

Abstract
G protein-coupled receptors (GPCRs) are the largest and most diverse superfamily of membrane proteins and mediate most cellular responses to hormones and neurotransmitters. Posttranslational modifications are considered the main regulators of all GPCRs. In addition to phosphorylation, glycosylation, and palmitoylation, increasing evidence as reviewed here reveals that ubiquitination also regulates the magnitude and temporospatial aspects of GPCR signaling. Posttranslational protein modification by ubiquitin is a key molecular mechanism governing proteins degradation. Ubiquitination mediates the covalent conjugation of ubiquitin, a highly conserved polypeptide of 76 amino acids, to protein substrates. This process is catalyzed by 3 enzymes acting in tandem: an E1, ubiquitin-activating enzyme; an E2, ubiquitin-carrying enzyme; and an E3, ubiquitin ligase. Ubiquitination is counteracted by deubiquitinating enzymes that deconjugate ubiquitin-modified proteins and rescue the substrate from proteasomal degradation. Although ubiquitination is known to target many GPCRs for lysosomal or proteasomal degradation, emerging findings define novel roles for the basal status of ubiquitination and for rapid deubiquitination and transubiquitination controlling cell surface expression and cellular responsiveness of some GPCRs. In this review, we highlight the classical and novel roles of ubiquitin in the regulation of GPCR function, signaling, and trafficking.
G protein-coupled receptors (GPCRs) are the largest superfamily of membrane proteins and mediate most cellular responses to hormones and neurotransmitters. In fact, GPCRs are a major target for drug discovery by virtue of their role in regulating a broad range of physiological responses and pathological conditions including cardiovascular and neurological disorders, immunological, endocrine, renal and pulmonary diseases, pain, and cancer (1). In vertebrates, GPCRs are now divided into 4 classes based on their sequence and primary structural similarity: rhodopsin (class A), secretin (class B), glutamate (class C), and frizzled (class 4) (2). GPCRs are among the essential nodes of communication between the internal and external environments of cells, transducing the information provided by stimuli into intracellular second messengers. On activation, GPCRs undergo conformational changes that facilitate activation of heterotrimeric guanine nucleotide binding proteins (G proteins) and also set in place a series of molecular interactions that allow for the following: 1) feedback regulation of G protein coupling; 2) receptor endocytosis; and 3) signaling through G protein-independent signal transduction pathways (3–9).
Ligand-stimulated GPCR activation, desensitization, internalization, and recycling proceed in a cyclical manner. This behavior provides a mechanism to protect cells against excessive stimulation, on the one hand, and guards cells against prolonged desensitization and hormone insensitivity on the other hand. Thus, the magnitude and duration of ligand-induced GPCR responses are tightly linked to the balance between signal generation and signal termination. The endocytic pathway tightly controls the activity of GPCRs. In most described situations this leads to terminating GPCR signaling. However, emerging information now reveals a new paradigm, whereby GPCR endocytosis promotes persistent signaling (10–15) and prolonged function (16).
Agonist-stimulated endocytosis can drive receptors toward divergent itineraries, including degradative routes that lead to GPCR down-regulation, and recycling pathways that promote receptor resensitization (17). Phosphorylation of the receptor and subsequent binding of β-arrestin prevent consequent interaction of receptors with G proteins, effectively terminating the G protein-mediated signal and initiating the endocytic process. GPCR phosphorylation occurs predominantly on Ser and Thr residues within the carboxyl receptor tail and the third intracellular loop. Agonist-activated GPCRs are rapidly phosphorylated by G protein-coupled receptor kinases (GRKs) (18) with ensuing translocation of arrestins to the plasma membrane (19). A conformational change in arrestin is induced on binding to GPCRs, exposing the carboxy-terminal domain, which interacts with clathrin and the β2-adaptin subunit of the clathrin adaptor adaptor protein complex-2, components of the endocytic machinery, thereby promoting endocytosis of arrestin-bound receptors (20–22). The endocytic activity of arrestin is also subject to dynamic regulation by dephosphorylation and ubiquitination as we discuss here.
Subsequent steps of endocytic sorting play a critical role in dictating the receptor signaling response after the initial desensitization event. The sorting of internalized receptors between lysosomal and recycling pathways produces essentially opposite effects on cell signaling. Endocytic trafficking to lysosomes represents a major mechanism by which many GPCRs and various other signaling receptors are down-regulated following ligand-induced activation (23–26). Some GPCRs are driven along an alternate route that facilitates the efficient recycling of plasma membrane receptors after ligand-induced endocytosis. The recycling pathway can promote rapid recovery (or resensitization) of cellular responsiveness (27–29). The molecular mechanisms that regulate trafficking of mammalian GPCRs are essential to receptor signaling, desensitization, and resensitization yet are incompletely understood. Posttranslational modifications are considered the primary regulator of all GPCRs. In addition to phosphorylation, glycosylation, and palmitoylation (30), emerging evidence increasingly identifies a role for posttranslational modification by ubiquitin that regulates the magnitude, duration, and spatial components of GPCR signaling. In this review, we discuss the classical and novel roles of ubiquitin in the regulation of GPCR function, signaling, and trafficking.
Posttranslational Modifications: Ubiquitination
Ubiquitination is the enzymatic posttranslational modification process that mediates the covalent conjugation of ubiquitin, a highly conserved polypeptide of 76 amino acids, to protein substrates (31). This reaction is catalyzed by 3 enzymes acting in tandem. First, ubiquitin is activated by an ubiquitin-activating enzyme (known also as E1) in an ATP-dependent process. The initial step involves production of a ubiquitin-adenylate intermediate, followed by the transfer of ubiquitin to the E1 active site cysteine residue and release of AMP. This step results in a thiol-ester linkage between the C-terminal carboxyl glycine of ubiquitin and the E1 cysteine sulfhydryl group. Second, a ubiquitin-conjugating enzyme or ubiquitin-carrier enzyme (UBCs or E2) accepts ubiquitin from E1 by a transthiolation reaction, again involving the carboxyl terminus of ubiquitin. Finally, an ubiquitin protein ligase (E3) transfers ubiquitin from the E2 enzyme to the ϵ-amino group of a lysine residue on the substrate (32, 33). Initially it was thought that each ubiquitin moiety attaches to a single internal Lys on the target protein. Later, Hershko (34) and then Varshavsky (35) and their respective colleagues demonstrated the formation of a polyubiquitin chain anchored to a single internal Lys residue (reviewed in Ref. 36) (Figure 1).
Figure 1.
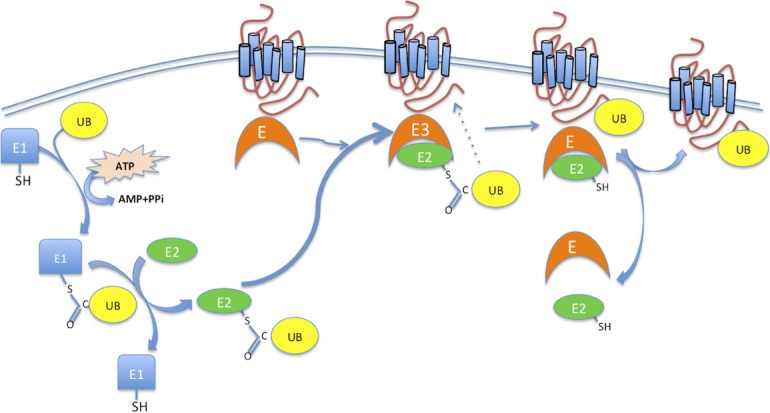
The posttranslational modification by ubiquitin is carried out by a set of 3 enzymes, E1, E2, and E3. Ubiquitin is first activated by ubiquitin-activating enzyme E1 and transferred to its active site. This transfer requires ATP. The ubiquitin molecule is then passed on to the second enzyme of the complex, E2 (ubiquitin-conjugating enzyme), before reaching the final enzyme, E3, the ubiquitin protein ligase, which recognizes, binds the target substrate, and labels it with the ubiquitin. The process can be repeated until a short chain is formed. PPi, pyrophosphate; SH, sulfydryl; UB, ubiquitin.
