

Abstract
Ischemia reperfusion injury is a major obstacle in liver resection and liver transplantation surgery. Understanding the mechanisms of liver ischemia reperfusion injury (IRI) and developing strategies to counteract this injury will therefore reduce acute complications in hepatic resection and transplantation, as well as expanding the potential pool of usable donor grafts. The initial liver injury is initiated by reactive oxygen species which cause direct cellular injury and also activate a cascade of molecular mediators leading to microvascular changes, increased apoptosis and acute inflammatory changes with increased hepatocyte necrosis. Some adaptive pathways are activated during reperfusion that reduce the reperfusion injury. IRI involves a complex interplay between neutrophils, natural killer T-cells cells, CD4+ T cell subtypes, cytokines, nitric oxide synthases, haem oxygenase-1, survival kinases such as the signal transducer and activator of transcription, Phosphatidylinositol 3-kinases/Akt and nuclear factor κβ pathways. Transgenic animals, particularly genetic knockout models, have become a powerful tool at elucidating mechanisms of liver ischaemia reperfusion injury and are complementary to pharmacological studies. Targeted disruption of the protein at the genetic level is more specific and maintained than pharmacological inhibitors or stimulants of the same protein. This article reviews the evidence from knockout models of liver IRI about the cellular and molecular mechanisms underlying liver IRI.
Keywords: Liver, Ischemia/reperfusion, Transgenic, Knockout, Nitric oxide synthase, Haem oxygenase, Mitogen-activated protein kinase, T cell receptor
INTRODUCTION
Ischemia reperfusion injury is a major cause of morbidity and mortality in liver resection and liver transplantation surgery. Prolonged organ ischemia is characterised reduced tissue oxygenation resulting in tissue adenosine triphosphate (ATP) depletion with a transition to activation of anaerobic metabolic pathways which cannot maintain cellular function for prolonged periods ultimately leading to cell death. Restoration of blood flow is necessary to restore cellular function, but paradoxically reperfusion can initiate a cascade of pathways that cause further cellular injury after prolonged ischaemia. Understanding the mechanisms of liver ischemia reperfusion injury (IRI) and developing strategies to counteract this injury will reduce acute complications in hepatic resection and transplantation, as well as expanding the potential pool of usable donor grafts.
The initial liver injury is initiated by reactive oxygen species (ROS) which cause direct cellular injury and also activate a cascade of mediators leading to microvascular changes, increased apoptosis and acute inflammatory changes with increased necrosis. Not all pathways activated are injurious and some adaptive pathways are activated during reperfusion that dampen the reperfusion injury. Classically two phases of liver injury have been described, an early (< 6 h) and late (> 12 h) phase of injury. In reality, this is a somewhat artificial distinction, as liver injury occurs as a continuum during reperfusion where pathways are activated at various often overlapping timepoints.
The extent of liver injury in IRI is normally measured by raised levels of serum liver enzymes, most commonly aspartate transaminase, alanine transaminase (ALT), lactate dehydrogenase and/or serum glutamic-oxaloacetic transaminase, and by histological assessment with the Suzuki classification, with or without modifications, being most widely used in liver IRI[1]. In this classification sinusoidal congestion, hepatocyte necrosis and ballooning degeneration are graded 0 to 5. No necrosis, congestion/centrilobular ballooning is given a score of 0 whereas severe congestion/ballooning degeneration, as well as > 60 % lobular necrosis is given a score of 5 (Table 1).
Table 1.
Suzuki classification of liver ischaemia reperfusion injury
| Numerical assessment | Sinusoidal congestion | Vacuolisation/ballooning | Necrosis |
| 0 | None | None | None |
| 1 | Minimal | Minimal | Single cell |
| 2 | Mild | Mile | < 30% |
| 3 | Moderate | Moderate | 30%-60% |
| 4 | Severe | Severe | > 60% |
Transgenic animals, particularly genetic knockout models, have become a powerful tool at elucidating mechanisms of liver ischaemia reperfusion injury and are complementary to pharmacological studies[2-9]. The mechanistic insights derived from transgenic knockout models of liver ischaemia reperfusion injury will be reviewed. Knockout models provide a very specific targeted disruption of a particular protein at the genetic level which is more informative than the use of “specific” pharmacological inhibitors or stimulants of the same protein are used.
REACTIVE OXYGEN SPECIES
Depletion of intracellular and extracellular ATP during ischaemia results in increased ATP degradation products, including adenosine, hypoxanthine and xanthine and a shift towards anaerobic metabolism. On reperfusion, initially the increase in oxygen delivery exceeds the rate at which cellular metabolism returns to aerobic pathways, which generates damaging free radicals. A wide variety of ROS are generated, the most widely implicated being superoxide, hydrogen peroxide and reactive nitrogen species, such as peroxynitrite.
There are thought to be three main pathways for the generation of ROS: conversion of xanthine dehydrogenase to xanthine oxidase during ischaemia, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation and uncoupling of the mitochondrial electron transport chain[10,11]. Although hepatocytes can directly produce ROS, physiologically Kupffer cells are thought to be the main source of ROS in the early stages of liver IRI with natural killer T-cells (NKT) cells generating ROS later and neutrophils being the main source in the very later stages[10,11]. The role of these various cells, NADPH oxidase and mitochondrial depolarisation have been supported by knockout animal models[12-15]. There are no xanthine oxidase knockout on liver IRI. These mice only survive up to 6 wk and are runted.
MICROCIRCULATORY DYSFUNCTION
Microcirculatory changes play an important part in hepatic IRI. Reduction in sinusoidal diameter and blood flow are among the earliest changes in reperfusion injury. This results from a combination of direct damage to sinusoidal endothelial cells (SECs), vasoconstriction and expression of adhesion molecules with accumulation of platelets and leucocytes.
Two of the key vasoactive substances that maintain sinusoidal vascular tone are endothelin-1 (ET-1), a vasoconstrictor, and nitric oxide (NO), a vasodilator and inhibitor of platelet aggregation. There appears to be a relative excess of ET-1 in the early stages of liver IRI.
Liver transplantation in pigs has provided evidence that after reperfusion Kupffer cell activation leads to increased release of ET-1 which binds to SEC and hepatocyte endothelin A (ETA) receptor, thereby reducing hepatic micro and macro-perfusion resulting in increased liver injury[16,17]. The activation of this pathway is associated with increased expression of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and endothelial NOS (eNOS)[16,17]. Knockout models for ETA receptor or heterozygote knockout for endothelin-1 have not been studied in liver ischaemia reperfusion injury. Double knockouts of ET-1, ET-2 and ETA receptor are lethal pre- or perinatally. It has become apparent products of heme oxygenase, namely carbon monoxide (CO) and biliverdin, and NO from nitric oxide synthase, which are all vasodilators, are likely play a role in reducing the severity of liver IRI in vivo.
CELL INJURY AND DEATH
Hepatocytes and SECs are the two main cell types that are injured in IRI. Hepatocytes are more sensitive to warm ischaemic injury (37 °C), while SECs are more sensitive to cold ischaemia (4 °C) found in cold preservation of donor liver grafts before transplantation. Physiologically, exclusive injury of one cell type is not found and there is evidence that both cell types have been injured directly in both cold and warm IRI.
There has been debate about what the primary mode of cell death is in liver IRI: apoptosis or necrosis. Apoptosis is an energy dependent process, so in theory when there is greater depletion of ATP, necrosis should dominate. Also, necrosis takes longer to become apparent, normally more than 3 h. This is challenging to show experimentally in vivo, as tissue ATP before and after reperfusion would need to be measured as well as the change in metabolic state of the cell. Many of the same initiators and pathways are involved for both types of cell death, so there is much overlap. Some authors refer to the process as neuroapoptosis.
