

Summary
Identification of complex molecular networks underlying common human phenotypes is a major challenge of modern genetics. In this study we develop a method for NETwork-Based Analysis of Genetic associations (NETBAG). We use NETBAG to identify a large biological network of genes affected by rare de novo CNVs in autism. The genes forming the network are primarily related to synapse development, axon targeting and neuron motility. The identified network is strongly related to genes previously implicated in autism and intellectual disability phenotypes. Our results are also consistent with the hypothesis that significantly stronger functional perturbations are required to trigger the autistic phenotype in females compared to males. Overall, the presented analysis of de novo variants supports the hypothesis that perturbed synaptogenesis is at the heart of autism. More generally, our study provides proof of the principle that networks underlying complex human phenotypes can be identified by a network-based functional analysis of rare genetic variants.
Identification of complex molecular networks underlying common human phenotypes is a major challenge of modern genetics. Recent evidence suggests that rare variants, including copy number variations (CNVs), play a significant role in the etiology of autism spectrum disorders (ASD). Although many such variants have been identified, the specific molecular networks associated with this complex disorder remain largely unknown. In this study we develop a method for NETwork-Based Analysis of Genetic associations (NETBAG). We use NETBAG to identify a large biological network of genes affected by rare de novo CNVs in autism. The genes forming the network are primarily related to synapse development, axon targeting and neuron motility. The identified network is strongly related to genes previously implicated in autism and intellectual disability phenotypes. Our results are also consistent with the hypothesis that significantly stronger functional perturbations are required to trigger the autistic phenotype in females compared to males. Overall, the presented analysis of de novo variants discovered through an unbiased genome-wide study supports the hypothesis that perturbed synaptogenesis is at the heart of autism. More generally, our study provides proof of the principle that networks underlying complex human phenotypes can be identified by a network-based functional analysis of rare genetic variants observed in a large collection of affected individuals.
Introduction
The ongoing revolution in genomic and sequencing technologies has allowed researchers to routinely perform genome-wide association studies (GWAS) for multiple common human diseases and phenotypes (Frazer et al., 2007; Hardy and Singleton, 2009). Although these studies have successfully identified hundreds of significant associations, common polymorphisms reaching genome-wide significance usually explain a relatively small fraction of disease heritability (Goldstein, 2009). There is a growing consensus in genetics that the most valuable contribution of GWAS studies will be in the identification of functional pathways underlying the observed phenotypes (Hirschhorn, 2009). In addition, it is likely that a significant fraction of so-called missing heritability (Manolio et al., 2009), which has eluded association studies, is accounted for by rare single nucleotide mutations and structural genomic variations (McClellan and King, 2010).
A notable example of a disease with a very complex allelic architecture is autism – one of the most common neurological disorders (Geschwind, 2008). Autism spectrum disorders are characterized by impaired social interactions, abnormal verbal communication, restricted interests and repetitive behaviors. Due in part to better detection strategies, the combined prevalence of ASD has been steadily increasing for several decades and is now approaching a staggering 1% in the human population. Although autism has a very strong genetic component, with an estimated heritability as high as 90% based on studies of monozygotic twins (Hyman, 2008), GWAS-based searches have implicated only a few genes that are associated with common polymorphisms reaching genome-wide significance (Wang et al., 2009; Weiss et al., 2009). In addition, the agreement between published findings remains poor (Manolio et al., 2009) and underlying genetic determinants for this disease still remain largely unknown. Importantly, there is growing evidence that rare sequence mutations and de novo copy number variations (CNVs) (Marshall et al., 2008; Moessner et al., 2007; Pinto et al., 2010; Sebat et al., 2007) significantly contribute to autism etiology (Zhao et al., 2007).
The main challenge in the analysis of rare genetic variations, such as de novo CNVs, is precisely their rarity, i.e. the fact that a vast majority of the observed genetic events are unique. Consequently, each rare variant by itself is not statistically significant, so an integrative conceptual framework is required to understand their overall functional impact. We hypothesized that recently obtained genome-wide de novo CNV data (Levy et al., 2011) could allow identification of the underlying biological pathways and processes if considered in the context of functional biological networks (Feldman et al., 2008; Iossifov et al., 2008). Here we develop a method for NETwork-Based Analysis of Genetic associations (NETBAG) and demonstrate its utility in autism. The presented approach can determine whether the observed rare events en masse affect a significantly interconnected functional network of human genes.
Results
NETBAG method overview
To implement our approach we first built a background network that connects any pair of human genes with a weighted edge encapsulating our a priori expectation that the two genes participate in the same genetic phenotype (see Experimental Procedures and Supplementary Experimental Procedures). This background network was based on a combination of various functional descriptors, such as shared gene ontology (GO) annotations (Ashburner et al., 2000), functional pathways in KEGG (Kanehisa and Goto, 2000), shared interaction partners and co-evolutionary patterns (see Experimental Procedures). Similar methods have been previously used to build functional networks in humans and several model organisms (Lee et al., 2004; Lee et al., 2008). In contrast to the aforementioned studies, edges in our network represent the likelihood that two genes participate in a similar genetic phenotype rather than necessarily share cellular functions. Importantly, no deliberate biases toward genes previously implicated in autism or biological functions related to nervous system were used in building the network. The likelihood network was assembled using a large set of known disease-gene associations that were carefully curated for our previous study (Feldman et al., 2008). This set contains 476 genes associated with 132 different genetic diseases (see Experimental Procedures).