E3 ubiquitin ligases and GPCRs
The human genome encodes 2 ubiquitin activating enzymes (E1), at least 38 ubiquitin-conjugating enzymes (E2), and 600 to 1000 ubiquitin protein ligases (E3) (37–39). Ubiquitination is a highly specific process at all levels. E1s select the correct ubiquitin-like protein for their respective pathways and display remarkable specificity. For example, despite the fact that ubiquitin and NEDD8 (ubiquitin-like protein) share 57% sequence identity and have markedly similar structures, NEDD8 is poorly employed by the ubiquitin conjugation system (40). This specificity is obvious because E1 also transfers the activated ubiquitin-like protein to its cognate E2, thereby coupling the ubiquitin-like protein to its cognate downstream pathway (33). E2 and E3 work in a combinatorial manner to generate different forms of substrate modification (32, 41). E3 ligases recognize the substrate protein and impart specificity to the reaction (38, 42). Many E2s have the capacity to determine ubiquitin chain topology. This probably requires a noncovalent interaction between E2 and the acceptor ubiquitin, which exposes a specific Lys on the acceptor ubiquitin to the active site of the E2 charged with the donor ubiquitin. Depending on the recognized surface of ubiquitin, chains of different linkage are assembled and the modified proteins will be assigned distinct fates (39). Based on the sequence homology of their E2-binding domains, there are 3 major types of E3s in eukaryotes, defined by the presence of either a homologous to the E6-AP carboxyl terminus (HECT), a really interesting new gene (RING) (38), or a U-box domain (43). To date, few E3 ligases attaching ubiquitin to specific GPCR have been identified. Nedd4 and especially Nedd4-2, HECT domain-containing ligases, are well known for their role in ubiquitinating ion channels, such as the amiloride-sensitive ENaC, and nonmembrane substrates mostly via direct interaction with the substrate. The substrate's specificity of the HECT-type E3 is dictated by protein-protein interaction domains (43). It is now known that Nedd4 ubiquitinates the β2-adrenergic receptor (B2AR) and regulates its lysosomal trafficking (44). However, B2AR ubiquitination by Nedd4 is effective only when β-arrestin2 is present as an adaptor, suggesting that β-arrestin2 binds at least 2 E3 ubiquitin ligases: Mdm2 (a RING E3 ubiquitin ligase) and Nedd4. These findings show dissimilar function in B2AR regulation: Mdm2 mediates β-arrestin ubiquitination (45), which defines the initial step in receptor endocytosis, whereas Nedd4 mediates receptor ubiquitination that targets the B2AR to lysosomal compartments (44). Other RING E3 ubiquitin ligases involved in GPCR ubiquitination include c-Cbl and AIP4. c-Cbl mediates ubiquitination, degradation, and down-regulation of the human protease-activated receptor (PAR) 2, while AIP4 is able to bind ubiquitin to C-X-C chemokine receptor-4 (CXCR4) (46) and δ-opioid receptors (DOR) (47). Interestingly, Hislop and colleagues uncovered a distinct function of AIP4-mediated ubiquitination in regulating the proteolytic processing of DOR without affecting its endocytic sorting to lysosomes (47).
Recently, the HECT E3 ubiquitin ligase WWP2 was identified as a critical regulator of sphingosine-1-phosphate receptor (S1PR) (lysophospholipid receptor) degradation induced by the inhibitor FTY720P (48). In the same way, RING E3 ubiquitin ligases and their role in the regulation of several GPCRs have been identified. Dorfin, for instance, mediates calcium-sensing receptor ubiquitination and degradation (49). Seven-in-absentia homolog 1A mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors (50), cullin3 with BTB Protein KLHL12 as adaptor targets the dopamine D4 receptor for ubiquitination (51), and mahogunin ubiquitinates the melanocortin 2 receptor (52).
Typical and atypical ubiquitin chains
Ubiquitin can be attached to proteins as a single unit on one or multiple sites, yielding monoubiquitinated and multimonoubiquitinated proteins, respectively (Figure 2). Ubiquitin contains 7 Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, or Lys63) and an amino-terminal Met (Met1) that can serve as acceptor sites for additional ubiquitin molecules generating polyubiquitinated proteins. Thus, ubiquitin chains can be connected by at least 8 different homotypic linkages, as well as by a range of atypical chains (heterologous, forked, or mixed) (39, 53–55) (Figure 2). However, the most common polyubiquitin chains are formed through linkages at K-48 and K-63. Extensive studies on Lys48-linked and Lys63-linked chains established essential roles in proteasomal degradation for Lys48 and in cell signaling for Lys63-linked chains and monoubiquitin conjugation. In comparison, rather little is known about the remaining atypical chain types (56). More recent reports show that Lys63-linked chains can drive degradation, whereas Lys48-linked chains may function nonproteolytically, for example, in the inactivation of the transcriptional activator Met-4 (57). This suggests that the functions of ubiquitination depend not only on chain topology but also on other factors such as the timing and reversibility of the reaction, enzyme or substrate localization, or interactions between E3s and effectors. Notably, ubiquitin can also link to the amino group in the N-terminal methionine (Met1) of the acceptor ubiquitin, leading to the formation of linear ubiquitin chains (58). It is clear that all ubiquitin linkage types coexist in all cell types analyzed to date (56). The different linkage types are classified as having either “compact” or “open” conformations. In the compact conformation of Lys6-linked, Lys11-linked, and Lys48-linked chains, the distal and the proximal ubiquitin moieties form an intramolecular interface, whereas in the open conformation of Lys63-linked and Met1-linked polymers, the only contact is the linkage point. Such conformation diversity may explain how proteins and enzymes discriminate between different linkage types (56). However, ubiquitination is a complex process and multiple acceptor sites could be ubiquitinated. Interestingly, the anchor of ubiquitin to Met1 facilitates the formation of Lys48 polyubiquitin chains in MyoD, a tissue-specific transcriptional activator (59). Met1, the acceptor site of the ubiquitin, generates a head-to-tail, or linear, interubiquitin linkage. The ubiquitin linkage in this specific acceptor site is crucial in various pathways. Furthermore, recent data suggest that physiological signaling requires cooperation between different ubiquitin linkage types (60). Lys63-linked or Lys48-linked polyubiquitin chains can be discriminated by different experimental techniques, using K63R and K48R mutants of ubiquitin, or specific Lys63 and Lys48 ubiquitin chain antibodies. These approaches were employed to determinate different linkage types and showed that Lys63-linked polyubiquitins are the dominant form in ubiquitinated human κ-opioid receptor (KOR) on agonist stimulation (61), and Lys48-linked chains were detected in the parathyroid hormone receptor (PTHR) on agonist and antagonist stimulation (62).
Figure 2.

Typical and atypical ubiquitin chains: ubiquitin can be attached to proteins as a single unit on one or multiple sites, yielding monoubiquitinated and multimonoubiquitinated proteins, respectively. Ubiquitin contains 7 Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, or Lys63) that can serve as acceptor sites for additional ubiquitin molecules generating poly-ubiquitinated proteins with distinct types of ubiquitin conjugation. The scheme shows the most representative ubiquitin chains.
In some instances, efficient protein polyubiquitination requires additional conjugation factors, so-called E4 enzymes, that support the elongation of ubiquitin chains (53, 63, 64). Although lysines are the most frequent substrate sites for attachment of ubiquitin and ubiquitin-like proteins in the case of GPCRs, conjugation can sometimes occur on other residues such as the free α-NH2 group of an N-terminal residue of a protein (65, 66). There are only a limited number of studies identifying the specific target sites of GPCR ubiquitination. It has been established that mutation of the 3 carboxyl tail lysines abrogated chemokine receptor CXCR4 ubiquitination and lysosomal sorting but did not affect receptor endocytosis (67). In the same way, the lysines residues (Lys338, Lys349, and Lys378) in the carboxy-terminal domain of the human KOR, and lysines residues (Lys421 and Lys422) in the carboxyl tail of the PAR1, are the sites of receptor ubiquitination. However, the carboxyl tail of receptors is not the only domain where GPCRs may be ubiquitinated. Mutation of a single lysine, located in the third intracellular loop of the vasopressin V2 receptor (V2R), abrogated its ubiquitination and degradation (68), and ubiquitin modification in μ-opioid receptors (MOR) was identified in the first cytoplasmic loop as being the critical location for ubiquitin-dependent control of MOR down-regulation (69). In the case of the B2AR, the lysines in its carboxyl tail were reported to be the main sites of protein ubiquitination (70). However, mutation of these lysines did not eliminate receptor ubiquitination. Recently, studies based on both molecular and proteomic approaches (mass spectrometry) show that ubiquitination of the B2AR occurs on both the third intracellular loop and the carboxyl tail (71).
