is likely contributed by a combined effect of a small percentage of cells transfected (∼10%–20%) and a marginal overlapping of ARF and AR proteins in cells. These results further confirmed that ARF functions as an AR corepressor to attenuate the expression of AR target genes.
Figure 4.

ARF suppresses PSA and NKX3.1 expression in human PCa cells. A, Relative expression of p14ARF in LNCaP, C4-2B, and CWR22Rv1 cells. B, Changes of PSA by ARF expression in LNCaP, C4-2B and CWR22Rv1 cells. C, Changes of NKX3.1 by ARF expression in LNCaP, C4-2B, and CWR22Rv1 cells. Cells were transfected with ARF plasmid (pCI-neo-p14ARF). After 18 hours, cells were replaced with androgen starvation medium and cultured in 10nM DHT for an additional 24 hours. The total RNA from cells was submitted to reverse transcription, followed by quantitative real-time PCR using SYBR Green dye. The relative mRNA levels of PSA and NKX3.1 are means (± SD) of triplicates.
Knockdown of endogenous ARF enhances AR activities in PCa cells
ARF interacts with more than 30 cofactors (22, 25, 38) with a distinct function to stabilize p53 protein by antagonizing the action of MDM2. Aberrant levels of p53 by induction or inactivation led to an inhibition of AR transactivation in PCa cells (39). This notion led us to reason that ARF down-regulation would result in an increased AR activity in a p53-dependent manner. We chose to use PC3 cells due to their high level of expression of endogenous p14ARF and their deficiency of p53, which allows us to assess the biological consequence of ARF knockdown. We first generated PC3 cells with p14ARF knockdown by shRNA (referred as PC3-shARF cells). As shown in Figure 5B, the ARF level was strikingly reduced upon ARF knockdown in PC3-shARF cells as compared with the control. AR activities were reversely correlated with the levels of p14ARF protein in PC3-scramble and PC3-shARF cells, indicating AR is regulated by ARF in a dose-dependent manner (Figure 5, A–C). We next sought to investigate whether the ARF-regulated AR activity is p53-dependent. Restoration of wild-type p53 in both PC3-scramble and PC3-shARF cells significantly decreased AR activities in a dose-dependent manner as compared with controls. This result is consistent with a previous report that p53 overexpression inhibits AR activities. However, p53 restoration failed to completely suppress the up-regulation of AR activities in the context of ARF knockdown (Figure 5, D and E), indicating ARF regulates AR activity in a manner different from p53. We further tested whether p53 mutant (V274L) had any effects on AR activities contributed by ARF knockdown. Our results showed that mutated p53 exhibited a less striking counteracting effect on the up-regulation of AR activities in the context of p14ARF loss as compared with wild-type p53 (Figure 5, D and E). Taken together, these data suggest that the attenuation of AR activities by p14ARF is regulated both in a p53-dependent and p53-independent manner.
Figure 5.

ARF knockdown enhances AR-mediated transactivation of ARR2-PB-Luc reporter gene in PC3 cells. A, Effects of p14ARF knockdown on AR activity in PC3 cells. PC3-scramble and PC3-shARF cells were transfected with ARR2-PB-Luc reporter, pRL-CMV reporter, and Flag-AR expression plasmids. B, Western blot results of p14ARF knockdown by shRNA in PC3 cells of A. C, AR protein level upon ARF knockdown. D, Effects of p53 restoration on AR activity in PC3 cells. PC3-scramble and PC3-shARF cells were transfected with the same set of reporter and AR plasmids as A, and 50 or 200 ng p53 wild-type, 200 ng p53 mutant, or 200 ng pcDNA3.1 as indicated. pcDNA3.1 was used to equalize the total amount of DNA for transfections. E, Western blot results on the levels of AR, p14ARF, and p53 proteins in cell lysates of D.