Using the constructed network, we searched for functionally connected clusters of human genes affected by de novo CNVs (Figure 1). The genes within the observed CNV regions were first mapped to the nodes corresponding to these genes in the network (Figure 1B). Clusters of genes were assigned scores based on the strength of their connections, and a greedy search algorithm (see Experimental Procedures) was then used to find high-scoring clusters of genes within the CNV regions (Figure 1C). In this search procedure genes from any CNV region could be selected to be members of the growing cluster (Figure 1C), but to prevent large CNV regions from dominating clusters, we allowed no more than one or two genes from a given CNV to participate in a cluster (Figure 2A and Figure 2B, respectively).
Figure 1.

Outline of the NETwork-Based Analysis of Genetic associations (NETBAG), the method used in our study to identify significant and functionally related gene networks affected by de novo CNV events. (A) A background network of human genes is constructed in which nodes indicate genes and edges represent the likelihood that two genes participate in the same genetic phenotype. (B) One or two genes are selected from each of de novo CNV region to form a cluster. The genes are mapped to the likelihood network and a combined score is calculated for each cluster based on interactions between its genes. (C) A greedy search procedure is used to identify the cluster with maximal score. (D) The significance of the cluster with maximum score is determined by comparing it to the distribution of maximal scores from randomly selected genomic regions with similar gene counts. See Figure S1 for a further description of the NETBAG approach.
Figure 2.

Gene clusters found using NETBAG analysis of de novo CNV regions observed in autistic individuals. A) The highest scoring cluster obtained using the search procedure with up to one gene per each CNV region. B) The cluster obtained using the search with up to two genes per region. In the figure genes (nodes) with known functions in the brain and nervous systems are colored in orange (see Table S2 for functional information about the genes forming the cluster). Node sizes represent the importance of each gene to the overall cluster score. Edge widths are proportional to the prior likelihood that the two corresponding genes contribute to a shared genetic phenotype. For clarity, we show only edges corresponding to the two strongest connections for at least one node.
A gene cluster was scored by considering the sum of log likelihood-based edges between all genes within the cluster. Such a scoring scheme is conceptually equivalent to calculating the expected likelihood that all genes within the cluster will participate in the same genetic phenotype. To account for the fact that functional interactions between genes in a cluster are not independent, we employed a previously developed de-weighting heuristic (Lee et al., 2004) described in the Experimental Procedures; similar results were obtained with or without the de-weighting procedure (see Table S1). To calculate the p-value for the resulting clusters, random events were generated with the same gene count, or alternatively with the same genomic length, as in the observed de novo CNV dataset. The greedy algorithm was then applied to search for high-scoring clusters formed by genes from these random events. P-values were assigned to clusters based on the distribution of scores in the randomized data clusters (see Experimental Procedures).
We and others have previously used various network-based methods to analyze genetic data from rare and common diseases (Feldman et al., 2008; Franke et al., 2006; Iossifov et al., 2009; Iossifov et al., 2008; Lango Allen et al., 2010; Raychaudhuri et al., 2009). NETBAG differs from the previous approaches in several important ways. Specifically, the underlying weighted network does not represent a molecular interaction network or a set of predefined functional pathways, but instead the prior likelihood that any pair of human genes is involved in the same genetic phenotype. NETBAG then defines a formal procedure for identifying strongly connected clusters among a large set of genetically perturbed genes and evaluating the genome-wide cluster significance. The relative importance of specific genes forming a cluster is then evaluated based on the contribution of genes to the overall cluster score. We are currently working on making the NETBAG method available as a web server; in the meantime, we will be happy to share the developed methodology with any interested parties.
Functional gene network identified from rare de-novo CNVs
The NETBAG approach was directly applied to the experimental CNV dataset described in the companion paper by Levy et al. This set contained 75 rare de novo CNVs encompassing 746 unique human genes. For our analysis, we combined all overlapping events into a single region and removed all events that did not intersect any genes; we also removed six very large CNV events (length >5 Mb). As a result, the final set used for our analysis contained 47 CNV regions from affected individuals intersecting 433 genes. In addition, Levy et al. also identified ultra-rare CNVs inherited by autistic subjects but not their siblings; inherited CNVs were classified as ultra-rare, if at least one of the corresponding genes was not affected by any other event from the study. Applying the same pre-processing procedure resulted in 156 CNV regions with 419 genes associated with rare inherited events in autistic children.
Using the data described above, we identified statistically significant gene clusters affected by de novo CNV events associated with autistic individuals. Significant clusters detected using either one-gene-per-CNV (p-value=0.02) or the two-genes-per-CNV (p-value=0.02) clustering are shown in Figure 2. If genes forming the high scoring clusters were masked, no other significant clusters were detected in the data.