Different assays have been used to implicate apoptosis, including activation of various genes such as caspase-3 which is thought to be a specific indicator of apoptosis, and Bax. One isolated ex vivo perfused liver model using knockout of Bax showed reduced liver IRI (Table 2), apoptosis and caspase-3 activation in the knockouts compared to the normal wild type livers[18]. The TUNEL assay has been used to indicate apoptosis, but it now appears that it does not specifically distinguish between apoptosis and necrosis. Varying degrees of necrosis and apoptosis have been shown in the literature for different ischaemia reperfusion protocols, but these conclusions on the different levels of necrosis versus apoptosis need to be interpreted with caution as the assays for apoptosis are relatively nonspecific.
Table 2.
Summary of knockout models of liver ischemia reperfusion injury pertaining to reactive oxygen species, cellular metabolism/adenosine and cells involved in the injurious mechanisms
| Ref. | Knockout model | IR protocol | Outcome measure | Agent | Adaptive responses | Injurious responses |
| Kuboki et al[29] | OTII; TCRd deficient | 70% I 90 min/R 4, 8 h | Histology; serum ALT; MPO | AntiCD1d Ab; anti NK1.1 Ab; anti CD25+ Ab | Antigen dependent CD4+ T cell activation via TCR and NKT cell activation increase IRI; GD T cell recruit PMN but not affect IRI | |
| Evans et al[2] | ob/ob or double knockout of leptin and UCP2 | Total hepatic ischaemia 15 min/R 1, 24 h | Histology (Neil and Hubscher scoring); serum ALT; WB; liver ATP assay; lipid peroxidation; 24 h survival | In steatotic livers of ob/ob mice only, UCP-2 depletes liver ATP which increases IRI 1 h onwards | ||
| Hanschen et al[12] | IL6 (-/-); CD4 (-/-); TNFR1 (-/-) | Left lobe I 90 min/R 30 min, 2, 3, 4 h | Kupffer cell activity (fluorescent latex beads and intravital microscopy, IVM); IH; serum AST and ALT | GdCl3 or glutathione to wild types (WT) only | Kupffer cells activation, ROS, IL6 and TNF-α increase SEC VAP-1 expression and CD4+ Tcell sinusoidal recruitment which increase IRI; CD4+ T cells inhibit Kupffer cell phagocytic activity | |
| Kim et al | Adenosine A1 receptor (A1AR) (-/-) | 70% I 1 h/R 24 h | Histology; serum ALT; IH; semiquantitative PCR; WB; TUNEL | CCPA (AIAR agonist); DPCPX (A1AR antagonist) | Endogenous adenosine via A1AR reduces IRI | Exogenous adenosine increase IRI most likely via a different adenosine receptor subtype to A1AR |
| Ben-Ari et al[18] | Bax (-/-); Bax (+/-) | Isolated liver perfused in environmental chamber: Global I 90 min/R 1 or 15 min | Histology (apoptosis features); serum ALT, AST, LDH; TUNEL and caspase-3 assay; WB | Bax activation after 15 min reperfusion activates caspase-3 which increases liver apoptosis | ||
| Lappas et al[30] | Rag1 (-/-), i.e., lack mature lymphocytes A2AR (-/-); IFNγ (-/-) | 70% I 72 min/R 2, 24 h | Histology; serum ALT; intracellular IFNγ | ip ATL146 (A2AR agonist); PK136 (NK1.1 depletion); CD1d Ab (inhibit NKT cell); NKT cell adoptive transfer from WT, A2AR and IFNγ KO to Rag1 KO | Exogenous and endogenous adenosine acts through A2AR to reduce NKT cell recruitment | NKT cell recruitment increases IRI through release of IFNγ from at least 2 h reperfusion onwards and increased neutrophil recruitment from at least 24 h after reperfusion |
| Shimamura et al[25] | Cd1d (-/-); nu/nu (no NKT cell, normal NK cells); perforin (-/-); gld/gld (Fas ligand deficient) | Total hepatic ischaemia 30 min/R 2 ,6, 12, 24, 48 h | Serum ALT; peroxide assay; cytotoxic assay; IH; ELISA | Anti-NK and anti-NKT Ab | NKT cell activation 1 to 24 h after reperfusion releases IFNγ and PMN activation 6 to 12 h after reperfusion with increased oxidative burst lead to increased apoptosis and necrosis in IRI | |
| Caldwell et al[28] | CD4 (-/-); B cell (-/-) | 70% I 90 min/R 1, 2, 4, 8 h | Histology; serum ALT; MPO | Adoptive transfer CD4+T cell to CD4(-/-); anti-IL17 Ab | CD4+ T cell only 1-4 h after reperfusion secrete IL17 releasing MIP-2 increasing neutrophil infiltration, but inhibiting their oxidative burst, and reducing necrosis 8 h reperfusion onwards | |
| Baskin-Bey et al | Cathepsin B (-/-) | Two weeks fed methionine choline deficient (MCD) diet to induce steatosis; liver stored 24 h 4 °C UWS then perfused in isolated apparatus at 37 °C for 1 h | Histology; electron microscopy (EM); TUNEL; IH; liver tissue ALT and LDH | R-3032 ip 2 h preop (cathepsin B inhibitor) | Reduced lysosomal integrity more pronounced in steatotic livers with increased cathepsin B release into cytosol associated with increased apoptosis and necrosis | |
| Khandoga et al[22] | ICAM (-/-) | Left lobe I 90 min/R 20 min | Serum AST and ALT; IH; caspase-3 assay; lipid peroxidation assay; IVM | Anti-fibronectin Ab | Platelets bind fibronectin deposited on ICAM-1 expressed on SECs, associated with reduced sinusoidal perfusion, increased lipid peroxidation and apoptosis | |
| Shen et al[33] | nu/nu; CD154 (-/-) | 70% I 90 min/R 4 h | Serum ALT; histology; MPO; WB | Anti-CD154 Ab to WT; adoptive transfer spleen lymphocytes into KO or Ab treated group | IRI induces HO-1 protein | CD4-CD154 T cell costimulation is associated with increased IRI |
| Wyllie et al[69] | Natural resistance associated macrophage protein 1 (Nramp) (-/-) | 70%I 45 min/R 30, 60 min | Plasma GOT and TNF-α; histology; WB; Northern Blot; IH; EMSA (NFκβ) | HO-1 expressed in this model is protective in IRI | Macrophage activation after reperfusion increases TNF-α release and NFκβ activity which increases IRI | |
| Young et al[21] | P-selectin/ ICAM-1 double KO | 70% I 90 min/R 1.5, 3, 6 h | Serum ALT; histology | P-selectin and ICAM-1 do affect the severity of IRI up to 6 h reperfusion in this model, although PMN infiltration is slightly increased in midzonal area | ||
| Ozaki et al[13] | gp91 phox component of phagocyte NADPH oxidase (-/-) | 70% I 60 min/R 5, 8, 24 h +/- iv injection 3 d preop of adenovirus | Serum GOT; histology (HE; ELISA for DNA histone fragments); TUNEL; IH; WB; assays for lipid peroxidation, hydrogen peroxide and superoxide; EMSA (NFκβ) | Replication deficient adenovirus encoding Rac1 (control: Adβgal) | Rac1 is activated in IRI and is protective | Liver tissue releases ROS within 5 min of reperfusion and PMN from 8 h onwards, associated with increased lipid peroxidation, apoptosis and necrosis. NFκβ DNA binding is associated with increased IRI; NADPH oxidase regulated by Rac1 small GTP binding protein is a source of ROS in IRI |
| Sawaya et al[19] | P-selectin (-/-) | Left lobe I 30 min/R 15, 30, 60, 120 min | Serum AST, ALT, LDH; histology; IVM in terminal hepatic venule (THV) | Radiolabelled anti P-selectin Ab | P selectin expression on SECs increases rolling, saltating and adherent leucocytes in THV peaking at 30 min reperfusion | |
| Singh et al[20] | P-selectin (-/-) | Left lobe I 30 min/R 20 min, 2, 5, 12, 24 h | Serum AST, ALT, LDH; histology; WB | Radiolabelled anti P-selectin Ab | P-selectin expression peaks at 20 min and 5 h after reperfusion and is associated with worse IRI |
KO: Transgenic knockout; I: Ischemia; R: Reperfusion; IR: Ischemia reperfusion; IRI: Ischemia reperfusion injury; ROS: Reactive oxygen species; ATP: Adenosine triphosphate; IH: Immunohistochemistry; HE: Hematoxylin and eosin; WB: Western blotting; MPO: Myeloperoxidase assay; PCR: Polymerase chain reaction; ELISA: Enzyme labelled immunosorbent assay; EMSA: Electrophoretic mobility shift assay; AST: Aspartate transaminase; ALT: Alanine transaminase; LDH; Lactate dehydrogenase; GOT: Glutamic oxaloacetic transaminase; NADPH; Nicotinamide adenine dinucleotide phosphate; IR: Ischemia reperfusion; IVM: Intravital microscopy; A2AR: Adenosine (subtype 2A) receptor; PMN: Polymorphonuclear cell; NKT: Natural killer T cell; NK: Natural killer cell; IFN: Interferon; Ab: Antibody; TNF: Tumour necrosis factor; TNFR1: Tumour necrosis factor receptor (subtype 1); TCR: T cell receptor; TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling (assay for cell death); IL: Interleukin; ICAM: Intercellular adhesion molecule; VAP: Vascular adhesion protein.