Ubiquitin-binding Domain Proteins: Chaperones for Trafficking Ubiquitinated GPCRs
Ubiquitination through alternative lysine sites is now considered a protein-modification code. Similar to the detection of phosphorylated proteins via phospho-binding domains recognizing phosphorylated Ser, Thr, or Tyr, ubiquitin modification of target proteins is recognized by a variety of ubiquitin-binding domain (UBD) -containing proteins or ubiquitin receptors that comprise at least one UBD within their structure. Effector proteins with UBDs translate the modifications into specific outcomes (72). The number of identified UBDs is growing, with more than 20 different families identified to date. Most of the UBDs fold into α-helical-based structures, including the ubiquitin-associated domain (UBA), ubiquitin interacting motif (UIM), DIUM (double-sided ubiquitin-interacting motif), MIU (motif interacting with ubiquitin), CUE (coupling of ubiquitin conjugation to ER degradation), and GAT (Golgi-associated γ-adaptin homologous and target of myb [TOM]) domains. Nonhelical UBDs are also frequent and include different ubiquitin-binding zinc fingers. Although structurally quite different, UBDs share a feature of noncovalent binding to the ubiquitin moiety formed by the hydrophobic patch including and surrounding Ile44 (73). The UBDs interact with monoubiquitinated or polyubiquitinated chains to recruit the ubiquitinated protein into cellular signaling networks. Some UBDs display selectivity toward different types of ubiquitin conjugations and function in multiple cellular processes, including protein degradation by proteasomes, protein trafficking to lysosomes, DNA repair, cell-cycle progression, autophagy, and gene transcription (72). On the basis of the apparent correlation between linkage specificity and cellular functionality, it has been proposed that different ubiquitin chain conformations can be recognized by distinct UBDs. However, in vitro experiments have been unable to identify selectivity of 30 UBA domains toward any type of linkage (74). The sorting of most ubiquitinated receptors from endosomes to lysosomes occurs via interaction with endosomal-sorting-complex-required-for-transport (ESCRT) proteins. The ESCRT machinery comprises 4 distinct ESCRT complexes that function sequentially to coordinate the sorting of ubiquitinated receptors into intraluminal vesicles (ILVs) of multivesicular bodies (MVBs) (75). ESCRT-dependent sorting of ubiquitin-modified receptors are recruited into an early endosomal subdomain enriched in hepatocyte growth factor-regulated tyrosine kinase substrate (HRS), a ubiquitin-binding component, also known as ESCRT-0, and clathrin (76). HRS interacts directly with polyubiquitinated receptors, monoubiquitinated receptors (23), and ESCRT-I, which also bind to ubiquitin and ESCRT-II. The ESCRT-III complex is then recruited, assembles on endosomal membranes, and facilitates ILV scission (77). Several ubiquitinated GPCRs, including the chemokine receptor CXCR4 (46), ocular albinism type 1 receptor (78), and PAR2 (79), are sorted by this mechanism. Furthermore, degradation of the DOR requires HRS and Vps4 functions (80). However, not all GPCRs require direct ubiquitination for MVB sorting and lysosomal degradation (81). For example, a ubiquitination-deficient mutant of DOR and the calcitonin-like receptor, a GPCR that is not ubiquitinated, traffic to MVBs/lysosomes like wild-type receptors (80, 82). Therefore, it remains to be determined whether a signaling receptor can bypass the requirement for both ubiquitination and ubiquitin-binding components of the ESCRT machinery and sort to MVBs/lysosomes (83). The association of other UBD-containing proteins with GPCRs has been studied. The Integrin-Associated Protein IAP (CD47) to cytoskeleton (PLIC-2), also called ubiquitilin 2, has been identified as a negative regulator of GPCR/arrestin clustering in clathrin-coated pits (CCPs). The PLIC proteins contain an amino-terminal ubiquitin-like domain and a carboxy-terminal UBA domain. PLIC-2 inhibited clustering of GPCR-arrestin complexes in CCPs, mediated by its ubiquitin-like domain and delayed endocytosis of the V2R and B2AR, but not of other membrane cargo like the epidermal growth factor receptor or transferrin receptor that are tyrosine-kinase receptors (84). It has been suggested that PLIC-2 could mediate this function by regulating the availability of a UIM-containing protein, such as Eps15 and/or epsin, that facilitates cargo recruitment into CCPs. Studies in yeast of UPDs epsin and Ede1 during receptor internalization showed that epsin and Ede1 ubiquitin-binding is dispensable for internalization of the GPCR Ste2, which uses ubiquitin internalization signals. However, variants of Ste2 carrying both ubiquitin-dependent and ubiquitin-independent internalization signals (Figure 3) were equally reliant on the epsin UIMs, indicating that UIM function is not restricted to ubiquitinated receptors (85). Conversely, recent studies in HEK293 cells show that ubiquitination of PAR1 and the UIM of epsin-1 are required for epsin1-dependent internalization of activated PAR1 (86). Furthermore, activation of PAR1 promotes epsin-1 deubiquitination, suggesting that epsin-1 deubiquitination may increase its endocytic activity toward ubiquitinated PAR1 to facilitate internalization.
Figure 3.

GPCR ubiquitin-dependent and ubiquitin-independent MVB sorting: ubiquitin targets agonist-activated GPCRs to lysosomes for degradation via the highly conserved ESCRT machinery. However, several GPCRs that are not ubiquitinated have been shown to sort to lysosomes independent of ubiquitination. The adaptor protein ALIX links the GPCR to ESCRT III and facilitates sorting to MVB/lysosomes. Ub, ubiquitin.
Ubiquitin controls the endocytic pathway and serves as an internalization signal
The plasma membrane abundance of GPCRs results from the balance between 2 pathways: one that delivers properly folded receptors to the cell surface, and the other that removes receptors by endocytosis, either temporarily (by internalization followed by recycling) or permanently (by internalization followed by subsequent degradation). It has recently been reported that the covalent modification of GPCRs by ubiquitin plays an important role on these processes.
Initial studies on yeast showed that GPCR internalization is mediated by direct ubiquitination (87, 88). A single ubiquitin moiety binding to Ste2 was sufficient to induce receptor internalization (89). Interestingly, ubiquitination of the cytoplasmic residues is not required for angiotensin 1 receptor internalization, in contrast to results obtained in yeast. These results suggest that endocytosis of mammalian angiotensin 1 receptor occurs by a different mechanism in mammals than in yeast (90).
However, the attachment of Lys63-linked ubiquitin chains facilitates endocytosis of many integral membrane proteins (91). Wolfe and colleagues revealed for the first time a direct and novel function for ubiquitination in internalization of a mammalian GPCR (92). They demonstrated that lysine-mutant PAR1, which is defective in ubiquitination, exhibited increased constitutive (ligand-independent) endocytosis. These results suggested that PAR1 is basally ubiquitinated and deubiquitinated after activation, and that ubiquitination negatively regulates PAR1 constitutive internalization. Recent studies with similar mutations of CXCR4, B2AR, and V2R demonstrate that mammalian GPCR ubiquitination is not required for receptor internalization (45, 67, 68).
Role of ubiquitin in the lysosomal sorting
As described above, abrogation of ubiquitination does not affect agonist-induced receptor internalization, but substantially increases receptor half-life (45, 67, 68). Although ubiquitination was originally characterized by its role in protein degradation carried out by the multiprotease complex, 26S proteasome (non-lysosomal-dependent), a nontraditional function of ubiquitin, especially in lysosomal degradation of membrane receptors, has been reported recently. CXCR4 provides a clear example of ubiquitin-dependent lysosomal sorting of a mammalian GPCR. A CXCR4 mutant defective in ubiquitination internalized at a normal rate but was not degraded in lysosomes (67). In the same way, it was observed that lysosomal trafficking correlated with B2AR ubiquitination. Agonist-stimulated B2AR sorted to late endosomes/lysosomes (71). Mutation of all lysines to arginines (zero-lysine β2-adrenergic receptor, 0K-B2AR) prevented the covalent attachment of ubiquitin, as well as receptor degradation (45). Only a negligible portion of the internalized 0K-B2AR localized to lysosomes, whereas most of the 0K-B2AR recycled to the plasma membrane (71).
Other well-studied GPCRs require agonist-dependent ubiquitination for subsequent steps of intracellular receptor trafficking involving proper receptor sorting to lysosomes. Both PAR2 and the neurokinin-1 receptor are ubiquitinated in response to chronic stimulation with the agonist and alterations in the correct ubiquitination prevent lysosomal trafficking and degradation (93, 94). Even neurokinin-1 receptor recycled to the plasma membrane suggests that receptors lacking ubiquitin tags preferentially traffic through recycling vesicles (94). Interestingly, ligand-stimulated lysosomal degradation of the platelet-activating factor receptor was reported to be ubiquitin-dependent, although receptor ubiquitination itself was not completely agonist dependent (95). Thus, ubiquitin is best known to target agonist-activated GPCRs to lysosomes for degradation via the highly conserved ESCRT machinery Figure 3. Nevertheless, as discussed earlier, a ubiquitination-deficient mutant of the murine DOR and the calcitonin-like receptor that is not ubiquitinated sort to lysosomes independent of ubiquitination. Surprisingly, these receptors require a ubiquitin-binding component of the ESCRT machinery, suggesting that not all GPCRs demand direct ubiquitination for interaction with the canonical ESCRT pathway. Conversely, PAR1 is ubiquitinated after agonist stimulation but is efficiently sorted from endosomes to MVBs/lysosomes and degraded independent of ubiquitination and the ubiquitin-binding ESCRT-0 and ESCRT-I proteins, HRS and Tsg101 (77, 92, 96). Further studies showed that PAR1 sorted to ILVs of MVBs through a pathway involving ESCRT-III components, which do not contain any known UBD (Figure 3). It was demonstrated that ALIX, an ESCRT-III-interacting protein, bound to a YPX3L motif within the second intracellular loop of PAR1, mediated ubiquitin-independent ESCRT-III/lysosomal sorting (83). In addition, 7 other mammalian GPCRs, such as α1B adrenergic receptor, angiotensin AT2, galanin (GAL2) histamine (H2), neuropeptide FF (NPFF2), neuropeptide S (NPS receptor), and P2Y1 purinergic receptors (P2YR), contain conserved YPX3L motifs within their second intracellular loop, raising the possibility that ALIX mediates lysosomal sorting without ubiquitin-protein modification, expanding the diversity of mechanism that control trafficking of mammalian GPCRs (76).