In contrast, no statistically significant clusters were obtained using ultra-rare inherited CNVs from affected individuals (the best cluster p-value=0.6) The absence of a significant cluster in the ultra-rare inherited dataset, which had a comparable number of genes to the de novo events, suggests that the inherited CNVs contain a significantly smaller fraction of casual genes, i.e. genes associated with autistic phenotype. This conclusion is also supported by the observation by Levy et al. that there is less bias in transmission of ultra-rare inherited events. In other words, autistic children were almost as likely as their unaffected siblings to inherit an ultra-rare event. This is in sharp contrast to the de novo events, which were nearly four times more frequent in the autistic children (7.9% in autistic children versus 2.0% in unaffected siblings).
The contribution of each gene to the cluster score, i.e. the functional connection to other cluster genes, is not uniform. To capture each gene’s contribution to the cluster score we performed Markov Chain Monte Carlo (MCMC) simulations, sampling clusters based on their scores. The size of each gene (node) in Figure 2 is proportional to the each gene’s membership in high-scoring clusters during the sampling simulations (see Experimental Procedures, Supplementary Experimental Procedures). Similar node sizes were also obtained based on the average connection strength from each gene to the other genes in the cluster (Pearson’s r= 0.8, p-value = 4*10−11).
Interestingly, we found that genes affected by de novo CNVs observed in females are significantly more important for the overall cluster score than genes affected by CNVs in males, i.e. female genes have stronger average connections with other genes from the identified network (see Figure S2, One-tail Mann-Whitney test, female > male, p-value=0.013). This observation is illuminating because one of the striking phenotypic characteristics of autism is the male-to-female incidence ratio of more than 5:1 for high-functioning ASD (Newschaffer et al., 2007). It has been previously suggested (Zhao et al., 2007) that stronger genetic perturbations are required, on average, to trigger an autistic phenotype in females than males due to currently unknown compensatory mechanisms. Two mechanisms may lead to stronger perturbations in females: CNVs encompassing a larger number of genes that are associated with ASD, and CNVs intersecting individual genes that produce a more deleterious impact when disrupted. The analysis of de-novo events in affected individuals lends support to both of these mechanisms: the CNVs in females are indeed significantly larger (with median of 10 genes per CNV in females, 3 genes per CNV in males, two-tail Mann Whitney P-value=0.02), and genes derived from female CNVs are more functionally important for the network shown in Figure 2. Using simulations of random CNVs we also confirmed that the difference in the relative importance of female versus male nodes is unlikely (P=0.024) to be a simple consequence of the larger CNV sizes in females (see Supplementary Materials, Figure S2, C). We believe that both of the aforementioned mechanisms are at play. Indeed, it would be surprising that stronger perturbation can be inflicted exclusively by larger CNVs and not disruption of high impact genes, and vice versa.
Functional characterization of the identified network
Analysis of the established annotation resources, such as Swiss-Prot (Consortium, 2007), GeneCards (www.genecards.org), WikiGenes (www.wikigenes.org), IHOP (Hoffmann and Valencia, 2004), suggests that a significant fraction of genes in the identified network either play a well-defined functional role in the brain or have been previously implicated in neurodegenerative and psychiatric disorders. Only ~25% (54 of a randomly selected 214) of all genes within the de novo CNV regions have been previously associated with brain-related phenotypes. However, when we consider genes in the identified clusters this proportion rises drastically (p-value < 10−3), to ~67% (Figure 2A, 30 out of 45) for the one-gene-per-CNV clustering or ~ 52% (Figure 2B, 38 out of 72) for the two-genes-per-CNV clustering (see Table S2 for functional description of cluster genes).
To characterize in more detail the specific biological processes related to the cluster in Figure 2A, we investigated the strength of functional interactions between the cluster genes and various Gene Ontology (GO) categories (Ashburner et al., 2000). GO categories represent a curated set of functionally related genes described by a controlled vocabulary. For human genes in each of 1454 GO categories we calculated their average log likelihood interaction score (using the background network) with the genes in the identified cluster (Figure 2). The GO-specific significance of these interaction scores was calculated by comparison with scores of randomly generated CNV events with the same gene count at in real data by Levy et al. A False Discovery Rate (FDR) procedure was used to correct for multiple hypothesis testing (see Experimental Procedures). The 25 GO categories with lowest Q-values, indicating the highest connection significance to the autism associated cluster, are shown in Table 1 (see Table S3 for other significant GO categories). These GO categories are primarily related to actin network dynamics and reorganization, synaptogenesis, axonogenesis, cell-cell adhesion, small GTPase signaling and neurite development. Consequently, the identified functional network is associated with a diverse collection of molecular and cellular processes essential for proper synaptogenesis and axon guidance. We note that is it not possible to obtain the same functional results by a statistical analysis of significantly overrepresented GO terms for all 433 gene within the de-novo CNVs from affected individuals (See Supplementary Experimental Procedures for details). The significant GO terms presented in Table 1 specifically describe the functional connection of the network in Figure 2.
Table 1.