ARF perturbs N-C interaction of AR
The interactions between N- and C-terminal domains of AR are essential for its transactivation activity. Because ARF binds with both N and C termini of AR, we hypothesized that ARF binding may interrupt the N-C interaction of AR. To test this, we performed reconstituted transcription assays similar to previous reports (40) in both HEK293T cells and PC3-shRNA cells. HEK293T cells were cotransfected with ARF, and Flag-ARNTD and/or ARDBD-H-LBD expression plasmids (Figure 6A), along with probasin or PSA promoter-luciferase reporter. After transfection, cells were treated with DHT and lysates were collected to determine luciferase activities. Because HEK293T cells have a nondetectable level of endogenous AR and p14ARF, the expression of ARNTD (1–551) or ARDBD-H-LBD (552–917) resulted in a minimal transactivation of AR in the presence of DHT (Figure 6B). However, coexpression of ARNTD with ARDBD-H-LBD dramatically enhanced the DHT-induced transactivation of probasin and PSA reporters (Figure 6B, lane N+C, upper panel). Upon ARF expression, the interaction of N and C termini of AR was significantly inhibited in the presence of DHT (Figure 6B). The expression level of N- and C termini of AR protein, alone and in combination with p14ARF, was further confirmed by Western blotting (Figure 6B, lower panel).
Figure 6.

ARF interrupts the N-C interaction of AR. A, A reconstituted transcription assay for the N-C interaction of AR. ARF disrupts the interaction between AR-NTD and AR-LBD. B, Effects of p14ARF overexpression on the N-C interaction of AR. HEK293T cells were transfected with pRL-CMV, ARR2-PB-Luc or PSA-Luc, AR mutants, and ARF expression plasmids. C, Effects of p14ARF knockdown on AR N-C interaction. PC3-scramble and PC3-shARF cells were transfected with ARR2-PB-Luc, pRL-CMV, and AR mutants. The luciferase activity (firefly luciferase light unit/Renilla luciferase) relative to AR-NTD with vehicle control was determined, and results were means (± SD) of triplicates. Levels of AR mutants and ARF proteins in lysates were analyzed by Western blot. D, ARF does not alter the nuclear shuttling of AR in LNCaP cells. LNCaP cells were transfected with p14ARF expression plasmids in androgen-starvation medium. After 24 hours, cells were treated with 10nM DHT for 20 minutes before harvesting. Cells were fixed and immunostained with anti-AR and anti-p14ARF antibodies and counterstained with DAPI to visualize nuclei. Scale bars, 10 μm.
Conversely, we examined the effects of ARF knockdown on the interaction of N and C termini of AR in cancer cells. As demonstrated, p14ARF knockdown in PC3 cells resulted in a slight up-regulation of overexpressed ARNTD or ARDBD-H-LBD-driven transactivation of ARR2-PB-Luc reporter, as compared with PC3-scramble cells (Figure 6C). Surprisingly, in the absence of DHT, a significant reduction of basal expression of ARR2-PB-Luc reporter activity was found with coexpression of ARNTD and ARDBD-H-LBD as compared with ARNTD or ARDBD-H-LBD alone (Figure 6C), indicating other factors may inhibit the N-and C-terminal interaction in androgen-insensitive PC3 cells (41). Remarkably, upon addition of DHT, the reporter activity was dramatically enhanced in PC3-shARF cells as compared with PC3-scramble cells (Figure 6C). These results suggest that p14ARF suppresses AR activity by affecting N and C-terminal interaction, and p14ARF knockdown revokes the inhibitory effects. Our data revealed that the perturbation of the AR N-C interaction by p14ARF results in a repression of ligand-induced AR transactivation, the destabilization of AR N-C interaction, and consequently blocking of the binding of other coactivators (40).
ARF does not alter nuclear translocation of AR
The nuclear translocation is a crucial step for androgen-induced AR activation, and AR binding to androgen-responsive elements in promoter regions of target genes. We further examined whether the inhibition of AR activity by ARF was the consequence of the suppressed compartment shuttling of AR protein. To do so, LNCaP cells were transfected with ARF and treated with 10 nM DHT, and then the localizations of AR and ARF proteins were determined. In the absence of DHT, endogenous AR (red) was found in both cytoplasm and nucleus, whereas the enforced overexpression of ARF (green) was found predominantly in nucleus (Figure 6D). Upon DHT treatment, AR protein accumulated primarily in nucleus, and ARF overexpression did not result in any changes on the distribution pattern of AR (Figure 6D, indicated by arrowhead). This result excludes the possibility that ARF represses AR activity through the alteration of AR nuclear trafficking.