Proteolytic functions of the ubiquitin modification: regulation of proteasomal degradation
The process of ubiquitin targeting to proteasomes for degradation of cytosolic proteins has been well studied. The 26S proteasome is a 2-component complex consisting of a 20S proteolytic core complex and two 19S regulatory complexes. The proteasome is able to cleave basic, acidic, and hydrophobic amino acids within proteins. The polyubiquitinated proteins are recognized by the 19S subunits. The ubiquitin units are recycled through the action of ubiquitin hydrolases, and the targeted protein substrates are degraded by the 20S catalytic core complex. A ubiquitin chain of at least 4 units in length is required for recognition (97). Lys48-linked chains are the most abundant linkage when the proteasome is inhibited, so their role in proteasomal targeting was first assigned to Lys48-linked chains. However, early experiments established that other linkages could be recognized by the proteasome.
Three types of opioid receptor, δ, μ, κ, have been cloned and characterized extensively (98–101). Li and associates showed that Lys63-linked polyubiquitination down-regulated the human KOR (hKOR) (61, 102), the demonstration of GPCR ubiquitination. They found that agonist-promoted Lys63-linked polyubiquitin was the dominant form of hKOR ubiquitination. Ubiquitin modification occurs after receptor phosphorylation but before internalization. Interestingly, agonist-triggered ubiquitination plays a facultative regulatory, but not obligatory, role in agonist-induced down-regulation because a lysine-deficient hKOR mutant still underwent down-regulation (102). Previously, it was reported that agonist-induced down-regulation of the hKOR was partially reduced by the proteasome inhibitor MG132 or the lysosome inhibitor chloroquine and virtually abolished by the combination of the two. These data indicate that lysosomes as well as proteasomes are involved in agonist-induced down-regulation of the hKOR (102). Studies of MOR and DOR opioid receptors using 4 different proteasome inhibitors confirmed the essential role of the proteasome in MOR and DOR basal turnover and agonist-induced down-regulation (104). This role of ubiquitin-dependent degradation leading to basal turnover of cell surface receptors via the proteasomal pathway has also been described for metabotropic glutamate receptors (mGlurR1 and mGluR5) (50) and the human follitropin receptor (105). Other GPCRs, such as rhodopsin and components of GPCR signaling including transducin, rhodopsin G protein, and G protein-coupled receptor kinase 2, are degraded by the ubiquitin/proteasome pathway (106, 107).
More recently it has been recognized that ubiquitination of GPCRs is involved in ER-associated protein degradation (ERAD) (104). In most such cases, ubiquitination contributes to quality control (QC) exerted in the ER by clearing newly synthesized but misfolded receptors. During biogenesis of GPCRs and other transmembrane proteins at the ER, proteins are folded and adopt distinct conformations, which are guided by interactions with chaperones before export to the Golgi for further processing. Misfolded or incompletely folded receptors like DOR, MOR, calcium-sensing receptor, and thyrotropin-releasing hormone receptor are retrotranslocated to the cytosolic side of the ER membrane, conjugated with ubiquitin, and degraded by the proteasome (49, 108, 109). ER-proteasome regulation could be demonstrated in both heterologous and physiologically relevant cell types, suggesting that proteasomes may play an important role in QC during biosynthesis and functions to sort misfolded receptors to the ERAD pathway, ensuring membrane delivery of only properly folded receptors. The relevance of this process was revealed after the discovery of naturally occurring point mutations in rhodopsin, V2R, and gonadotropin hormone-releasing hormone receptors that cause misfolding and loss of cell surface expression and contribute to disease progression (110). Pharmacological agents that bind specifically to misfolded gonadotropin-releasing hormone receptor have been discovered and by inducing proper receptor folding they are able to rescue defects in sorting at the ER, thereby resulting in increased cell surface expression (111). In addition to ubiquitination, deubiquitination of GPCRs during biosynthesis regulates ER QC and increases receptor surface. The A2A adenosine receptor (A2AR) binds directly to ubiquitin-specific protease 4 (USP4), a deubiquitinating enzyme, which results in enhanced cell surface expression of functionally active receptor (112).
GPCR Transubiquitination and Role of Ubiquitination on GPCR-associated Proteins
GPCRs are known to transactivate growth factor tyrosine kinase receptors, such as the epidermal growth factor receptor, platelet-derived growth factor receptor, and fibroblast growth factor receptor, by releasing membrane-anchored ligands or through modulation of receptor cytoplasmic domains (113), leading to both Ras-dependent MAPK activation and stimulation of PI3K (114). Recently, it has been observed that signaling by GPCRs can promote transubiquitination of other GPCRs, which can change their cell and tissue responses. Transubiquitination was first described for the angiotensin-II type 1 receptor (AT1R) in response to dopamine D5 receptor (D5R) activation (115). Dopamine and angiotensin-II have counterregulatory effects on cellular signal transduction, production of reactive oxygen species, renal sodium excretion, and blood pressure, where AT1R signaling is prohypertensive and D5R signaling is antihypertensive. Li et al found that activation of the D5R decreased AT1R protein abundance by promoting AT1R degradation in proteasomes via an ubiquitin/proteasome pathway, suggesting a possible mechanism for the regulation of both receptors and therefore their counterregulatory effects.
GPCRs can form functional homodimers or heterodimers. The regulation and trafficking of dimerized GPCRs after activation of one receptor protomers are of considerable interest. A recent study showed that, depending on the activating ligand, internalized DOR and MOR heterodimers are either recycled or sorted to lysosomes for degradation (116). The stimulation with a DOR-specific agonist induces ubiquitination but not phosphorylation of MOR, causing degradation of both DOR and the nonstimulated MOR. Disruption of the heterodimers rescued MOR cell surface expression and increased the sensitivity to opiate agonists. These results underscore the potential importance of heterodimer-mediated GPCR transubiquitination on therapies that depend on trafficking of GPCRs. Further, in addition to transubiquitination between GPCRs, recent studies suggest that different receptors and antagonists can stimulate GPCR transubiquitination and degradation. For example, the orexin receptor OX2, which participates in regulating the sleep/wake cycle, is ubiquitinated and degraded in response to signaling by the proinflammatory cytokine TNF-α (117). Similarly, the S1PR inhibitor FTY720 (also known as fingolimod) induces phosphorylation of the carboxy-terminal domain of S1P1 receptor at multiple sites, promoting receptor internalization, polyubiquitination, and degradation by proteasomes. The ubiquitin E3 ligase WWP2 was also identified as a critical requirement for polyubiquitination of the S1PR carboxy-terminal domain induced by FTY720 (48).
The discussion thus far focused on the following 3 modes of GPCR ubiquitination: 1) constitutive receptor ubiquitination; 2) rapid, direct agonist-induced GPCR ubiquitination followed by proteasomal degradation; and 3) transubiquitination. In addition to these modes of regulation, ubiquitin modification of GPCR-associated proteins and GPCR-initiated signaling pathways provides another mechanism by which ubiquitination regulates GPCR trafficking and function. As noted above, internalization of mammalian GPCRs does not require ubiquitin posttranslational modification, in contrast to the paradigm in yeast. Shenoy and coworkers discovered that agonist-stimulated B2AR internalization required polyubiquitination of β-arrestin2 and MDM2, an E3 ubiquitin ligase that mediates β-arrestin2 ubiquitination (45). This ubiquitination appeared crucial for the proper function of arrestins (44, 118–120). β-Arrestins regulate the fate of the receptor by acting on the balance between receptor recycling to the plasma membrane or its lysosomal degradation. Interestingly, β-arrestin is able to recruit ubiquitin ligases and promote receptor ubiquitination, acting as adaptor proteins. In this case, β-arrestin2 interacts with the ubiquitin ligase Nedd4, which promotes B2AR ubiquitination at endosomes, leading to its lysosomal targeting (44). Ubiquitin ligases of the Need4 family harbor WW domains that can interact with specific proline-rich motifs. Surprisingly, no such PPxY motif has been identified in β-arrestin, and polyproline regions are not involved in Nedd4 interactions. In addition, Nedd4 recruitment to the B2AR was not affected by mutations in Nedd4 WW domains, indicating that the interaction between Nedd4 and β-arrestin involves noncanonical binding (121). Another notable feature of β-arrestin ubiquitination is the distinct kinetics that correspond to a particular receptor type (122). Although stimulation of the B2AR leads to transient β-arrestin ubiquitination, stimulation of the AT1aR angiotensin receptor results in a relatively sustained β-arrestin ubiquitination (123). An additional layer of complexity emerges from observations that β-arrestin interacts with deubiquitinase enzymes that regulate their ubiquitination status as well as receptor ubiquitination. In fact, USP33 (a deubiquitinase enzyme) was first identified as a β-arrestin interactant (120), suggesting that β-arrestin2 could be involved in USP33 recruitment to the B2AR. However, Berthouze and coworkers demonstrated that USP33 is transferred from agonist-activated B2AR to β-arrestin2, triggering its deubiquitination and dissociation from the receptor, once internalized (124). These data suggest that the association and dissociation of receptor from β-arrestin may coordinate the ubiquitin conjugation and deconjugating activities toward B2AR to tune the balance between receptor degradation and recycling.