Gene Ontology (GO) terms highly connected to the functional network in Figure 2A. We defined a score that reflects the degree to which the network genes interact with gene sets assigned to various GO categories (1454 in total). For every GO category, a background distribution was built by generating clusters from randomly chosen CNV regions with the same gene count as observed in real data by Levy et al. These distributions were used to derive False Discovery Rate (FDR) corrected q-values accounting for the multiple hypothesis tests (one test for every GO gene set, see Experimental Procedures).
| Gene Ontology Term | GO Category | q-value |
|---|---|---|
| GO:0007015: actin filament organization | Biological process | < 0.01 |
| GO:0030424: axon | Cellular component | < 0.01 |
| GO:0048469: cell maturation | Biological process | < 0.01 |
| GO:0007611: learning and or memory | Biological process | < 0.01 |
| GO:0044456: synapse part | Cellular component | < 0.01 |
| GO:0045202: synapse | Cellular component | < 0.01 |
| GO:0007163: establishment and or maintenance of cell polarity | Biological process | 0.01 |
| GO:0045216: intercellular junction assembly and maintenance | Biological process | 0.01 |
| GO:0019201: nucleotide kinase activity | Molecular function | 0.01 |
| GO:0005912: adherens junction | Cellular component | 0.01 |
| GO:0007409: axonogenesis | Biological process | 0.01 |
| GO:0016323: basolateral plasma membrane | Cellular component | 0.01 |
| GO:0030041: actin filament polymerization | Biological process | 0.01 |
| GO:0051258: protein polymerization | Biological process | 0.01 |
| GO:0021700: developmental maturation | Biological process | 0.01 |
| GO:0030863: cortical cytoskeleton | Cellular component | 0.01 |
| GO:0000904: cellular morphogenesis during differentiation | Biological process | 0.01 |
| GO:0005925: focal adhesion | Cellular component | 0.01 |
| GO:0030427: site of polarized growth | Cellular component | 0.01 |
| GO:0032271: regulation of protein polymerization | Biological process | 0.01 |
| GO:0031175: neurite development | Biological process | 0.01 |
| GO:0048666: neuron development | Biological process | 0.01 |
| GO:0030055: cell matrix junction | Cellular component | 0.01 |
| GO:0030832: regulation of actin filament length | Biological process | |
| GO:0030036: actin cytoskeleton organization and biogenesis | Biological process | 0.01 |
Using the same methodology, we found that the cluster in Figure 2A is strongly related to the set of genes previously implicated in autism (p-value=0.001, see Supplementary Experimental Procedures) and genes associated with intellectual disability phenotypes (p-value=0.017). The collections of genes responsible for these phenotypes were manually compiled recently by Pinto et al. (Pinto et al., 2010) through an extensive review of the literature and available databases. In spite of strong functional connections, the overlap between genes in the aforementioned sets and the genes identified in our analysis is relatively small (~3%). Thus, our study significantly expands the collection of genes implicated in ASD. The cluster genes are also strongly connected (p-value=0.013) to proteins identified experimentally by recent proteomic profiling of postsynaptic density (PSD) from human neocortex (Bayes et al., 2011).
Discussion
At the core of the processes listed in Table 1 is the development and maturation of synaptic contacts in the brain. The functional relationships between proteins in the identified cluster can be better appreciated if considered in the context of molecular interactions involved in formation and maturation of the excitatory (glutamatergic) synapse (Figure 3). The excitatory synaptic connections are formed between axons and dendritic spines, which are complex and dynamic post-synaptic structures containing thousands of different proteins (Alvarez and Sabatini, 2007; Tada and Sheng, 2006). The formation, maturation and elimination of dendritic spines lie at the core of synaptic transmission and memory formation (Roberts et al., 2010; Yang et al., 2009). In Figure 3 the genes that are members of the identified network are shown in yellow, other functionally related genes within rare de novo CNV regions from Levy et al. are in blue and genes previously implicated or discussed in the context of autism are highlighted using orange borders. Although the picture shows a dense and interconnected web of molecular interactions, the processes depicted in the figure can be understood in terms of several signaling and structural pathways. Many of these pathways ultimately converge on the regulation of the growth and branching of the actin filament network, which is essential for spine structural remodeling and morphogenesis.
Figure 3.