ADHESION MOLECULES
The adhesion to the hepatic sinusoidal endothelial cells and transmigration into liver tissue require sequential steps in which many molecules are involved. The selectin family (P-, E- and L-selectin) of adhesion molecules are expressed by SECs early in reperfusion. They mediate loose or rolling adhesion of platelets and leucocytes. Knockout models indicate that there is an initial peak of P-selectin expression 20 to 30 min after reperfusion which is required for early IRI[19,20]. Functionally, some groups have found that E-selectin expression, and not P-selectin, is required for IRI to occur[21]. This is followed by firmer adhesion of leucocytes on SECs by upregulation of integrins, such as anti-CD11a and anti-CD11b, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecules (ICAM-1), respectively (Table 2).
PLATELETS
Platelets and leucocytes begin to adhere to SECs within 5 min of reperfusion (Table 2). Khandoga et al[22] used an ICAM-1 knockout model of early liver IRI and showed reduced IRI in the knockouts. Although ICAM-1 deficiency attenuated postischemic adherence of both platelets and leukocytes, the application of an anti fibrinogen antibody selectively reduced the number of adherent platelets but did not influence leukocyte adhesion, which significantly reduced liver IRI. The study concluded that the very early phase of IRI is characterised by increased lipid peroxidation, apoptosis and reduced sinusoidal perfusion, depends on platelet rather than leucocyte adhesion on SEC’s and that this is mediated by fibrinogen deposited on the adhesion molecules: E-selectin, VCAM-1 and ICAM-1.
NEUTROPHILS
Neutrophils are important cellular mediators of liver IRI after 6 h of reperfusion as demonstrated using partial hepatic ischemia reperfusion (IR) models with histology and MPO assay of liver samples (myeloperoxidase, an enzyme expressed most abundantly in neutrophils) as endpoints[23,24]. The neutrophil oxidative burst is the main source of reactive oxygen species in the later stages of IRI and contributes directly to hepatocellular injury. This has been supported by immunologically deficient knockout models of liver IRI using nude (nu/nu) mice which lack a thymus so cannot generate mature T lymphocytes and a knockout for gp91 phox, the glycosylated subunit of the heterodimer phagocyte NADPH oxidase(-/-). The knockouts have shown reduced liver IRI, reduced neutrophil infiltration and reduced oxidative burst (Table 2)[13,25].
Leucocyte transmigration across endothelial and extracellular matrix (ECM) barriers is a complex process. Leukocyte migration across ECM proteins is dependent on matrix degradation, not only by increasing matrix permeability, but also for generating ECM-derived fragments, which are highly chemotactic for leukocytes. Matrix metalloproteinase (MMP)-9 is one of two major gelatinases in the MMP family responsible for the turnover and degradation of several ECM proteins, including fibronectin, a key ECM protein expressed by SEC’s in the early phase of IRI.
An MMP-9-/- knockout model of liver IRI showed reduced liver damage compared to normal mice and that neutrophil transmigration within liver sinusoids occurs over fibronectin in an MMP-9 dependent manner[26]. These conclusions were based on correlations between assays of MMP-9 activity and liver histology from in vivo experiments and in vitro but not in vivo studies of neutrophil transmigration induced by fibronectin. The limitations of this study are that it did not assess other ECM proteins and that the conclusions are based on in vitro studies which may not reflect the in vivo mechanism of neutrophil transmigration. For instance, the role of SECs in leucocyte migration was not considered in this knockout model of IRI. SEC activation and injury has an important role in liver IRI as discussed earlier, by contributing to microcirculatory dysfunction. Neutrophil recruitment is mediated, at least in part, by macrophage inflammatory protein-2 (MIP-2) binding to the chemokine receptor (CXCR2) on neutrophils, supported by a study using a CXCR2 knockout model which showed reduced neutrophil transmigaration and hepatocellular injury in the knockout animals[27]. These responses are coordinated by a complex mixture of substances including cytokines, chemokines and adhesion molecules produced by other leucocytes and various liver cell types. These will be discussed further.
T CELL RESPONSES IN LIVER WARM IRI
CD4+ T cells are activated and recruited into liver sinusoids in liver IRI (Figure 1). They have a dual role either contributing to injury or reducing the extent of injury depending on the CD4+ subtype and mechanism of cellular activation. The majority of CD4+ T cells can be subdivided into αβ TCR (the most common subtype) expressing cells, γδ TCR expressing cells, NKT cells and regulatory T cells (Tregs). B cells, CD8+ T cells[28] and NK cells[25,29] do not have an important role in modulating IRI.
Figure 1.
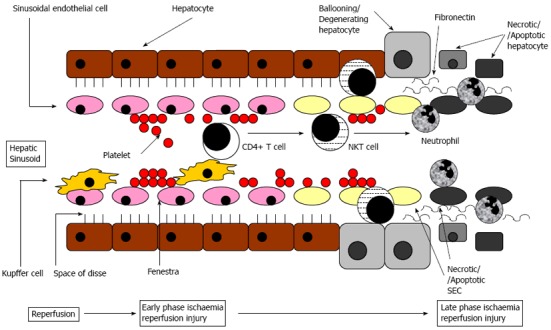
Schematic diagram of cellular mechanisms of liver ischaemia reperfusion injury within a liver sinusoid and the surrounding area containing hepatocytes. Initial sinusoidal perfusion failure from platelet plugging, then Kupffer cells activate CD4+ T cells that activate natural killer T (NKT) cell which cause sinusoidal endothelial cells (SEC) and hepatocyte injury, followed by neutrophil activation, adhesion and transmigration causing more cell injury.