Several recent studies have demonstrated that GPCR-associated proteins and β-arrestin are also regulated by ubiquitination. The number of these proteins seems to be relatively limited, but the effects of ubiquitination are diverse. Both yeast and mammalian stimulatory G protein α-subunits and the effectors they activate are dynamically ubiquitinated and degraded by the 26S proteasome (125–130). Other G protein subunits, such as the olfactory (Gαo), inhibitory (Gαi), Gt, and rhodopsin βγ, are degraded by proteasomal-dependent pathways. Ubiquitin ligases involved in G protein ubiquitination have yet to be identified with the exception of Gαi3, which is regulated by the RING domain E3 ligase GIPN (131). Other signal transduction proteins, including regulator of G protein signaling proteins and G protein-coupled receptor kinase 2, are also ubiquitinated, although the significance of this process is undefined (107, 132). Immediate downstream effector molecules of the GPCR pathway have also been shown to be regulated by ubiquitination. The most representative example is the inositol 1,4,5-trisphosphate receptor, Ins(1,4,5)P3R, that is ubiquitinated on activation of phospholipase C-linked receptors (133). Initial studies showed that Ins(1,4,5)P3R levels were reduced by chronic activation of certain phospholipase C-linked GPCRs such as muscarinic acetylcholine receptors. This down-regulation was due to increased receptor degradation, and the receptors were processed by the ubiquitin proteasome pathway. The ubiquitination and degradation of Ins(1,4,5)P3Rs represent the first example of an intracellular protein being processed by the ERAD pathway following GPCR activation.
Balanced Ubiquitination and Deubiquitination Determine GPCR Fate
GPCR ubiquitination is dynamic and can be reversed. Similar to phosphorylation and dephosphorylation, which are carried out by large families of kinases and phosphatases, ubiquitination is counteracted by deubiquitinating enzymes (DUBs) that deconjugate ubiquitin-modified proteins and rescue the substrate from proteasomal degradation. There are some 90 DUBs in the human proteome. DUBs can be divided into 5 different classes: ubiquitin C-terminal hydrolases, USPs, Machado-Joseph disease protein domain proteases, ovarian tumor proteases, and JAMM motif proteases. All DUBs are cysteine proteases, with the exception of JAMM motif proteases, which are metalloproteases (134). DUBs can display specificity at multiple levels, thus distinguishing between the many ubiquitin-like molecules, isopeptides (using an ϵ-amino group), and linear peptides (using an α-amino group), and between different types of ubiquitin linkage and chain structure (135). Ligand-induced deubiquitinases have been shown only for a few GPCRs. Adenosine A2AR receptors are deubiquitinated by USP4 (112); the β2-AR undergoes increased agonist-stimulated ubiquitination, lysosomal trafficking, and degradation after knockdown of USPs 20 and 33 (124). PAR1 is tightly regulated in a spatiotemporal manner through the actions of ubiquitin ligases and DUBs (86). Similarly, the balance between ubiquitination and deubiquitination of Frizzled-4, a 7-transmembrane receptor for Wnt ligands, is also important for regulating surface expression and cellular responsiveness (136). Constitutive ubiquitination of Frizzled-4 promotes internalization and lysosomal degradation, whereas deubiquitination mediated by USP8 leads to recycling and increased surface expression that occurs in a Wnt-independent manner (136). DUBs are remarkably specific. Overexpression of USP4, which promotes cell surface targeting of the A2AR (112), does not affect B2AR ubiquitination (124). Thus, receptor deubiquitination involves specific USP isoforms. Berthouze and coworkers proposed that fast recycling from early endosomes mainly involves dephosphorylation of nonubiquitinated receptors, whereas slow recycling of ubiquitinated receptors from deeper subcellular compartments is regulated by DUBs and protein phosphatases (124). Thus, the DUBs switch the receptor's fate from lysosomal/proteasomal degradation to recycling and enhanced cellular resensitization (Figure 4).
Figure 4.
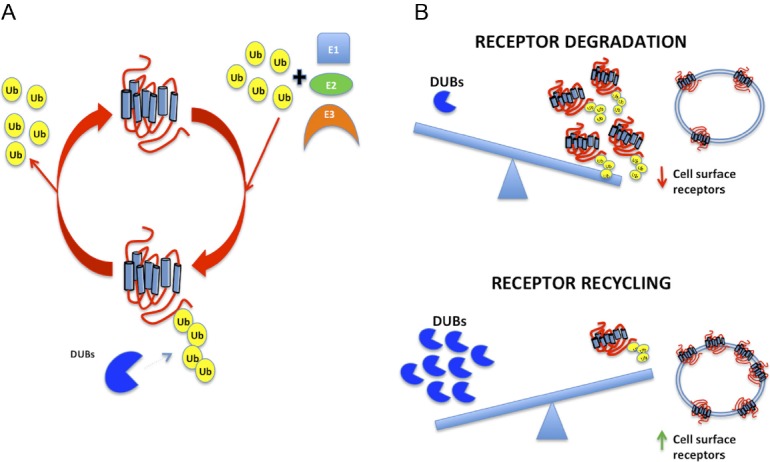
A, Ubiquitination is a reversible process. DUBs deconjugate ubiquitin-modified proteins and rescue the substrate from proteasomal degradation. B, The balance between GPCR ubiquitination and deubiquitination regulates their surface expression and cellular responsiveness. Greater receptor ubiquitination results in fewer cell surface receptors by virtue of degradation and down-regulation. Conversely, with more DUBs, the final number of receptors that are ubiquitinated is diminished, with less attendant degradation, consequently more receptor recycling, and greater numbers of membrane-delimited GPCRs.
Other GPCRs regulated by ubiquitination and deubiquitination include the CXCR4. CXCR4 undergoes CXCL12-induced ubiquitination, resulting in lysosomal degradation (137). Overexpression of USP14 provoked deubiquitination of CXCR4, and therefore, its escape from degradation (138). Furthermore, the deubiquitinating enzyme USP8 has been shown to participate indirectly in CXCR4 regulation by modulating the dynamics of signaling endosomes (139). CXCR7 binds CXCL12 with high affinity. Canals and coworkers showed that in the basal state, CXCR7 is ubiquitinated and the Lys residues on its carboxy-terminus are the site of ubiquitination (140). They observed that receptor activation by CXCL12 results in reversible deubiquitination because subsequent removal of the chemokine partially restored the ubiquitinated receptor to levels detected in the basal state. These results contrast to what has been described for CXCR4 and highlight the differences that could potentially underlie distinct functions and/or patterns of expression of the 2 CXCL12-binding receptors (140). Thus, the same ligand can exert dissimilar effects on different receptors. In the same manner, the activation of a single GPCR by 2 or more distinct ligands can elicit distinct responses. This phenomenon is known as “biased agonism” (141) and knowledge of the molecular basis for biased agonism has important implications on drug development in pharmacology. Our recent work revealed that ubiquitination is important in determining differential GPCR regulation induced by biased agonists. The PTHR regulates bone growth and mineral ion balance and is responsive to distinct activating [PTH(1–84), PTH(1–34)] and nonactivating [PTH(7–84), PTH(7–34)] ligands. Stimulation with the activating ligand PTH(1–34) elicited transient PTHR ubiquitination (62). The receptor is internalized and then recycled following deubiquitination. In contrast, nonactivating PTH(7–34) triggered continuous PTHR ubiquitination and receptor degradation. The different trafficking behaviors exhibited by the PTHR appear to be regulated by the deubiquitinating enzyme USP2. PTH(1–34) increase USP2 levels, favoring the balance toward rapid deubiquitination and recycling of the receptor. However, PTH(7–34) does not increase or activate USP2. The slow deubiquitination induced by PTH(7–34) leads to an accumulation of ubiquitin tags on the PTHR and subsequent receptor degradation by the proteasomal pathway (62). These findings indicate that specific PTHR ligands distinctly modulate the activity of the ubiquitination machinery, resulting in differential receptor ubiquitination and trafficking and provide a novel paradigm for ligand biased GPCR ubiquitination.
Ubiquitination, GPCRs, and Disease
Dysregulated GPCR trafficking and the deregulation of ubiquitin pathways (142), as well as defective endocytosis, are associated with several human diseases, including many tumor types (143). GPR55 in mouse skin, for example, drives tumor development and is up-regulated in human squamous cell carcinomas (144). In this context, recent studies show that CXCR7 expression increases tumor formation and metastasis for some cancers (145, 146). The data also suggest that an important function of CXCR7 is to prevent degradation of CXCR4 (147). Therefore, high expression of CXCR7 in tumor cells may contribute to disproportionate signaling through CXCR4, a landmark of the pathophysiology of WHIM syndrome, a rare congenital immunodeficiency disorder that is also associated with tumor growth and metastasis (148). The ubiquitination state of CXCR7 under these pathophysiological conditions could help explain this scenario; however, it remains to be explored. Additionally, disrupted trafficking of CXCR4 and PAR1 within the endosomal-lysosomal system contributes to increased surface expression of receptors in breast cancer cells and cancer progression (149, 150). Tumor cells display aberrant PAR1 trafficking, which causes persistent signaling and cellular invasion. These defects appear to be specific to breast cancer cells, because PAR1 in normal human mammary epithelial cells displays appropriate trafficking and signal termination. It remains to be determined if the ubiquitin pathway is the mechanism responsible for defective PAR1 trafficking in breast carcinoma (149).