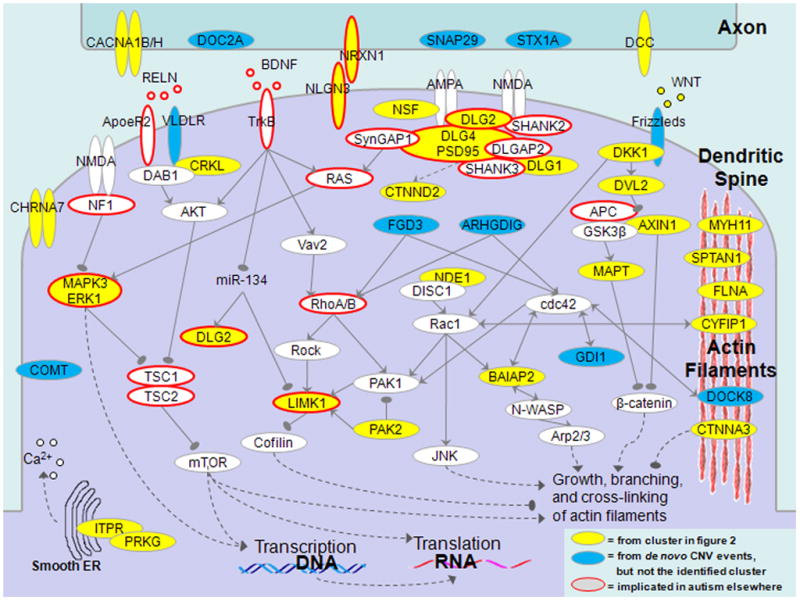
Genes associated with the morphogenesis of dendritic spines. Dendritic spines are dynamically forming protrusions from a neuron’s dendrite which mediate excitatory connection to axons and determine synaptic strength. The proteins shown in the figure play crucial roles in formation of physical contacts between axons and dendrites, organization of postsynaptic density (PSD), and signaling processes controlling spine morphology. Many of the signaling pathways ultimately converge on the regulation of the growth and branching of the actin filament network, which is essential for spine structural remodeling. The proteins encoded by genes from the identified functional cluster (Figure 2) are shown in yellow, other genes hit by de novo CNV from Levy et al., in blue, and genes previously implicated or discussed in the context of autism are highlighted using orange borders.
The initial contacts between axons and dendrites are mediated by specific adhesion-related proteins, such as neurexin and neuroligin (e.g. NRXN1 and NLGN3, genes perturbed by rare de novo CNVs associated with ASD are underlined here and below) (Sudhof, 2008). On the postsynaptic side of an excitatory synapse, the initial axon-dendrite contacts ultimately develop into a complex and dense structure, the postsynaptic density (PSD), dominated by several types of glutamate receptors (such as AMPA and NMDA), various scaffolding proteins (DLG4/PSD95, DLG2, SHANK2/3, SynGAP1, DLGAP2) and trafficking/signaling proteins (CTNND2). In total, the PSD contains many hundreds of distinct proteins (Bayes et al., 2011; Sheng and Hoogenraad, 2007). Information for activity-dependent regulation of spine morphology is passed through an intermediate level of signaling protein, such as Rho family (Linseman and Loucks, 2008) of small GTPases (RhoA/B, Cdc42, Rac1) to downstream targets (LIMK1 and PAK1/2/3) connected to proteins modifying morphology of the actin network (cofilin and Arp2/3) (Blanchoin et al., 2000). The activity of the GTPases is regulated pre- or post-synaptically by many guanine exchange factors (GEFs), GDP dissociation inhibitors (GDIs, such as GDI1) and GTP-activating proteins (GAPs). Many other proteins shown in Figure 3, such as FLNA, CTNNA3, DOCK8, SPTAN1, CYFIP1, either bind directly to the actin network or mediate interaction of actin filaments with other proteins.
The WNT signaling pathway plays a crucial role in diverse processes associated with formation of neural circuits (Salinas and Zou, 2008). This pathway is also known to be directly involved in the regulation of dendrite morphogenesis (Rosso et al., 2005; Salinas et al., 1994). WNT signaling is accomplished through the canonical branch (DVL, AXIN1, beta-catenin) and the non-canonical branch (DVL1/2/3, Rac1 and JNK); both of these pathway branches converge on regulation of actin network morphogenesis. Similar to WNT, the reelin signaling also plays a prominent role in the context of autism phenotype and specifically dendritic spine morphogenesis (Fatemi et al., 2005; Niu et al., 2008). Signaling by secreted extracellular RELN protein acts though VLDR and Apoer2 receptors and the PI3K/Akt pathway (Jossin and Goffinet, 2007) regulating the mammalian target of rapamycin (mTOR) pathway (Kumar et al., 2005; Shaw and Cantley, 2006). Another important pathway converging on mTOR involves MAPK3/ERK, which can be activated by Ras and NF1. mTOR integrates various inputs from upstream growth-related pathways, and is also known to regulate dendrite morphogenesis (Tavazoie et al., 2005).
Although we discussed proteins in the context of dendritic spine development (Figure 3 and text above), many of the aforementioned proteins also participate in diverse cellular processes and are reused in the context of axon guidance and neuron motility (Shen and Cowan, 2010). Such recycling of proteins is natural because actin network dynamics are essential for such processes as growth of axonal filopodia, which are used in searching for growth cone guidance cues (Tessier-Lavigne and Goodman, 1996). The presence of DCC protein in the identified network (Figure 2 and Figure 3), also suggests an important role of perturbed axonal guidance in autism. Although DCC is also involved in dendrite development (Suli et al., 2006), this receptor and its signaling protein, netrin, are primarily essential for guiding axons to their final destinations (Tessier-Lavigne and Goodman, 1996). Several signaling pathways highlighted in Figure 3, such as the WNT and reelin pathways, also play prominent roles in neuron motility (Reiner and Sapir, 2005; Salinas and Zou, 2008). In addition, several specific proteins, such as PAKs and LIMK, which regulate the dynamics of actin network, are reused in axonal morphogenesis. Consequently, malfunction of many proteins shown in Figure 3 may influence autistic phenotypes through their role in either dendrite or axon signaling, or possibly a combination of these processes.