NKT cells contribute to liver injury in the early stages from 1 h of reperfusion onwards. This has been supported by immunologically deficient knockout models, such as nu/nu, CD1d-/- (a non classical MHC that presents glycolipid and phospholipid to NKT cell TCR activating NKT cells) and RAG-1-/- (recombination activation gene-1 required for the maturation of lymphocytes) knockout mice with up to 50% reduction in liver injury in the knockouts[25,29,30]. NKT cells are also thought to contribute to neutrophil activation mediated by cytokines which NKT cells release, such as interferon gamma[30]. A study using T cell subtype specific knockouts showed Treg cells are not involved in IRI. γδ TCR T cells recruit neutrophils but this does not affect the severity of IRI[29].
The CD4+ T cell activation in IRI is by an antigen independent pathway[31]. This is supported by knockout models of the Toll-like receptor 4 (TLR 4) in which TLR 4 knockouts show reduced IRI and in normal animals the TLR 4 is activated on Kupffer cells resulting in IRI[32].
One study on CD4+ T cell related liver IRI suggested co-stimulatory activation of the CD4 with the CD154 receptor on activated T cells[33]. There is emerging evidence that there are also antigen dependent pathways activated in liver IRI. One partial hepatic IR (ischaemia reperfusion) model with 90 min ischaemia and 8 h reperfusion using a knockout for a TCR specific for ovalbumin self antigen showed reduced IRI in the knockout, with serum ALT reduced by 15% and reduced histological injury although this was not quantified, indicating that a small subset of T cells sensitised to self antigen contribute directly to liver IRI at least up to 8 h into reperfusion[29].
CD4+ T cells of the αβ TCR variety are recruited into liver sinusoids within 1 h of reperfusion. CD4+ T cell knockout models of liver IRI with adoptive transfer of functional CD4+ T cells into the knockout mice indicate that these cells are involved in neutrophil recruitment via cytokines such as interleukin 17 (IL 17) and MIP-2, these T cells inhibit the neutrophil oxidative burst. In a model of partial hepatic ischaemia for 90 min followed by reperfusion, CD4+ knockouts showed greater IRI than normal mice, with serum ALT approximately 25% higher in the knockouts and more severe histological injury in the knockouts although this was not numerically quantified[28]. They reduce the extent of liver IRI both indirectly via cytokines they release affecting other leucocytes and directly acting on hepatocytes[28].
CYTOKINES AND CHEMOKINES
The complex interplay between cytokines and chemokines in liver IRI is not fully understood. The most extensively studied cytokines are TNF-α, interferon (IFN)-β, IFN-γ and IL-6 (Figure 2).
Figure 2.

Cytokine and downstream signalling pathways in liver ischaemia reperfusion injury. Following liver ischaemia reperfusion, there is activation of tumor necrosis factor-α (TNF-α) is by chemokine (CXCL) 10, interferon regulatory factor (IRF) and toll-like receptor (TLR) 4 in parallel. TNF-α activates downstream hepatocyte/sinusoidal endothelial cells (SEC) nuclear factor κβ (NFκβ) and CD4+ T cells separately which activate c-Jun N-terminal protein kinase-2 (JNK-2) and signal transducer activator of transcription-4 (STAT4), respectively leading to increased cell injury. A parallel pathway of cell injury occurs where TLR4 activation stimulates interferon (IFN)-β and IFN-γ expression, which acting through their receptor IFN receptor subtype (AR) activate interferon regulatory element (IRE)-1 which in turn activate CD4+ T cells. CXCR: Chemokine receptor.
TNF-α is raised in serum within 30 min of reperfusion and persists for up to 8 h[34,35]. TNF-α has ischaemic but not normal liver tissue[36]. Release of TNF-α is stimulated by a cytokine cascade involving activation of interferon regulatory factor-1 (IRF-1), as shown using a double knockout of this factor in a partial hepatic IR (ischaemia reperfusion) model with 60 min ischaemia and 6 h reperfusion, where hepatocellular injury was 60% less in the IRF-/- knockout[37].
There is some evidence from knockout studies that antigen independent macrophage/Kupffer cell TLR-4 activation stimulates TNF-α secretion[32,38]. The effects of TNF-α are mediated by binding to its receptor Tumour necrosis factor receptor subtype 1 (TNFR1) leading to increased apoptosis[39,40] and increased CD4+ T cell sinusoidal recruitment within 30 min of reperfusion[12]. One TNFR1-/- knockout model of mouse liver transplantation with liver transplantation of either normal or TNFR1-/- livers into normal or TNFR-/- mice showed the deleterious effects of TNF-α are mediated by TNFR outside the liver, most likely infiltrating leucocytes, but TNFR on liver cells appear to reduce IRI in this model (Table 3).
Table 3.
Cytokine, chemokine, tolllike receptor, complement knockout models of liver ischemia reperfusion injury
| Ref. | Knockout model | IR protocol | Outcome measure | Agent | Adaptive responses | Injurious responses |
| Kuboki et al[26] | CXCR2 | 70% I 90 min/R 12, 24, 48, 96 h | Histology; MPO; serum ALT, TNF-α, IL6; WB and NFκβ activity | CXCR2 activates STAT3 hepatocyte proproliferative pathway | MIP2 activates CXCR2 which increases neutrophil recruitment and IRI. Nuclear factor (NF) κβ activity reduced in IRI | |
| Zhai et al[44] | IFNAR type1 (-/-); IFNAR type 2 (-/-) | 70% I 90 min/R 6 h | Histology; quantitative PCR | IFNβ (not IFNγ) mediates IRI by binding to IFNAR type 1 | ||
| Zhao et al | CXCL10 (-/-) | 70% I 90 min/R 1, 2, 4, 8 h | Histology; serum ALT; IH; quantitative PCR; WB | CXCL10 activation increases TNF-α, IL6, IL1b, iNOS, MIP-2 mRNA and PMN and Kupffer cell activation contributes to IRI | ||
| Fondevilla et al | C6 deficient rats | Donor/recipient: WT/WT, KO/WT, WT/KO, KO/KO; | Serum GOT; histology; MPO; IH; TUNEL; WB; PCR; ELISA | Membrane attack complex (C5b-C9) activation in this OLT model of cold/warm IRI increases apoptosis, necrosis, PMN and macrophage infiltration and TNF-α, IFNγ and IFNβ expression | ||
| OLT and organ storage 24 h 4 °C UWS | ||||||
| Shen et al[32] | Toll like receptor 4 (TLR4) (-/-) | Donor/recipient: WT/WT, KO/WTWT/KO, KO/KO; OLT with dearterialisation, organ stored 24 h 4 °C UWS | Histology; IH; MPO; quantitative PCR; capsase-3 activity; WB | TLR4 activation increases IL4 and IL10, but inhibits HO-1 | TLR4 activation increases TNF-α, IL1b, IL2, IFNγ, ICAM1, CXCL10, PMN and CD4+ T cell recruitment leading to increased liver necrosis and apoptosis | |
| Conzelmann et al | TNFR (-/-) | Donor/recipient: WT/WT, KO/WTWT/KO, KO/KO; organ storage 12 h 4 °C UWS; 8 h graft harvest | Histology; serum ALT; MPO; TUNEL and caspase-3 assay; IH | TNFR within liver mediates reduced IRI | TNFR outside liver increases IRI in terms of necrosis, apoptosis and neutrophil infiltration | |
| Tsung et al[37] | Interferon regulatory factor-1 (IRF-1) (-/-) | 70% I 60 min/R 1, 3, 6, 12 h | Histology; serum ALT; WB; PCR | Adenovirus IRF-1 vector | IFNγ, IFNβ, TNF-α, IL1β all activate IRF-1 which increase JNK (not p38 MAPK) and TNF-α and iNOS expression in IRI | |
| Tian et al[40] | TNFR1 (-/-); IL6 (-/-) | Donor/recipient: WT/WT, KO/WT, WT/KO, KO/KO; OLT: 50% or small for size 30% arterialised graft | Histology; serum AST; portal flow measurement; IVM; IH; PCR; 30 d mortality | GdCl3 (ip to donor); pentoxifylline (to donor and recipient sc); recombinant IL6 to KO only | Increased IL6 | Increased activation of Kupffer cells and TNF-α mediated activation of IFNR1 from 3 h reperfusion onwards increases liver necrosis, nonperfused sinusoids, adherent leucocytes and reduces hepatocyte regeneration |