ARF expression is reversely correlated with AR activity in human prostate tissues
To determine whether ARF expression correlates with AR activity in human PCa, we performed and quantified the IHC staining of ARF, AR, and PSA in human prostate tissue microarrays containing 35 PCa and 5 normal cases in 80 cores. Our results showed that ARF protein is largely found in the nucleus, whereas PSA protein is localized to the cytoplasm of epithelial cells (Figure 7, A and B, and Supplemental Figure 6). The correlation between the averaged ARF and PSA staining scores were determined by Pearson correlation test, with grades taken as continuous variable. We also assessed the correlation between the expressions of ARF and PSA proteins using the χ2 test. Results from both statistical analyses revealed that ARF expression is reversely correlated with PSA in PCa (Pearson correlation coefficient = −0.49, P = .0029; χ2 test, P = .036 [Figure 7C and Supplemental Table 2]) when graded in intensity staining scores. A similar result was found when the IHC staining of ARF and PSA proteins was graded in percentage scores (Pearson correlation coefficient = −0.47, P = .0041; χ2 test, P = .014 [Supplemental Figure 7 and Supplemental Table 3]). Furthermore, we examined the expression profile of ARF, AR, and PSA proteins on adjacent sections of the same specimens. Notably, cancer cells with a high level of ARF normally showed a high level of AR, but a low level of PSA in the same lesions (Figure 7B and Supplemental Figure 6). This set of clinical evidence supports that the low level of PSA is likely a result of a decreased activity of AR by p14ARF in human prostate tissues.
Figure 7.

Expression of ARF is reversely correlated with PSA level in human prostate tissue. A, Representative images of immunohistochemical staining on human prostate array tissues. The sections were stained with anti-p14ARF and anti-PSA antibodies. Magnification, ×4. B, Representative images showing the association of the expression of ARF, AR, and PSA on adjacent tissue sections of the same human prostate specimen by IHC. Magnification, ×40. C, Statistical analysis of the tissue array stained with anti-p14ARF and anti-PSA antibodies. The percentages of different PSA levels were calculated for each level of p14ARF protein in 35 cases of human PCa specimens. p14ARF and PSA levels are graded as 0, 1, 2, and 3 by intensity scores. The PSA grades are color-coded. Numbers in parentheses represent sample sizes. The statistical significances were determined by χ2 test (Supplemental Table 2).
Discussion
AR signaling plays a vital role in PCa, and transactivation and biological functions of AR in cells are highly mediated by many coregulators including coactivators and corepressors in a spatial-temporal manner (4, 12). A large number of publications have focused on AR coactivators; however, limited reports are found on AR corepressors. Our report is the first study to identify a novel role of ARF to function as an AR corepressor in PCa.
AR contains several functional domains/motifs through which coregulators interact to regulate AR transactivation (42). The transactivation property of AR is predominantly executed through the NTD in which the activation function 1 (AF-1) region contributes most of the transcriptional activity, whereas the CTD displays some supplementary ligand-dependent transcriptional activity (3). Coregulators mediate AR activity preferentially through either the NTD or CTD of the AR. Interestingly, ARF binding to the NTD and LBD of the AR results in a remarkable inhibition of several AR-mediated genes. Several corepressors, including glycogen synthase kinase-3 β (GSK3β), the orphan nuclear receptor small heterodimer partner (SHP), and silencing mediator for retinoid and thyroid receptors (SMRT) have been reported to regulate AR activity at multiple levels, such as the N-C interaction, chromatin modification, nuclear translocation, DNA binding, or competition with coactivators (42–46). Among these, the N-C interaction is thought to be the major site for AR corepressors (42). Upon androgen induction, the 23FXXLF27 motif at the NTD interacts with a hydrophobic cleft of the LBD to stabilize the ligand binding of AR (47). The N-C interaction contributes to the modulation, activity, and stability of AR binding to chromatin and consequently recruitment of coregulators (47, 48). In this study, we find that ARF negatively regulates AR activity by blocking the interaction between the N and C terminus of AR (Figure 6, B and C). These results suggest the repression mechanism of AR-mediated activities by ARF may be caused by impairing the ligand binding of AR and the recruitment of coactivators. It is still unclear whether ARF binds AR on chromatin to attenuate the AR transcription activity due to lack of evidence, yet it is worthy for further investigation. Together, our findings provide mechanistic insights into the suppressive effect of ARF on AR activity in PCa.