Conversely, the efficient coupling of ER QC and proteasomal activity may well be necessary to avert inherited diseases, such as retinitis pigmentosa, male pseudohermaphroditism, and nephrogenic diabetes insipidus, that result from ER retention of mutated rhodopsin (151, 152), luteinizing hormone receptors (153, 154), and V2R receptors (155).
It is increasingly apparent that ubiquitination is crucial to cell integrity. This awareness has informed the development of ubiquitin-proteasome pathway inhibitors as potential therapeutic agents, culminating in the recent approval for patients with refractory multiple myeloma of bortezomib, a potent proteasome inhibitor that blocks the degradation of ubiquitinated proteins (156). Moreover, proteasomal inhibitors have anti-inflammatory effects and as such their short-term use is proposed to exert beneficial actions in pathologic conditions associated with acute inflammation, including myocardial infarction or stroke (157–160). Thus, it is also likely that inhibitors of either ubiquitinating or DUBs regulating various cardiovascular proteins may have similar therapeutic utility. These studies suggest that the development of pharmacological agents capable of directly or indirectly inducing ubiquitination and down-regulation of targeted GPCRs could be applied in particular disease scenarios.
Conclusion and Future Directions
The ubiquitin system has integral roles in most cellular events and is therefore an important therapeutic target, for example, for the development of treatments against cancer and neurodegenerative disorders, as well as for inflammatory and infectious diseases (161, 103). In an interesting way, it revealed an unanticipated role for ubiquitination in regulating mammalian GPCR longevity and lysosomal sorting. Similarly astonishing is the function that modification by ubiquitin plays in the modulation of GPCR regulators such as GRKs, MAPKs, and β-arrestins. Although the ubiquitin posttranslational modification is carried out by the addition of the same ubiquitin chemical tag, this modification is capable of mediating discrete and very different molecular consequences. Our understanding of the regulation of GPCR signaling and trafficking by ubiquitination and deubiquitination is extraordinarily limited. Thus, studies are needed to explore new functions of ubiquitin in GPCR signaling. This field is an emerging area at the nexus of endocrinology and pharmacology that has important implications in drug development with fewer adverse side effects.
Acknowledgments
Original work conducted in the authors' laboratories was supported by Grants DK054171 and DK069998 (P.A.F.) from the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- A2AR
- A2A adenosine receptor
- ALIX
- an ESCRT-III-interacting protein
- AT1R
- angiotensin-II type 1 receptor
- B2AR
- β2-adrenergic receptor
- CCPs
- clathrin-coated pit
- CXCR4
- C-X-C chemokine receptor-4
- D5R
- D5 receptor
- DOR
- δ-opioid receptors
- DUB
- deubiquitinating enzymes
- ER
- endoplasmic reticulum
- ERAD
- ER-associated protein degradation
- ESCRT
- endosomal-sorting-complex-required-for-transport
- GPCR
- G protein-coupled receptors
- GRK
- G protein-coupled receptor kinases
- HECT
- homologous to the E6-AP carboxyl terminus
- HRS
- hepatocyte growth factor-regulated tyrosine kinase substrate
- Ins(1,4,5)P3R
- inositol 1,4,5-trisphosphate receptor
- 0K-B2AR
- zero-lysine β2-adrenergic receptor
- KOR
- κ-opioid receptor
- mGlurR1
- metabotropic glutamate receptor 1
- mGlurR5
- metabotropic glutamate receptor 5
- MOR
- μ-opioid receptor
- MVB
- multivesicular bodies
- PAR
- protease-activated receptor
- PLIC-2
- protein linking IAP to cytoskeleton
- PTHR
- parathyroid hormone receptor
- QC
- quality control
- RING
- really interesting new gene
- S1PR
- sphingosine-1-phosphate receptor
- UBA
- ubiquitin-associated domain
- UBC
- ubiquitin-carrier enzyme
- UBD
- ubiquitin-binding domain
- UIM
- ubiquitin interacting motif
- USP
- ubiquitin-specific protease
- V2R
- vasopressin receptor 2.
References
- 1. Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260 [DOI] [PubMed] [Google Scholar]
- 2. Sharman JL, Mpamhanga CP, Spedding M, et al. , NC-IUPHAR IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 2011;39:D534–D538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefkowitz RJ. G protein-coupled receptor kinases. Cell. 1993;74:409–412 [DOI] [PubMed] [Google Scholar]
- 4. Ferguson SSG, Barak LS, Zhang J, Caron MG. G-protein-coupled receptor regulation: role of G-protein-coupled receptor kinases and arrestins. Can J Physiol Pharmacol. 1996;74:1095–1110 [DOI] [PubMed] [Google Scholar]
- 5. Ferguson SS, Caron MG. G protein-coupled receptor adaptation mechanisms. Semin Cell Dev Biol. 1998;9:119–127 [DOI] [PubMed] [Google Scholar]
- 6. Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319 [DOI] [PubMed] [Google Scholar]
- 7. Hall RA, Premont RT, Lefkowitz RJ. Heptahelical receptor signaling: beyond the G protein paradigm. J Cell Biol. 1999;145:927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183 [DOI] [PubMed] [Google Scholar]
- 9. Schoneberg T, Schultz G, Gudermann T. Structural basis of G protein-coupled receptor function. Mol Cell Endocrinol. 1999;151:181–193 [DOI] [PubMed] [Google Scholar]
- 10. Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434 [DOI] [PubMed] [Google Scholar]
- 12. Calebiro D, Nikolaev VO, Lohse MJ. Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J Mol Endocrinol. 2010;45:1–8 [DOI] [PubMed] [Google Scholar]
- 13. Feinstein TN, Wehbi VL, Ardura JA, et al. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. Endocytosis promotes rapid dopaminergic signaling. Neuron. 2011;71:278–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vilardaga JP, Gardella TJ, Wehbi VL, Feinstein TN. Non-canonical signaling of the PTH receptor. Trends Pharmacol Sci. 2012;33:423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okazaki M, Ferrandon S, Vilardaga JP, Bouxsein ML, Potts JT, Jr, Gardella TJ. Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation. Proc Natl Acad Sci USA. 2008;105:16525–16530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568 [DOI] [PubMed] [Google Scholar]
- 18. Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534 [DOI] [PubMed] [Google Scholar]
- 19. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517 [DOI] [PubMed] [Google Scholar]
- 20. Goodman OB, Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450 [DOI] [PubMed] [Google Scholar]
- 21. Laporte SA, Oakley RH, Zhang J, et al. The β2-adrenergic receptor/barrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007;8:462–470 [DOI] [PubMed] [Google Scholar]
- 23. Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106:145–155 [DOI] [PubMed] [Google Scholar]
- 24. Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905 [DOI] [PubMed] [Google Scholar]
- 25. Hicke L. Gettin' down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999;9:107–112 [DOI] [PubMed] [Google Scholar]
- 26. Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18 [DOI] [PubMed] [Google Scholar]
- 27. von Zastrow M. Mechanisms regulating membrane trafficking of G protein-coupled receptors in the endocytic pathway. Life Sci. 2003;74:217–224 [DOI] [PubMed] [Google Scholar]
- 28. Lefkowitz RJ, Pitcher J, Krueger K, Daaka Y. Mechanisms of β-adrenergic receptor desensitization and resensitization. Adv Pharmacol. 1998;42:416–420 [DOI] [PubMed] [Google Scholar]
- 29. Mohan ML, Vasudevan NT, Gupta MK, Martelli EE, Naga Prasad SV. G-protein coupled receptor resensitization-appreciating the balancing act of receptor function. Curr Mol Pharmacol. 2012;5:350–361 [PMC free article] [PubMed] [Google Scholar]
- 30. Torrecilla I, Tobin AB. Co-ordinated covalent modification of G-protein coupled receptors. Curr Pharm Des. 2006;12:1797–1808 [DOI] [PubMed] [Google Scholar]
- 31. Wojcikiewicz RJ. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol Sci. 2004;25:35–41 [DOI] [PubMed] [Google Scholar]
- 32. Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178 [DOI] [PubMed] [Google Scholar]
- 33. Lee I, Schindelin H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell. 2008;134:268–278 [DOI] [PubMed] [Google Scholar]
- 34. Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A. 1980;77:1783–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583 [DOI] [PubMed] [Google Scholar]
- 36. Hershko A, Ciechanover A, Varshavsky A. Basic Medical Research Award. The ubiquitin system. Nat Med. 2000;6:1073–1081 [DOI] [PubMed] [Google Scholar]
- 37. Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434 [DOI] [PubMed] [Google Scholar]
- 39. Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitby FG, Xia G, Pickart CM, Hill CP. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem. 1998;273:34983–34991 [DOI] [PubMed] [Google Scholar]
- 41. Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007;14:941–948 [DOI] [PubMed] [Google Scholar]
- 42. Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21 [DOI] [PubMed] [Google Scholar]
- 44. Shenoy SK, Xiao K, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J Biol Chem. 2008;283:22166–22176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated ß2-adrenergic receptor and ß-arrestin. Science. 2001;294:1307–1313 [DOI] [PubMed] [Google Scholar]