| Shen et al[38] | TLR4 (-/-); TLR2 (-/-) | 70% I 90 min/R 6 h | Histology; serum ALT; MPO; WB; PCR | Snpp (inhibit HO-1); CoPP | HO-1 is expressed which inhibits TLR4 | TLR4 activation increases TNF-α expression associated with increased IRI |
| Lagoa et al[81] | PAI-1 (-/-) | MAP 25-30 mmHg for 2.5 h (2.25 mL/100 g blood withdrawn)/Resuscitation MAP > 80 mmHg for 4 h (30 min with shed blood and crystalloid) | Serum ALT, IL6, IL10; histology; Electron microscopy; IH; zymography for plasminogen activators; DNA microarray; PCR; WB | PAI-1 to PAI-1 (-/-) mice | PAI-1 expression in SEC contributes to IRI with periportal/pericentral injury, loss of sinusoidal fenestra and prominent SEC injury; PAI-1 inhibits u-PA which reduces formation of active HGF and increases active TGF-β1, but no effect on IL6 or IL10; this is associated with reduced activation of ERK-1/-2 pathway. | |
| Teoh et al[36] | TNF-α (-/-) | 70%I 90 min/R 2, 4, 24 h | Serum ALT; IH; serum TNF-α; EMSA (NFκβ); WB | Low dose or high dose TNF-α ip | TNF-α from at least 2 h reperfusion onwards is injurious to ischaemic but not normal liver, increasing NFκβ DNA binding | |
| Inderbitzin et al[57] | CI inhibitor overexpressed | Total hepatic ischaemia 30 min/R 2 h | Endothelial permeability index (measured using radiolabelled albumin iv into inferior vena cava) of liver, lung and gut | C1 inhibitor overexpression is protective in IRI | Classical complement pathway is activated in IRI; liver ischaemia and reperfusion causes liver and gut, but not lung, IRI in this model | |
| Zhai et al | TLR4 (-/-); TLR2 (-/-) | 70%I 90 min/R 6 h | Serum ALT; histology; PCR | TLR4 activation increases expression of IRF3 which upregulates IFNβ associated with increased IRI | ||
| Rudiger et al[39] | TNFR (-/-); Fas (-/-); FasL (-/-) | 70% I 75 min/R 3 h | Serum AST; TUNEL; caspase-3 assay; ELISA; WB | Pentoxifylline | TNF-α binds to TNFR1 which increases apoptosis in IRI; fas and FasL not involved in this model | |
| Kato et al[82] | IL1R (-/-) | 70%I 90 min/R 1, 2, 4, 8, 16, 24 h | Serum ALT, IL1β, TNF-α and MIP-2; histology (PMN score); MPO; EMSA (NFκβ); PCR | IL1R not involved in IRI | ||
| Calmargo et al | IL6 (-/-) | Median lobe (45%) I 90 min/R 30, 60, 90, 120 min | Serum AST and ALT; histology; PCR | Recombinant IL6 | IL6 released in IRI is protective | TNF-α expression during reperfusion is associated with worse IRI |
KO: Transgenic knockout; WT: Wild type (normal animals); IH: Immunohistochemistry; WB: Western blotting; MPO: Myeloperoxidase assay; PCR: Polymerase chain reaction; ELISA: Enzyme labelled immunosorbent assay; EMSA: Electrophoretic mobility shift assay; AST: Aspartate transaminase; ALT: Alanine transaminase; GOT: Glutamic oxaloacetic transaminase; I: Ischemia; R: Reperfusion; IR: Ischemia reperfusion; IRI: Ischemia reperfusion injury; IVM: Intravital microscopy; IFN: Interferon; Ab: Antibody; TNF: Tumour necrosis factor; TNFR1: Tumour necrosis factor receptor (subtype 1); TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling (assay for cell death); IL: Interleukin; CXCR: Chemokine receptor; PAI: Plasminogen activator inhibitor; NF: Nuclear factor; C1-9: Complement protein 1 to 9; MAP: Mean arterial pressure; MIP: Major intrinsic protein.
IFN-β is a cytokine which is involved throughout the reperfusion period in liver IRI, a finding substantiated by work using knockout mice (Table 3). The damaging effects of IFN-β are mediated by binding to the interferon receptor subtype IFN AR (Type 1)[41] with no hepatocellular liver injury demonstrated in IFN AR-/- knockouts (Figure 2)[37]. Knockout models support other studies showing that IFN-γ produced by NKT cells contribute to liver IRI from early on in reperfusion[25,27,30,32,37,42,43]. Activation of innate immune pathways via TLR 4 stimulate release of IFN-β and IFN-γ in liver IRI, confirmed by TLR 4 knockout models[32].
Some cytokines released during liver IRI appear to reduce the severity of injury. The best evidence for this from knockout studies is for IL6[34]. Camargo et al[34] showed worse IRI in livers of IL6 knockout mice than wild type mice, which was restored to the wild type injury patterns by administration of recombinant IL6 to the knockout mice before ischaemia. There is evidence that IL4 and IL10 may also be protective in IRI, but knockouts of these cytokines have not been used in liver IRI models to substantiate this.
Chemokines (CXCL), like cytokines, are a family of locally produced factors, but chemokines are smaller molecules, which act locally forming concentration gradients that guide leucocyte chemotaxis. A CXCL 10 knockout model found that this chemokine contributed to liver IRI (Figure 2 and Table 3) from 1 h of reperfusion onwards with associated activation of neutrophils, Kupffer cells and increased TNF-α and IL 1β release[44]. A study using knockouts of chemokine receptor 2 (CXR 2) showed CXCR 2 activation contributes to liver IRI and neutrophil recruitment[26].
Plasminogen activator inhibitor type-1 (PAI-1) inhibits plasmin generation by inhibiting activation of plasminogen activators which play a role in diverse proteolysis related processes. One group used a model of liver ischemia reperfusion using wild type normal and PAI-1 knockout mice based on controlled intravenous haemorrhage maintaining MAP 25-30 mmHg for 2.5 h followed by controlled resuscitation intravenously with shed blood/Ringer’s lactate maintaining MAP > 80 mmHg for 4 h. They demonstrated liver IRI in wild type animals reflected by raised serum ALT, histological periportal and pericentral injury (zone 2) and electron microscopic SEC injury with loss of fenestra. DNA microarray showed PAI-1 mRNA was most elevated after haemorrhage-resuscitation and immunohistochemistry showed PAI-1 expression was localised to SEC. PAI-1 knockouts had no liver injury following haemorrhage-resuscitation. In normal mice PAI-1 mediated IRI was associated with reduced urokinase plasminogen activator (u-PA) in zymogens of hepatocytes, reduced hepatocyte growth factor (HGF) and increased TGF-β1, but no differences in IL6 and IL10. These changes with haemorrhage-resuscitation are associated with reduced phosphorylation activation of the ERK-1/-2 MAP kinase pathway which were reversed in PAI-1 knockouts. Based on these results, the study concluded that liver IR activates SEC PAI-1 which inhibits u-PA which reduces levels of active HGF and increases levels of TGF-β1 that together reduce activation of downstream ERK MAP kinase pathway, mediating liver IRI. The group used a systemic model of liver IRI (systemic hypotension) rather than direct liver ischaemia and reperfusion so PAI-1 may not have the same role in liver IRI resulting from total or partial inflow occlusion. The knockout animals which showed no liver IRI with heamorrhage-resusciatation, were anaesthetised with isoflurane, while wild type animals which did show liver IRI with haemorrhage-resuscitation were anaesthetised with sodium pentobarbital. These results need to be interpreted with caution as isoflurane preconditions against liver IRI, so the protective effect may not be due to lack of PAI-1.