Our study on mapping of the ARF and AR domains that interact revealed that ARF binds to AR via its N-terminal HDM2-binding motif (A2) (Figure 1F). HDM2 is an E3 ubiquitin ligase that may degrade AR upon AKT phosphorylation, through its interaction with the N terminus and DBD of AR (49). One could reason that ARF binding to AR via the ARF HDM2-binding motif should stabilize AR protein by preventing the HDM2-mediated AR degradation. Surprisingly, our findings showed that ARF overexpression doesn't result in a significant increase of AR protein, and the inhibitory effect of ARF on AR was not executed by the reduced level of AR protein. Further investigation will be needed to address the mechanism of action.
The mechanisms leading to the recurrent growth of CRPC are complicated by the heterogeneity of cancerous cells and the differential activation of oncogenic pathways during cancer progression. AR signaling has been long recognized as an essential role in the development and progression of PCa including CRPC (21, 41). Increased AR activity due to gene amplification or mutations may provide a mechanistic explanation for continued AR activation when androgen levels are low or inhibited (2, 7, 10). Yet PCa cells tolerate only a narrow range of AR activities, and overactivation of AR may lead to growth arrest (41). Paradoxically, emerging evidence reported the selective down-regulation of AR signaling is associated with the progression and metastasis of PCa (17, 18). In addition, epithelial cells showed an increase of apoptosis and the epithelial basal cells displayed an increase of proliferation in AR-deficient mice (21). This notion suggests that the modulation of AR signaling by other factors may occur in the advanced stages of PCa. For example, AR inhibition contributes to the hyperactivation of the AKT pathway via an impaired feedback of FK506 binding protein 5-PH domain and leucine rich repeat protein phosphatase (FKBP5-PHLPP) signaling in PCa (19, 20). Recent reports show that the p14ARF level is positively correlated with the malignance grade of PCa (28), and high-grade PCa actually secretes less PSA than low-grade PCa (50). Our results demonstrate that ARF inhibits AR transactivation activity, which provides an alternative explanation on the down-regulation of AR signaling in some clinical cases, particularly in the context of phosphatase and tensin homologs deleted on chromosome 10 (PTEN) loss. Down-regulation of AR activity by ARF may be involved in progression of PCa to CRPC.
Taken together, we have demonstrated a novel role of ARF as an AR corepressor in PCa. Our results show that the HDM2-binding motif of ARF is essential for the interaction with the N and C termini of AR, and the AR N-C interaction is suppressed by ARF. Our findings underscore a novel mechanism for the role of ARF on AR function, implying the possibility of creating an ARF motif-based peptide that could be used to inhibit AR transactivation in PCa (51, 52). Further investigation on the ARF and AR oncogenic potentials may provide additional insights into their regulatory role in advanced PCa.
Acknowledgments
We thank Dr Shaoyong Chen for pCMV3.1-3xFlag-hAR plasmid; Dr Marc Rodriguez-Niedenfuhr for pGFP-p14ARF-pcDNA4/TO plasmid; Dr Wendell G. Yarbrough for pCI-Neo-p14ARF, Myc-p14ARF, and mutant plasmids; Dr Ifeanyi J. Arinze for pCI-neo plasmid; Dr LaMonica V. Stewart for LNCaP cells; and Dr Renjie Jin for CWR22Rv1 cells. We thank Dr J. Shawn Goodwin for his excellent assistance on microscopy.
Microscopy experiments and data analysis were performed through the use of Meharry Medical College Morphology Core supported in part by National Institutes of Health Grants U01NS041071 and S10RR0254970. This work was supported in part by National Institute on Minority Health and Health Disparities (NIMHD) Grant MD004038 (to Z.C.), DK055748 (to R.J.M.), NCI U54 Grant CA091408, CA163069, G12 MD007586, Vanderbilt CTSA Grant UL1 RR024975-01 and 2 UL1 TR000445-06 from National Center for Research Resources (NCRR)/NIH.