- 46. Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003;5:709–722 [DOI] [PubMed] [Google Scholar]
- 47. Hislop JN, Henry AG, Marchese A, von Zastrow M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J Biol Chem. 2009;284:19361–19370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oo ML, Chang SH, Thangada S, et al. Engagement of S1P(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121:2290–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang Y, Niwa J, Sobue G, Breitwieser GE. Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J Biol Chem. 2006;281:11610–11617 [DOI] [PubMed] [Google Scholar]
- 50. Moriyoshi K, Iijima K, Fujii H, Ito H, Cho Y, Nakanishi S. Seven in absentia homolog 1A mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2004;101:8614–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rondou P, Haegeman G, Vanhoenacker P, Van Craenenbroeck K. BTB Protein KLHL12 targets the dopamine D4 receptor for ubiquitination by a Cul3-based E3 ligase. J Biol Chem. 2008;283:11083–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cooray SN, Guasti L, Clark AJ. The E3 ubiquitin ligase Mahogunin ubiquitinates the melanocortin 2 receptor. Endocrinology. 2011;152:4224–4231 [DOI] [PubMed] [Google Scholar]
- 53. Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. 2011;12:295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ’Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NF-kappaB activation. EMBO Rep. 2009;10:706–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523 [DOI] [PubMed] [Google Scholar]
- 57. Flick K, Ouni I, Wohlschlegel JA, et al. Proteolysis-independent regulation of the transcription factor Met4 by a single Lys 48-linked ubiquitin chain. Nat Cell Biol. 2004;6:634–641 [DOI] [PubMed] [Google Scholar]
- 58. Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012;31:3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ciechanover A, Breitschopf K, Hatoum OA, Bengal E. Degradation of MyoD by the ubiquitin pathway: regulation by specific DNA-binding and identification of a novel site for ubiquitination. Mol Biol Rep. 1999;26:59–64 [DOI] [PubMed] [Google Scholar]
- 60. Rieser E, Cordier SM, Walczak H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci. 2013;38:94–102 [DOI] [PubMed] [Google Scholar]
- 61. Li JG, Haines DS, Liu-Chen LY. Agonist-promoted Lys63-linked polyubiquitination of the human κ-opioid receptor is involved in receptor down-regulation. Mol Pharmacol. 2008;73:1319–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alonso V, Magyar CE, Wang B, Bisello A, Friedman PA. Ubiquitination–deubiquitination balance dictates ligand-stimulated PTHR sorting. J Bone Miner Res. 2011;26:2923–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644 [DOI] [PubMed] [Google Scholar]
- 64. Hoppe T. Multiubiquitylation by E4 enzymes: ’one size’ doesn't fit all. Trends Biochem Sci. 2005;30:183–187 [DOI] [PubMed] [Google Scholar]
- 65. Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 2004;14:103–106 [DOI] [PubMed] [Google Scholar]
- 66. Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180 [DOI] [PubMed] [Google Scholar]
- 67. Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512 [DOI] [PubMed] [Google Scholar]
- 68. Martin NP, Lefkowitz RJ, Shenoy SK. Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J Biol Chem. 2003;278:45954–45959 [DOI] [PubMed] [Google Scholar]
- 69. Hislop JN, Henry AG, von Zastrow M. Ubiquitination in the first cytoplasmic loop of mu-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J Biol Chem. 2011;286:40193–40204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liang W, Hoang Q, Clark RB, Fishman PH. Accelerated dephosphorylation of the β2-adrenergic receptor by mutation of the C-terminal lysines: effects on ubiquitination, intracellular trafficking, and degradation. Biochemistry. 2008;47:11750–11762 [DOI] [PubMed] [Google Scholar]
- 71. Xiao K, Shenoy SK. β2-adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J Biol Chem. 2011;286:12785–12795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains—-from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Grabbe C, Dikic I. Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem Rev. 2009;109:1481–1494 [DOI] [PubMed] [Google Scholar]
- 74. Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708–714 [DOI] [PubMed] [Google Scholar]
- 75. Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat Rev Mol Cell Biol. 2010;11:556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–398 [DOI] [PubMed] [Google Scholar]
- 77. Dores MR, Paing MM, Lin H, Montagne WA, Marchese A, Trejo J. AP-3 regulates PAR1 ubiquitin-independent MVB/lysosomal sorting via an ALIX-mediated pathway. Mol Biol Cell. 2012;23:3612–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Giordano F, Simoes S, Raposo G. The ocular albinism type 1 (OA1) GPCR is ubiquitinated and its traffic requires endosomal sorting complex responsible for transport (ESCRT) function. Proc Natl Acad Sci U S A. 2011;108:11906–11911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hasdemir B, Bunnett NW, Cottrell GS. Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J Biol Chem. 2007;282:29646–29657 [DOI] [PubMed] [Google Scholar]
- 80. Hislop JN, Marley A, Von Zastrow M. Role of mammalian vacuolar protein-sorting proteins in endocytic trafficking of a non-ubiquitinated G protein-coupled receptor to lysosomes. J Biol Chem. 2004;279:22522–22531 [DOI] [PubMed] [Google Scholar]
- 81. Henry AG, White IJ, Marsh M, von Zastrow M, Hislop JN. The role of ubiquitination in lysosomal trafficking of delta-opioid receptors. Traffic. 2011;12:170–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cottrell GS, Padilla B, Pikios S, et al. Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J Biol Chem. 2007;282:12260–12271 [DOI] [PubMed] [Google Scholar]
- 83. Dores MR, Chen B, Lin H, et al. ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol. 2012;197:407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. N'Diaye EN, Hanyaloglu AC, Kajihara KK, et al. The ubiquitin-like protein PLIC-2 is a negative regulator of G protein-coupled receptor endocytosis. Mol Biol Cell. 2008;19:1252–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dores MR, Schnell JD, Maldonado-Baez L, Wendland B, Hicke L. The function of yeast epsin and Ede1 ubiquitin-binding domains during receptor internalization. Traffic. 2010;11:151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen B, Dores MR, Grimsey N, Canto I, Barker BL, Trejo J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J Biol Chem. 2011;286:40760–40770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287 [DOI] [PubMed] [Google Scholar]
- 88. Roth AF, Davis NG. Ubiquitination of the yeast a-factor receptor. J Cell Biol. 1996;134:661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Terrell J, Shih S, Dunn R, Hicke L. A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol Cell. 1998;1:193–202 [DOI] [PubMed] [Google Scholar]
- 90. Mihalik B, Gaborik Z, Varnai P, Clark AJ, Catt KJ, Hunyady L. Endocytosis of the AT1A angiotensin receptor is independent of ubiquitylation of its cytoplasmic serine/threonine-rich region. Int J Biochem Cell Biol. 2003;35:992–1002 [DOI] [PubMed] [Google Scholar]
- 91. Galan JM, Haguenauer-Tsapis R. Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J. 1997;16:5847–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. 2007;177:905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076–16087 [DOI] [PubMed] [Google Scholar]
- 94. Cottrell GS, Padilla B, Pikios S, et al. Ubiquitin-dependent down-regulation of the neurokinin-1 receptor. J Biol Chem. 2006;281:27773–27783 [DOI] [PubMed] [Google Scholar]
- 95. Dupre DJ, Chen Z, Le Gouill C, et al. Trafficking, ubiquitination, and down-regulation of the human platelet-activating factor receptor. J Biol Chem. 2003;278:48228–48235 [DOI] [PubMed] [Google Scholar]
- 96. Gullapalli A, Wolfe BL, Griffin CT, Magnuson T, Trejo J. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol Biol Cell. 2006;17:1228–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061 [PubMed] [Google Scholar]
- 98. Yasuda K, Raynor K, Kong H, et al. Cloning and functional comparison of κ and δ opioid receptors from mouse brain. Proc Natl Acad Sci U S A. 1993;90:6736–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12 [PubMed] [Google Scholar]
- 100. Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The δ-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci U S A. 1992;89:12048–12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955 [DOI] [PubMed] [Google Scholar]
- 102. Li JG, Benovic JL, Liu-Chen LY. Mechanisms of agonist-induced down-regulation of the human κ-opioid receptor: internalization is required for down-regulation. Mol. Pharmacol. 2000;58:795–801 [PubMed] [Google Scholar]
- 103. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444 [DOI] [PubMed] [Google Scholar]
- 104. Chaturvedi K, Bandari P, Chinen N, Howells RD. Proteasome involvement in agonist-induced down-regulation of mu and delta opioid receptors. J Biol Chem. 2001;276:12345–12355 [DOI] [PubMed] [Google Scholar]
- 105. Cohen BD, Bariteau JT, Magenis LM, Dias JA. Regulation of follitropin receptor cell surface residency by the ubiquitin-proteasome Pathway. Endocrinology. 2003;144:4393–4402 [DOI] [PubMed] [Google Scholar]