Considering the genes hit by rare de novo variants from the perspective of the functional molecular network (Figure 3) allowed us to investigate the likely morphological consequences of some CNVs. There is growing evidence that changes in dendritic spine morphology contribute to a number of neurological disorders (Halpain et al., 2005). A decrease in the density of dendritic spines in regions of the cerebral cortex has been linked to schizophrenia (Blanpied and Ehlers, 2004; Garey et al., 1998; Glantz and Lewis, 2000). On the other hand, an increase in spine size or density has been also connected to Fragile X syndrome, a disorder frequently associated with autism (Fiala et al., 2002; Kaufmann and Moser, 2000). Following the logic that CNV deletions should decrease while duplications increase the dosage of the affected genes, we can infer – based on the structure and regulatory logic of the functional network in Figure 3 – the morphological effects of 13 gene perturbations on dendritic spines. Specifically, we found that in 11 out of 13 cases (~85%) the gene perturbations caused by the observed CNV events should increase either dendritic spine growth or their density (see Table S4). This result is consistent with recent findings that autistic individuals have increased spine density in portions of their cerebral cortex (Hutsler and Zhang, 2010; Woolfrey et al., 2009) and possibly a local brain over-connectivity (Scott-Van Zeeland et al., 2010).
Overall, the results of this study, the first to our knowledge, demonstrate that autism-associated rare de novo CNVs, observed in an unbiased genome-wide study, form a large and statistically significant functional network responsible for synaptogenesis, axon guidance and related molecular processes. Therefore, our analysis strongly supports the hypothesis that autism is primarily a disease of synaptic and neuronal connectivity malfunction (Zoghbi, 2003). The identified functional network also reveals a striking genetic complexity of autism. The genetic events we observe affect the whole arc of molecular processes essential for proper synapse formation and function. Similar genetic complexity is already apparent in many cancers (Network, 2008; Wood et al., 2007) and - as we and others believe - will be a hallmark of many other common human phenotypes and maladies (Wang et al., 2010). In spite of the observed complexity, our study provides an important proof of the principle that underlying functional networks responsible for common phenotypes can be identified by an unbiased analysis of multiple rare genetic perturbations from a large collection of affected individuals.
The functional network presented in Figure 3 contains approximately 70 genes, with about 40% of them perturbed by rare de novo CNVs observed by Levy et al. As more genetic data are analyzed it is likely that the network will grow in size and significance. Considering that up to a thousand (Sheng and Hoogenraad, 2007) distinct proteins are associated with postsynaptic density or that hundreds of different GAPs/GEFs modify activity of Rho GTPases that are associated with actin network remodeling, it is likely that many hundreds of genes could ultimately contribute to the autistic phenotype. This estimate, based on the functional network, is consistent with independent estimates based on recurrent mutations and the overall incidence of autism in the human population (Zhao et al., 2007; Levy et al., 2011). Deleterious variants in different genes contributing to autistic phenotype will almost certainly have different penetrance and vulnerabilities. The identification of the complete set of genes responsible for ASD and understanding their respective contributions to the phenotype will require analyses of next generation sequencing data coupled with investigation of underlying molecular networks.
Experimental Procedures
Copy Number Variation (CNV) dataset
In our analysis we used the CNV dataset obtained in a companion study by Levy et al. The dataset contained 75 rare de novo CNV events from autistic children. Six very large CNV events, spanning more than 5 Mb each, were not considered in our analysis. The initial CNV dataset contained several overlapping events, including a set of 10 events all within the region 16p11.2. Any overlapping CNVs were collapsed into single events to avoid double counting of genes. We ignored all CNV events that did not contain any annotated human gene based on the NCBI genome build 36. After aforementioned preprocessing steps, our final CNV set from autistic children contained 47 loci in total affecting 433 human genes; the average number of genes within each de novo CNV region was ~9, with the median of 3 genes per regions. Levy et al. also identified 157 ultra-rare inherited CNVs transmitted between parents and autistic children. Inherited CNVs were classified as ultra-rare if the genes within these events were not affected by any other inherited events in the study. We applied the same pre-processing steps to these inherited CNVs, resulting in 156 regions affecting 418 genes.
Building the background likelihood network
To perform the NETBAG analysis, we built a background network connecting all pairs of human genes. Every gene pair in this network was assigned a score proportional to the log of the ratio of the likelihood that the two genes participate in the same genetic phenotype to the likelihood that they do not (see Supplementary Experimental Procedures). Importantly, although similar in spirit to integrative methods that have been used previously to build functional networks in several model species (Lee et al., 2004; Lee et al., 2008), the edges in our network represent the likelihood to participate in the same genetic phenotype rather than share a functional and molecular interaction.