Disclosure Summary: The authors have nothing to declare.
NURSA Molecule Pages†:
Nuclear Receptors: GR;
Ligands: Dihydrotestosterone.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AR
- androgen receptor
- ARF
- alternative reading frame
- CRPC
- castration-resistant PCa
- CT
- cycle threshold
- DAPI
- diamidino-2-phenylindole
- DBD
- DNA-binding domain
- DHT
- dihydrotestosterone
- FBS
- fetal bovine serum
- H
- hinge
- HDM2
- human double minute 2
- IHC
- immunohistochemistry
- LBD
- ligand-binding domain
- MDM2
- mouse double minute 2
- NTD
- N-terminal domain
- PCa
- prostate cancer
- RNAi
- RNA interference
- shRNA
- short hairpin RNA
- ULF
- ubiquitin ligase for ARF.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29 [DOI] [PubMed] [Google Scholar]
- 2. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39 [DOI] [PubMed] [Google Scholar]
- 3. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8:440–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808 [DOI] [PubMed] [Google Scholar]
- 5. Rahman M, Miyamoto H, Chang C. Androgen receptor coregulators in prostate cancer: mechanisms and clinical implications. Clin Cancer Res. 2004;10:2208–2219 [DOI] [PubMed] [Google Scholar]
- 6. Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–490 [DOI] [PubMed] [Google Scholar]
- 7. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555 [PubMed] [Google Scholar]
- 8. Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406 [DOI] [PubMed] [Google Scholar]
- 9. Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Curr Genomics. 2009;10:18–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45 [DOI] [PubMed] [Google Scholar]
- 11. Mulholland DJ, Jiao J, Wu H. Hormone refractory prostate cancer: lessons learned from the PTEN prostate cancer model. Adv Exp Med Biol. 2008;617:87–95 [DOI] [PubMed] [Google Scholar]
- 12. Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer. 2007;120:719–733 [DOI] [PubMed] [Google Scholar]
- 13. Fu M, Wang C, Wang J, et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol. 2002;22:3373–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002;277:25904–25913 [DOI] [PubMed] [Google Scholar]
- 15. Liu P, Li S, Gan L, Kao TP, Huang H. A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 2008;68:10290–10299 [DOI] [PubMed] [Google Scholar]
- 16. Lakshmikanthan V, Zou L, Kim JI, Michal A, et al. Identification of βArrestin2 as a corepressor of androgen receptor signaling in prostate cancer. Proc Natl Acad Sci U S A. 2009;106:9379–9384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hendriksen PJ, Dits NF, Kokame K, et al. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66:5012–5020 [DOI] [PubMed] [Google Scholar]
- 18. Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51 [DOI] [PubMed] [Google Scholar]
- 19. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niu Y, Altuwaijri S, Lai KP, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105:12182–12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673 [DOI] [PubMed] [Google Scholar]
- 23. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000 [DOI] [PubMed] [Google Scholar]
- 24. Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222 [PubMed] [Google Scholar]
- 25. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734 [DOI] [PubMed] [Google Scholar]
- 26. Kuo ML, Duncavage EJ, Mathew R, et al. Arf induces p53-dependent and -independent antiproliferative genes. Cancer Res. 2003;63:1046–1053 [PubMed] [Google Scholar]
- 27. Yarbrough WG, Bessho M, Zanation A, Bisi JE, Xiong Y. Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Res. 2002;62:1171–1177 [PubMed] [Google Scholar]
- 28. Chen Z, Carracedo A, Lin HK, et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci Signal. 2009;2:ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424 [DOI] [PubMed] [Google Scholar]
- 30. Zhang Z, Rosen DG, Yao JL, Huang J, Liu J. Expression of p14ARF, p15INK4b, p16INK4a, and DCR2 increases during prostate cancer progression. Mod Pathol. 2006;19:1339–1343 [DOI] [PubMed] [Google Scholar]