COMPLEMENT SYSTEM
Activation of the complement system has been shown to occur in local and remote IRI in many organs[45]. This system consists of around 30 soluble and membrane bound proteins which are activated by one of three pathways: the antibody dependent classical pathway, the alternate pathway and the mannose binding lectin pathway. Activated complement acts both directly through the formation and deposition of membrane attack complexes (C5b-C9) and indirectly following activation by cytokines and chemokines in IRI[46]. Knockout models have been used to consolidate the results of other studies showing activation of complement by all three pathways having direct and indirect effects on cellular reperfusion injury. The best examples of these complement knockout studies of IRI are in the gut[47-51], kidneys[52,53] and heart[54]. Studies on the liver have looked at nonspecific blockade of all parts or the final common pathways of the complement system, so based on current evidence there is little understanding of the relative importance of the different pathways of complement activation in liver IRI.
Inhibition of complement formation before hepatic ischaemia in studies of liver IRI have shown a reduction in the severity of injury within an 1 h of reperfusion when cobra venom factor (to inhibit all parts of the complement system)[11] was used, but liver IRI and polymorphonuclear cell accumulation were also reduced in the late phase of IRI 24 h following ischaemia when animals were pretreated with sCR1 (a complement inhibitor derived from the family of complement regulatory glycoproteins and inhibits activation of C3, which is common to all pathways of complement activation, and so blocks the generation of both C3a and C5a and the MAC)[11]. Scozaec et al[11] found local complement activation in human liver allografts correlated to cell injury. One pharmacological in vivo and in vitro study of a warm liver IRI model in a rat showed that complement is involved in the induction of Kupffer cell-induced oxidant stress, the priming of Kupffer cells and neutrophils for enhanced reactive oxygen generation, and the continuous accumulation of neutrophils in the liver during reperfusion. In a pig whole liver to canine left liver xenotransplantation model where a control group was compared against a gadolinium chloride (GdCl3) (which depletes Kupffer cells) and cobra venom treated group, there was less liver injury following transplantation in those with compliment inhibition using the cobra venom. This provided a large animal model supporting the role of complement in activation of Kupffer cells in liver ischaemia reperfusion injury. The groups were small (3 animals each) and the conclusions would be more robust if there had been groups treated with only GdCl3 or cobra venom[55].
One group used wild type rats and rats deficient in C6 in all combinations of donor and recipient in a liver transplantation model to show that there was less injury in recipients of C6-/- grafts, implicating that the membrane attack complex (C5b-9) is involved in cold ischaemia related liver IRI in this model[56]. One model of liver IRI using mice overexpressing the inhibitor of the classical complement pathway, C1 inhibitor, showed C1 inhibitor reduces liver IRI and remote IRI in the lungs and gut followed liver ischaemia and reperfusion[57]. This would appear to indicate that the classical pathway of complement activation is the most important complement activation pathway involved in liver IRI (Table 3), although further studies using C1 inhibitor knockouts and mannose binding lectin knockouts to substantiate the relative contribution of the various activation pathways, as well as specific knockouts of other complement components to assess the significance of different complement mediated injurious pathways.
MATRIX METALLOPROTEINASE-9
MMP-9 is a zinc dependent secreted gelatinase which catalyses degradation of type IV collagen and gelatin. MMP-9 knockout models of liver IRI have shown liver IRI is reduced by up to 80% in MMP-9-/- knockouts and that in normal animals increased expression of MMP-9 on macrophages and neutrophils occurs during reperfusion (Table 4) which increases neutrophil transmigration over fibronectin in liver sinusoids and increases TNF-α and interferon γ secretion and CD4+ T cell activation by mechanisms that remain to be elucidated[12,27]. The mechanisms by which MMP-9 expression is increased in liver IR were not investigated in these knockout studies, but a possible pathway involves induction of MMP-9 by ROS and TNF-α.
Table 4.
Nitric oxide synthase, HSP/heme oxygenase-1, matrix metalloproteinase knockout models of liver ischemia reperfusion injury
| Ref. | Knockout model | IR protocol | Outcome measure | Agent | Adaptive responses | Injurious responses |
| Hamada et al[26] | iNOS (-/-); MMP-9 (-/-) | 70% I 90 min/R 3, 6, 24 h | Histology; serum ALT, NO2-/NO3-; myeloperoxidase activity (MPO); immunohistochemistry; PCR, Western blotting; MMP-9 activity assay; MMP-9 protein levels; neutrophil (PMN) migration assay; TUNEL and caspase-3 activity | ONO-1714 (iNOS inhibitor); NO donor (DETA NONOate) | Increased macrophage iNOS producing NO increases PMN MMP-9 and PMN transmigration over fibronectin | |
| Hamada et al[42] | MMP-9 (-/-) | 70% I 90 min/R 6, 24 h | Histology; serum GPT and GOT; MPO; IH; PCR | Anti MMP-9 iv; MMP-2/9 inhibitor; anti MMP-2 (all to WT only) | MMP-9 (not MMP-2) increase TNF-α, IFNg, IL2, IL6 and increase PMN and CD4+ T cell recruitment leading to increased liver necrosis | |
| Kuboki et al[73] | HSP70 (-/-) | 70% I 90 min/R 1, 8 h | Histology; serum AST; TNF-α; IL6; MIP-2; MPO; WB; EMSA (NFκβ) | Sodium arsenite iv to induce HSP70; recombinant HSP70 | No involvement of HSP70 in IRI; NFκβ activity associated with IRI | |
| Theruvath et al[59] | eNOS (-/-) | Donor (WT/KO) to WT recipient; organ stored 18 h, 4 °C, UWS | Histology; serum ALT; IVM; TUNEL; IH (macrophage infiltration) | eNOS activation reduces necrosis and apoptosis, with associated inhibition of macrophage infiltration, increased sinusoidal diameter and blood flow | ||
| Tsuchiashi et al | HO-1 (+/-); HO-1 (-/-) | 70% I 90 min/R 6 h | Histology; serum GOT; MPO; quantitative real time RT-PCR; WB; TUNEL | CoPP (induces HO-1) 24 h preop | HO-1 upregulated which inhibits expression of cytokines TNF-α and IFNγ | TNF-α and IFNγ expression increased overall in IRI associated with increased apoptosis and necrosis |
| Hines et al[23] | eNOS (-/-); iNOS (-/-) | 70% I 45 min/R 1, 3 h | Serum ALT; histology; PCR | Increased eNOS expression in IRI inhibits TNF-α and IL12 expression; iNOS activates eNOS in this model | No PMN infiltration at 3 h reperfusion | |
| Lee et al[58] | eNOS (-/-); iNOS (-/-) | 70% I 1 h/R 1, 3, 6 h | Serum ALT and AST; perfusion studies; PCR | eNOS activated during IRI is protective | Increased iNOS mRNA expression from 3 h reperfusion onwards regulates reperfusion and is associated with worse IRI | |
| Hines et al[60] | iNOS (-/-) | 70% I 45 min/R 1, 3, 6 h | Serum ALT; histology; MPO | L-NIL (iNOS inhibitor) | Reduced IRI in iNOS (-/-), but no iNOS mRNA or L-NIL effect in WT; may be genetic compensation effect in KO | |
| Kawachi et al[24] | eNOS (-/-); iNOS (-/-) | 70% I 45 min/R 5 h | Serum ALT; histology; MPO | eNOS is activated in IRI and is protective | There is no PMN infiltration up to 5 h reperfusion and iNOS is not activated in IRI in this model |
IH: Immunohistochemistry; WB: Western blotting; MPO: Myeloperoxidase assay; RT-PCR: Reverse transcriptase polymerase chain reaction; ELISA: Enzyme labelled immunosorbent assay; EMSA: Electrophoretic mobility shift assay; AST: Aspartate transaminase; ALT: Alanine transaminase; GOT: Glutamic oxaloacetic transaminase; I: Ischemia; R: Reperfusion; IR: Ischemia reperfusion; IRI: Ischemia reperfusion injury; IVM: Intravital microscopy; IFN: Interferon; Ab: Antibody; TNF: Tumour necrosis factor; TNFR1: Tumour necrosis factor receptor (subtype 1); TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling (assay for cell death); IL: Interleukin; NF: Nuclear factor; MMP: Matrix metalloproteinase; HSP: Heat shock protein; eNOS: Endothelial nitric oxide synthase; iNOS: Inducible nitric oxide synthase; HO-1: Heme oxygenase (subtype 1).