The likelihood network was build using, as a positive gold standard, the carefully curated set of human genes compiled recently by Feldman et al. (Feldman et al., 2008). This set contains 476 human genes associated with 132 different genetic phenotypes. As a negative gold standard we used a set of randomly selected pairs of human genes that are not known to be associated with identical diseases phenotypes. Importantly, no genes previously implicated in ASD or any biologically related functions were used in the network construction. The likelihood score was derived based on naive Bayesian integration of various descriptors of proteins function: shared GO annotations, participation in the same KEGG pathways, shared protein domains in InterPro, direct protein-proteins interactions and shared interaction partners from multiple databases (BIND, BioGRID, DIP, HPRD, InNetDB, IntAct, BiGG, MINT, and MIPS), sequence homology between the gene pair calculated using BLAST (Altschul et al., 1997), and two measures of similarity in co-evolutionary patterns: phylogenetic profile similarity and chromosomal co-clustering across genomes (Chen and Vitkup, 2006). We cross-validated the quality of the background network by showing that it can be successfully used to prioritize (rank) genes, located within a chromosomal region, across a variety of genetic phenotypes (see Supplementary Experimental Procedures for details).
Search for high scoring clusters affected by CNVs
To score a cluster of genes in the network (Figure 1) we combined the scores for all gene pairs forming the cluster. The direct multiplication of the corresponding likelihoods (network edges) is conceptually equivalent to assuming that all connections within the cluster are independent; we refer to this procedure as the naïve scoring scheme. Second, we applied a simple de-weighting scheme used previously for functional data integration (Lee et al., 2004). For each gene forming the cluster the de-weighting scheme scores the strongest connection in full, and then decreases the other connections in order of their strength in a linear fashion (see Supplementary Experimental Procedures). Effectively, the de-weighting scheme gives more weight to strongest gene-gene connections within the cluster. The detected functional clusters were significant under both scoring schemes.
A greedy growth algorithm was used to find strongly connected clusters of genes located within CNV regions (Figure 1). Specifically, the search algorithm was started from every possible gene in CNV regions, then the gene with the strongest connection to the first gene was added. At all subsequent iterations, genes located within CNV regions that most increased the cluster score were added. Only one (results in Figure 2A) or two (Figure 2B) genes per each CNV region were allowed in the growing cluster. This growth procedure was run until no further genes could be added. For each cluster size, clusters obtained by starting with each gene within CNV regions were compared and the cluster with the highest score was selected.
Evaluating significance of cluster scores
We first determined the p-value for the best cluster at each cluster size; we refer to this as the local p-value. Local p-values were calculated based on re-running the greedy search algorithm using random human genome regions identical (either in length or gene number) to those observed by Levy et al. Second, to determine the most significant cluster across sizes, we compared the lowest local p-value obtained from the real data, to the distribution of lowest local p-values obtained in the 10,000 trails from the randomized regions. Effectively, this allowed us to assign a p-value to our local p-value; we refer to this as the global p-value. The global p-value is more stringent because it accounts for multiple hypotheses testing, arising due to different cluster sizes; in our manuscript we refer to global p-value simply as p-value. In the aforementioned calculation of local and global p-values we used two alternative randomization procedures for human genomic regions: we either preserved the genomic size of CNVs or the gene counts to the values observed in the real data. All randomized regions were generated using the NCBI human genome build 36 (hg18). The functional cluster identified in our work was significant under both randomization schemes (preserving length of CNVs or gene counts) and cluster scoring methods (naïve and de-weighted). The p-values for different randomization procedures are given in Table S1.
In addition to the randomization of genomic regions we wanted to ensure that our results were not due to some general topological features of the background network. To explore this possibility, we randomly shuffled the background network while preserving the distribution of connection strengths for each gene (see Supplementary Experimental Procedures). We then repeated the NETBAG search using the de novo CNVs from affected children. This search using the shuffled network identified no significant clusters or GO terms.
Analysis of genes contribution to cluster score
Contributions of different genes to the score of the identified functional cluster (Figure 2) vary substantially. To capture that, we devised a formal method to assign weights to individual genes reflecting their contribution to high scoring clusters. The method is based on two distributions over clusters: p(C), in which clusters with high scores are assigned a high probability, and a uniform distribution, pu(C), in which all clusters are equally likely (See Supplementary Experimental Procedures). Each individual gene was then given a score equal to the ratio of the number of clusters that contain the gene sampled from p(C) to the number sampled from pu(C). As a result, the genes which were more frequently included in high-scoring clusters were assigned higher ratios. We used Markov-Chain Monte Carlo (MCMC) to sample 5 million clusters from each of the two distributions.
Calculation of Functional Relationships between the cluster and various GO categories
To characterize the identified cluster we investigated its interactions with a collection of a-priori defined functional sets of human genes. For this purpose, we utilized the 1454 gene sets corresponding to the Gene Ontology (GO) categories used in the MSigDB database (Subramanian et al., 2005). Using the background likelihood network, we calculated, for each gene set, its average interaction to the identified cluster shown in Figure 2. To determine the significance of the calculated interaction scores we built gene set-specific background distributions by generating random clusters from the randomized genomic regions with the same gene count as in Levy et al. We used the background distribution to assign an empirical p-value for every gene set, and then applied the FDR procedure to address the multiple hypotheses involved in testing all gene sets within the collection (see Supplementary Experimental Procedures).
Supplementary Material
Highlights.