- 106. Obin MS, Jahngen-Hodge J, Nowell T, Taylor A. Ubiquitinylation and ubiquitin-dependent proteolysis in vertebrate photoreceptors (rod outer segments). Evidence for ubiquitinylation of Gt and rhodopsin. J Biol Chem. 1996;271:14473–14484 [DOI] [PubMed] [Google Scholar]
- 107. Penela P, Ruiz-Gómez A, Castaño JG, Mayor F., Jr Degradation of the G protein-coupled receptor kinase 2 by the proteasome pathway. J Biol Chem. 1998;273:35238–35244 [DOI] [PubMed] [Google Scholar]
- 108. Petaja-Repo UE, Hogue M, Laperriere A, Bhalla S, Walker P, Bouvier M. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem. 2001;276:4416–4423 [DOI] [PubMed] [Google Scholar]
- 109. Cook LB, Zhu C-C, Hinkle PM. Thyrotropin-releasing hormone receptor processing: role of ubiquitination and proteasomal degradation. Mol Endocrinol. 2003;17:1777–1791 [DOI] [PubMed] [Google Scholar]
- 110. Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225–250 [DOI] [PubMed] [Google Scholar]
- 111. Conn PM, Ulloa-Aguirre A. Pharmacological chaperones for misfolded gonadotropin-releasing hormone receptors. Adv Pharmacol. 2011;62:109–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Milojevic T, Reiterer V, Stefan E, et al. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol Pharmacol. 2006;69:1083–1094 [DOI] [PubMed] [Google Scholar]
- 113. Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4:651–657 [DOI] [PubMed] [Google Scholar]
- 114. Gavi S, Shumay E, Wang HY, Malbon CC. G-protein-coupled receptors and tyrosine kinases: crossroads in cell signaling and regulation. Trends Endocrinol Metab. 2006;17:48–54 [DOI] [PubMed] [Google Scholar]
- 115. Li H, Armando I, Yu P, et al. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Invest. 2008;118:2180–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. He SQ, Zhang ZN, Guan JS, et al. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron. 2011;69:120–131 [DOI] [PubMed] [Google Scholar]
- 117. Zhan S, Cai GQ, Zheng A, et al. Tumor necrosis factor-alpha regulates the Hypocretin system via mRNA degradation and ubiquitination. Biochim Biophys Acta. 2011;1812:565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of ß-arrestin directs assembly and targeting of 7TM receptor-signalosomes. J Biol Chem. 2005;280:15315–15324 [DOI] [PubMed] [Google Scholar]
- 119. Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell. 2008;135:714–725 [DOI] [PubMed] [Google Scholar]
- 120. Shenoy SK, Modi AS, Shukla AK, et al. ß-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci USA. 2009;106:6650–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Becuwe M, Herrador A, Haguenauer-Tsapis R, Vincent O, Leon S. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem Res Intl. 2012;2012:242764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shenoy SK, Lefkowitz RJ. Trafficking patterns of β-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506 [DOI] [PubMed] [Google Scholar]
- 123. Shenoy SK. Seven-transmembrane receptors and ubiquitination. Circ Res. 2007;100:1142–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Berthouze M, Venkataramanan V, Li Y, Shenoy SK. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009;28:1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Marotti LA, Jr, Newitt R, Wang Y, Aebersold R, Dohlman HG. Direct identification of a G protein ubiquitination site by mass spectrometry. Biochemistry. 2002;41:5067–5074 [DOI] [PubMed] [Google Scholar]
- 126. Wang Y, Marotti LA, Jr, Lee MJ, Dohlman HG. Differential regulation of G protein α subunit trafficking by mono- and polyubiquitination. J Biol Chem. 2005;280:284–291 [DOI] [PubMed] [Google Scholar]
- 127. Obin M, Lee BY, Meinke G, et al. Ubiquitylation of the transducin βγ subunit complex. Regulation by phosducin. J Biol Chem. 2002;277:44566–44575 [DOI] [PubMed] [Google Scholar]
- 128. Hamilton MH, Cook LA, McRackan TR, Schey KL, Hildebrandt JD. γ2 subunit of G protein heterotrimer is an N-end rule ubiquitylation substrate. Proc Natl Acad Sci U S A. 2003;100:5081–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mouledous L, Neasta J, Uttenweiler-Joseph S, et al. Long-term morphine treatment enhances proteasome-dependent degradation of Gβ in human neuroblastoma SH-SY5Y cells: correlation with onset of adenylate cyclase sensitization. Mol Pharmacol. 2005;68:467–476 [DOI] [PubMed] [Google Scholar]
- 130. Naviglio S, Pagano M, Romano M, et al. Adenylate cyclase regulation via proteasome-mediated modulation of Gαs levels. Cell Signal. 2004;16:1229–1237 [DOI] [PubMed] [Google Scholar]
- 131. Fischer T, De Vries L, Meerloo T, Farquhar MG. Promotion of Gαi3 subunit down-regulation by GIPN, a putative E3 ubiquitin ligase that interacts with RGS-GAIP. Proc Natl Acad Sci U S A. 2003;100:8270–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim E, Arnould T, Sellin L, et al. Interaction between RGS7 and polycystin. Proc Natl Acad Sci U S A. 1999;96:6371–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wojcikiewicz RJ, Xu Q, Webster JM, Alzayady K, Gao C. Ubiquitination and proteasomal degradation of endogenous and exogenous inositol 1,4,5-trisphosphate receptors in alpha T3–1 anterior pituitary cells. J Biol Chem. 2003;278:940–947 [DOI] [PubMed] [Google Scholar]
- 134. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286 [DOI] [PubMed] [Google Scholar]
- 135. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563 [DOI] [PubMed] [Google Scholar]
- 136. Mukai A, Yamamoto-Hino M, Awano W, Watanabe W, Komada M, Goto S. Balanced ubiquitylation and deubiquitylation of Frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 2010;29:2114–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem. 2007;282:36971–36979 [DOI] [PubMed] [Google Scholar]
- 138. Mines MA, Goodwin JS, Limbird LE, Cui FF, Fan GH. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J Biol Chem. 2009;284:5742–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Berlin I, Schwartz H, Nash PD. Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8.STAM complex. J Biol Chem. 2010;285:34909–34921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Canals M, Scholten DJ, de Munnik S, Han MK, Smit MJ, Leurs R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS One. 2012;7:e34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Urban JD, Clarke WP, von Zastrow M, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13 [DOI] [PubMed] [Google Scholar]
- 142. Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6:776–788 [DOI] [PubMed] [Google Scholar]
- 143. Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60 [DOI] [PubMed] [Google Scholar]
- 144. Perez-Gomez E, Andradas C, Flores JM, et al. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas [published online July 2, 2012]. Oncogene. doi:10.1038/onc.2012.278 [DOI] [PubMed] [Google Scholar]
- 145. Miao Z, Luker KE, Summers BC, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zabel BA, Lewen S, Berahovich RD, Jaen JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol Cancer. 2011;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sanchez-Alcaniz JA, Haege S, Mueller W, et al. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron. 2011;69:77–90 [DOI] [PubMed] [Google Scholar]
- 148. Balabanian K, Lagane B, Pablos JL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449–2457 [DOI] [PubMed] [Google Scholar]
- 149. Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004;24:1990–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Li YM, Pan Y, Wei Y, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer cell. 2004;6:459–469 [DOI] [PubMed] [Google Scholar]
- 151. Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277:34150–34160 [DOI] [PubMed] [Google Scholar]
- 152. Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918 [DOI] [PubMed] [Google Scholar]
- 153. Apaja PM, Tuusa JT, Pietila EM, Rajaniemi HJ, Petaja-Repo UE. Luteinizing hormone receptor ectodomain splice variant misroutes the full-length receptor into a subcompartment of the endoplasmic reticulum. Mol Biol Cell. 2006;17:2243–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Richter-Unruh A, Verhoef-Post M, Malak S, Homoki J, Hauffa BP, Themmen AP. Leydig cell hypoplasia: absent luteinizing hormone receptor cell surface expression caused by a novel homozygous mutation in the extracellular domain. J Clin Endocrinol Metab. 2004;89:5161–5167 [DOI] [PubMed] [Google Scholar]
- 155. Sadeghi HM, Innamorati G, Birnbaumer M. An X-linked NDI mutation reveals a requirement for cell surface V2R expression. Mol Endocrinol. 1997;11:706–713 [DOI] [PubMed] [Google Scholar]
- 156. Mitchell BS. The proteasome—an emerging therapeutic target in cancer. N Engl J Med. 2003;348:2597–2598 [DOI] [PubMed] [Google Scholar]
- 157. Bao J, Sato K, Li M, et al. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am J Physiol Heart Circ Physiol. 2001;281:H2612–2618 [DOI] [PubMed] [Google Scholar]
- 158. Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med (Berl). 2003;81:235–245 [DOI] [PubMed] [Google Scholar]
- 159. Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21:30–36 [DOI] [PubMed] [Google Scholar]
- 160. Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res. 2006;70:410–421 [DOI] [PubMed] [Google Scholar]
- 161. Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 2010;143:686–693 [DOI] [PubMed] [Google Scholar]