NITRIC OXIDE SYNTHASE
NOS catalyses formation of NO from L-arginine. NO is a versatile molecule which is vasoactive, is involved in activating molecular signalling pathways in cell survival, has immunological effects as well as directly injurious effects in high levels as a free radical itself. There are three isoforms of NOS: constitutive calcium (Ca2+) dependent forms which are eNOS and neuronal NOS (nNOS), and an inducible calcium independent form, namely inducible NOS (iNOS). Only eNOS and iNOS are expressed in liver. Most studies agree that eNOS is upregulated in liver IRI and this reduces the severity of IRI. This is confirmed by knockout models of IRI (Table 4), where eNOS expression is related to reduced liver necrosis, apoptosis, leucocyte infiltration and increasing liver sinusoidal diameter and liver blood flow[23,24,58,59].
The role of iNOS is more controversial (Table 4)[58-64]. Some knockout models of iNOS show no role for iNOS in liver IRI[24,60,61]. One knockout study of iNOS showed in the liver IRI model used that iNOS was protective which was at least partly mediated by activation of iNOS by eNOS[23]. Yet another set of knockout studies conclude iNOS contributes to IRI[27,37,58]. Hamada et al[27] used separate iNOS and MMP-9 knockout mice to show in their model of liver IRI that iNOS is upregulated in macrophages which increases IFN-γ release and NO which increases MMP-9 expression on the macrophages and neutrophils. This signalling cascade contributes to increased liver IRI. However, one study using the partial (70%) hepatic IR model with 45 min lobar ischaemia found increased IR injury in iNOS-/- mice demonstrated by increased hepatocellular and histological injury and increased liver sinusoid neutrophil infiltration compared to wild type animals, but they found no iNOS mRNA expression in wild type livers[24]. This led the authors to conclude that there may be genetic compensation in iNOS-/- animals, although there was no reference to which genes may have been involved in this compensation. Genetic compensation is a rare phenomenon where after a gene is mutated and its function is lost, compensatory genes are upregulated, although the mechanisms for this is unclear[65].
Large animal studies using pigs and various inhibitors of iNOS (AG, ONO-1714) on both a warm liver ischaemia reperfusion model and an orthotopic liver transplantation model have shown that iNOS expression is stimulated by IR in Kupffer cells and neutrophils in the centrilobular region resulting in higher levels of serum nitrite/nitrate, reduced capillary perfusion with more thrombi and ultimately increased liver injury and increased mortality[62-64].
The conflicting results of the role of iNOS in early phase liver IRI may reflect different roles of iNOS and the regulation of its function in liver IRI depending on the duration of the liver ischaemia. Partial hepatic IR models with prolonged ischaemia of 60 min or longer have found that iNOS does have a role in liver IRI[27], while models using shorter ischaemia times of 45 min have shown no role of iNOS[24]. Several studies that have used iNOS-/- knockout animal models of hepatic IR, but there is evidence, that iNOS-/- knockouts show genetic compensation[24], so conclusions based on models of liver IR using iNOS-/- animals need to be interpreted with caution.
The overall conclusions from the current literature is that eNOS and iNOS are both induced during liver IRI from 1 h reperfusion onwards (for mRNA and 2 h reperfusion onwards for protein), eNOS reduces injury and low levels of iNOS induction are probably protective while high expression of iNOS contributes to increased injury and the overall effect of iNOS physiologically depends on how ischaemia and reperfusion is produced.
HAEM OXYGENASE-1
Haem oxygenase-1 (HO-1 or heat shock protein 32, HSP 32) is the inducible isoform of HO-1, the constitutive isoform being HO-2. This enzyme catalyses the formation of CO, biliverdin and Fe2+ from haem degradation. HO-1 has been implicated as having a protective role in IRI[66] through CO and biliverdin being vasodilators and reducing apoptosis and necrosis[67]. HO-1 is typically expressed three or more h after liver reperfusion[38,68]. The protective effects of HO-1 are supported by knockout models of liver IRI, where knockouts of HO-1 have more severe IRI than normal animals (Table 4)[38,43]. These knockout models also provide evidence that HO-1 acts at least partly by inhibiting TLR4 (Toll-like receptor 4) activation and the resulting release of TNF-α and IFN-γ[38,43,69].
DOWNSTREAM PATHWAYS
A wide range of downstream pathways have been studied in liver IRI. The majority of systems which are activated in ischaemia reperfusion are effective through these pathways. Some of the key mediatoare TNF-α, IFN-β, IFN-γ and CXCL10 (Figure 2). In particular, the roles of the transcription factors nuclear factor κβ (NFκβ)[70], the survival kinases (JNK, MAPK’s, PKC, PI3K/Akt), signal transducer and activator of transcription (STAT’s)[71], poly ADP ribose polymerase (PARP), peroxisome proliferator-activated receptor (PPAR) from pharmacological studies have been supported by knockout models of liver IRI (Table 5).
Table 5.