Rare de novo CNVs associated with autism contain functionally connected genes
NETBAG method identifies a significant functional network affected by rare variants
Identified network is related to synaptogenesis, axon guidance, and neuronal motility
Genes perturbed in females carry more weight in the network than genes in males
Acknowledgments
This work was supported in part by a grant from the Simons Foundation (SFARI award number SF51 to MW), the National Centers for Biomedical Computing (MAGNet) grant U54CA121852 to Columbia University. SRG was supported in part by the training grant T32 GM082797. We are grateful to all of the families at the participating SFARI Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, D. Grice, A. Klin, R. Kochel, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, B. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, E. Wijsman).
We would also like to sincerely thank Simons Foundation Autism Research Initiative for generous financial support, Linda Van Aelst, Thomas Jessell, Gerald Fischbach, Marian Carlson, Alan Packer, Barry Honig, Itsik Pe’er, Lauren DeMaria, and Stephen Johnson for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, Grant SG. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD, Mullins RD. Interactions of ADF/cofilin, Arp2/3 complex, capping protein and profilin in remodeling of branched actin filament networks. Curr Biol. 2000;10:1273–1282. doi: 10.1016/s0960-9822(00)00749-1. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55:1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Chen L, Vitkup D. Predicting genes for orphan metabolic activities using phylogenetic profiles. Genome Biol. 2006;7:R17. doi: 10.1186/gb-2006-7-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TU. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007;35:D193–197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Reutiman TJ, Lee S, Brooks AI, Pearce DA. Reelin signaling is impaired in autism. Biol Psychiatry. 2005;57:777–787. doi: 10.1016/j.biopsych.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci U S A. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Franke L, van Bakel H, Fokkens L, de Jong ED, Egmont-Petersen M, Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am J Hum Genet. 2006;78:1011–1025. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Halpain S, Spencer K, Graber S. Dynamics and pathology of dendritic spines. Prog Brain Res. 2005;147:29–37. doi: 10.1016/S0079-6123(04)47003-4. [DOI] [PubMed] [Google Scholar]
- Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Valencia A. A gene network for navigating the literature. Nat Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Hyman SE. A glimmer of light for neuropsychiatric disorders. Nature. 2008;455:890–893. doi: 10.1038/nature07454. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Rodriguez-Esteban R, Mayzus I, Millen KJ, Rzhetsky A. Looking at cerebellar malformations through text-mined interactomes of mice and humans. PLoS Comput Biol. 2009;5:e1000559. doi: 10.1371/journal.pcbi.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Zheng T, Baron M, Gilliam TC, Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res. 2008;18:1150–1162. doi: 10.1101/gr.075622.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 2007;27:7113–7124. doi: 10.1128/MCB.00928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- Lee I, Lehner B, Crombie C, Wong W, Fraser AG, Marcotte EM. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet. 2008;40:181–188. doi: 10.1038/ng.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, et al. Rare de novo and transmitted copy number variation in autistic spectrum disorders. Neuron. 2011 doi: 10.1016/j.neuron.2011.05.015. (this issue) [DOI] [PubMed] [Google Scholar]
- Linseman DA, Loucks FA. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Front Biosci. 2008;13:657–676. doi: 10.2741/2710. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, Mandell DS, Miller LA, Pinto-Martin J, Reaven J, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health. 2007;28:235–258. doi: 10.1146/annurev.publhealth.28.021406.144007. [DOI] [PubMed] [Google Scholar]
- Niu S, Yabut O, D’Arcangelo G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J Neurosci. 2008;28:10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Plenge RM, Rossin EJ, Ng AC, Purcell SM, Sklar P, Scolnick EM, Xavier RJ, Altshuler D, Daly MJ. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O, Sapir T. Similarities and differences between the Wnt and reelin pathways in the forming brain. Mol Neurobiol. 2005;31:117–134. doi: 10.1385/MN:31:1-3:117. [DOI] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature. 2010;463:948–952. doi: 10.1038/nature08759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005;8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- Salinas PC, Fletcher C, Copeland NG, Jenkins NA, Nusse R. Maintenance of Wnt-3 expression in Purkinje cells of the mouse cerebellum depends on interactions with granule cells. Development. 1994;120:1277–1286. doi: 10.1242/dev.120.5.1277. [DOI] [PubMed] [Google Scholar]
- Salinas PC, Zou Y. Wnt signaling in neural circuit assembly. Annu Rev Neurosci. 2008;31:339–358. doi: 10.1146/annurev.neuro.31.060407.125649. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, Bookheimer SY. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010;2:56ra80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shen K, Cowan CW. Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol. 2010;2:a001842. doi: 10.1101/cshperspect.a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suli A, Mortimer N, Shepherd I, Chien CB. Netrin/DCC signaling controls contralateral dendrites of octavolateralis efferent neurons. J Neurosci. 2006;26:13328–13337. doi: 10.1523/JNEUROSCI.2858-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Wang K, Bucan M, Grant SF, Schellenberg G, Hakonarson H. Strategies for genetic studies of complex diseases. Cell. 2010;142:351–353. doi: 10.1016/j.cell.2010.07.025. author reply 353–355. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci. 2009;12:1275–1284. doi: 10.1038/nn.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat J, et al. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci U S A. 2007;104:12831–12836. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.