- 31. Humbey O, Pimkina J, Zilfou JT, et al. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008;68:9608–9613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen S, Kesler CT, Paschal BM, Balk SP. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J Biol Chem. 2009;284:25576–25584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464:624–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bothner B, Lewis WS, DiGiammarino EL, Weber JD, Bothner SJ, Kriwacki RW. Defining the molecular basis of Arf and Hdm2 interactions. J Mol Biol. 2001;314:263–277 [DOI] [PubMed] [Google Scholar]
- 35. Heery DM, Hoare S, Hussain S, Parker MG, Sheppard H. Core LXXLL motif sequences in CREB-binding protein, SRC1, and RIP140 define affinity and selectivity for steroid and retinoid receptors. J Biol Chem. 2001;276:6695–6702 [DOI] [PubMed] [Google Scholar]
- 36. Wang J, He X, Luo Y, Yarbrough WG. A novel ARF-binding protein (LZAP) alters ARF regulation of HDM2. Biochem J. 2006;393:489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu J, Zhang SS, Saito K, et al. PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 2009;28:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pomerantz J, Schreiber-Agus N, Liegeois NJ, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723 [DOI] [PubMed] [Google Scholar]
- 39. Cronauer MV, Schulz WA, Burchardt T, Ackermann R, Burchardt M. Inhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell lines. Oncogene. 2004;23:3541–3549 [DOI] [PubMed] [Google Scholar]
- 40. Wang L, Hsu CL, Ni J, et al. Human checkpoint protein hRad9 functions as a negative coregulator to repress androgen receptor transactivation in prostate cancer cells. Mol Cell Biol. 2004;24:2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tararova ND, Narizhneva N, Krivokrisenko V, Gudkov AV, Gurova KV. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate. 2007;67:1801–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burd CJ, Morey LM, Knudsen KE. Androgen receptor corepressors and prostate cancer. Endocr Relat Cancer. 2006;13:979–994 [DOI] [PubMed] [Google Scholar]
- 43. Wang L, Lin HK, Hu YC, Xie S, Yang L, Chang C. Suppression of androgen receptor-mediated transactivation and cell growth by the glycogen synthase kinase 3 beta in prostate cells. J Biol Chem. 2004;279:32444–32452 [DOI] [PubMed] [Google Scholar]
- 44. Gobinet J, Auzou G, Nicolas JC, Sultan C, Jalaguier S. Characterization of the interaction between androgen receptor and a new transcriptional inhibitor, SHP. Biochemistry. 2001;40:15369–15377 [DOI] [PubMed] [Google Scholar]
- 45. Agoulnik IU, Krause WC, Bingman WE, 3rd, et al. Repressors of androgen and progesterone receptor action. J Biol Chem. 2003;278:31136–31148 [DOI] [PubMed] [Google Scholar]
- 46. Liao G, Chen LY, Zhang A, et al. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278:5052–5061 [DOI] [PubMed] [Google Scholar]
- 47. He B, Minges JT, Lee LW, Wilson EM. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem. 2002;277:10226–10235 [DOI] [PubMed] [Google Scholar]
- 48. Li J, Fu J, Toumazou C, Yoon HG, Wong J. A role of the amino-terminal (N) and carboxyl-terminal (C) interaction in binding of androgen receptor to chromatin. Mol Endocrinol. 2006;20:776–785 [DOI] [PubMed] [Google Scholar]
- 49. Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21:4037–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Partin AW, Carter HB, Chan DW, et al. Prostate specific antigen in the staging of localized prostate cancer: influence of tumor differentiation, tumor volume and benign hyperplasia. J Urol. 1990;143:747–752 [DOI] [PubMed] [Google Scholar]
- 51. Johansson HJ, El-Andaloussi S, Holm T, et al. Characterization of a novel cytotoxic cell-penetrating peptide derived from p14ARF protein. Mol Ther. 2008;16:115–123 [DOI] [PubMed] [Google Scholar]
- 52. Reeb CA, Gerlach C, Heinssmann M, et al. A designed cell-permeable aptamer-based corepressor peptide is highly specific for the androgen receptor and inhibits prostate cancer cell growth in a vector-free mode. Endocrinology. 2011;152:2174–2183 [DOI] [PubMed] [Google Scholar]