Knockouts of downstream mediators and models of liver ischemia reperfusion injury
| Ref. | Knockout model | IR protocol | Outcome measure | Agent | Adaptive responses | Injurious responses |
| Theruvath et al[14,15] | JNK (-/-) | 70% I 1 h/R 4, 8 h | Histology; serum ALT; IVM (dyes to probe mitochondrial function and cell death); survival 14 d | JNK2 activation leads to mitochondrial depolarization and increased necrosis only | ||
| Theruvath et al[14,15] | JNK (-/-) | WT or KO donor; 30 h, 4 °C, UWS preservation; WT recipient | Histology 8 h posttransplant; serum ALT, TUNEl and caspase-3 assay; IVM; IH; lipid peroxidation | JNK2 activation leads to caspase-3 activation, mitochondrial depolarisation and release of cytochrome c, lipid peroxidation which all translate into reduced survival posttransplant | ||
| Beraza et al[72] | Conditional hepatocyte specific NEMO knockout | 70% I 1 h (caudate lobe resected)/R 3, 6 h | TUNEL and caspase-3 assay; WB; IH; Southern blotting; EMSA (NFκβ) | NFκβ activity reduces necrosis and apoptosis, inhibits TNF-α, JNK and iNOS | ||
| Okaya et al[35] | PPARα (-/-) | 70%I 90 min/R 4, 8 h | Serum ALT; TNF-α; MIP-2; MPO; liver NO2/NO3; WB; EMSA (AP-1, NFκβ) | WY14643 iv (PPARα agonist) | PPARα protective in IRI | PPARα independent release of TNF-α and MIP-2 and increased NO2/NO3 associated with IRI |
| Shen et al[78] | STAT4 (-/-); STAT6 (-/-); nu/nu | 70%I 90 min/R 6 h | Serum ALT; histology; MPO; WB; PCR | Adoptive transfer of CD4+ T cells from WT or other KO to nu/nu; SnPP ip | HO-1 expressed at very low levels after 6 h in this model, but protective in IRI | CD4+T cell activation involving T cell STAT4 activation, but not STAT6 associated with increased IRI |
| Khandoga et al[61] | PARP (-/-) | Left lobe I 90 min/R 30 min | Serum ALT; IVM; IH; PCR | PARP activation in IRI upregulates E-selectin, ICAM1 and VCAM1, associated with increased platelet and leucocyte endothelial interaction and reduced sinusoidal perfusion | ||
| Kato et al[82] | P50 NFκβ (-/-) | 70% I 90 min/R 1, 8 h | Serum ALT; histology; MPO WB; EMSA (p50 and p65 subunits of NFκβ) | No effect of p50 subunit deletion, but increased p50/p65 heterodimer in WT and some p65 in KO, so there may be some functional redundancy of NFκβ subunits | ||
| Kato et al[83] | STAT4 (-/-) | 70% I 90 min/R 30 min, 1, 2, 4, 8 h | Serum ALT; histology; MPO; WB | Anti IL12 Ab | IL12 expression associated with IRI. STAT4 not activated in IRI in this model | |
| Kato et al[70] | STAT6 (-/-) | 70% I 90 min/R 1, 4, 8 h | Serum ALT, TNF-α; MPO; PCR; EMSA (NFκβ) | IL4 or IL13 iv | STAT6 is not activated in IRI in this model, although STAT6 activation by iv IL4 or IL13 is protective. IRI is associated with increased NFκβ DNA binding |
KO: Transgenic knockout; WT: Wild type (normal animals); IH: Immunohistochemistry; WB: Western blotting; MPO: Myeloperoxidase assay; PCR: Polymerase chain reaction; EMSA: Electrophoretic mobility shift assay; ALT: Alanine transaminase; I: Ischemia; R: Reperfusion; IR: Ischemia reperfusion; IRI: Ischemia reperfusion injury; IVM: Intravital microscopy; IFN: Interferon; Ab: Antibody; TNF: Tumour necrosis factor; TNFR1: Tumour necrosis factor receptor (subtype 1); TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling (assay for cell death); IL: Interleukin; NF: Nuclear factor; STAT: Signal Transducer and Activator of Transcription; JNK: A survival kinase; PARP: Poly (ADP-ribose) polymerase.
Most studies show that NFκβ DNA binding increases after liver ischaemia and contributes to liver IRI[13,36,67,70,71]. TNF-α increases NFκβ activity[36,70]. One knockout study, in contrast, showed a CXCR2 dependent fall in NFκβ activity following liver reperfusion[26], although a longer period of ischaemia and reperfusion was used than other studies. Another group used a conditional NFκβ knockout to study liver IRI and showed NFκβ activity was protective, reducing necrosis, apoptosis, JNK expression and TNF-α expression. Unlike the other studies, the caudate lobe was resected in their protocol, which alters how IR occurs compared to a model of hepatic IR without resection. The role of NFκβ, therefore, is unclear in liver IRI, but it may be that the specific way in which IR is executed modulates NFκβ activity and the resulting activation of downstream signalling pathways in IRI.
Theruvath et al[14,15] used a JNK 2 knockout to show JNK 2 contributes to IRI in both a mouse liver transplantation model and warm ischaemia reperfusion model of liver IRI by increasing mitochondrial depolarisation and caspase-3 activity leading to liver injury manifested as hepatocyte cell membrane lipid peroxidation, necrosis and apoptosis. A knockout model has provided evidence that JNK activity, but not p38 MAPK contributes to liver IRI after reperfusion (Table 5)[37].
Knockout models of PI3K and Protein kinase C (PKC) have been used in cardiac IRI, but not in the liver IRI. One group used a porcine liver transplant model using chelerythrine (a PKC inhibitor) and/or ischaemic preconditioning (IPC) of the donor liver before cold storage to show that PKC activity was not affected by IRI alone, although PKC was strongly activated by IPC reducing the severity of IRI[73-79].
In a rat model of warm partial hepatic ischaemia reperfusion using a caspase-3 inhibitor (Z-Aspemk iv 2 min before 2 h of ischaemia) it was found that IRI was associated with reduced PI3K/Akt activity and increased hepatocyte apoptosis, with the converse pattern in the caspase-3 inhibitor treated group[77-79]. In contrast, in another rat model of IRI, hepatocytes isolated from rats which underwent partial warm hepatic ischaemia/reperfusion or sham laparotomy cultured and treated with IL-1β provided evidence that during IR, IL-1β binding to IL-1β receptor-1 increased NFκβ activity and phosphorylated Akt which acted in parallel to increase iNOS expression (mRNA and protein) with a resulting increase in NO release[77-79].
Activation of the STAT family of transcription factors is mediated by extracellular signalling molecules such as cytokines which bind to membrane receptors which activate intracellular Janus kinases on the cytoplasmic face of the plasma membrane, which in turn activate a STAT protein which is then transported to the nucleus where they bind DNA to affect gene expression (Jak/STAT signalling pathway). STAT6 activation does not appear to be involved in liver IRI based on results from knockout models[71,72,80]. Kato et al[72] found in their model of IRI using STAT4 knockouts that STAT4 did not affect the extent of liver injury after 8 h reperfusion. In contrast, Shen et al[80] showed STAT4 expression was related to IRI after 6 h of reperfusion, although it was specifically its expression within CD4+ T cells that mediated the liver injury (Table 5). This was demonstrated by reduced IRI in mice lacking mature lymphocytes (nu/nu mice), which was restored to normal animal IRI severity in the nu/nu mice by adoptive transfer of CD4+ T cells from spleens of normal mice but not from spleens of STAT4 knockouts. The reasons for this discrepancy between Kato et al[72] and Shen et al[80] are not clear, as both groups used a partial (70%) hepatic IR model of 90 min ischaemia and reperfusion including the same timeframe, the same endpoints and double knockouts. The transgenic knockouts had different wild type backgrounds in the two studies of C57Bl6 or Balb/c wild types by Kato et al[72] and Shen et al[80] respectively, which may have affected the IRI results. Genetic compensation in the knockouts of either study is a possibility that may explain the discrepancy, although STAT4 protein expression was not assessed by Shen et al[80] and there is little detail on how the knockouts were generated in both studies.
A knockout of PARP has been used to show that PARP activation contributes to early liver IRI (Table 5) and activates signalling pathways increasing expression of adhesion molecules on SECs[61]. A liver IRI model using a PPAR knockout demonstrated background PPAR activity reduces the severity of liver reperfusion injury acting via signalling pathways that remain to be elucidated but appear not to involve NO or TNF-α, both of which act independently of PPAR in this model[35].
CONCLUSION
Liver IRI is a clinically relevant phenomenon in a wide range of settings including trauma surgery, hepatic resection and transplantation, affecting clinical outcome. Laboratory work using knockout models and large animal studies have provided insights into the mechanisms of liver IRI. Liver IRI occurs as a continuum beginning from the moment of reperfusion onwards for up to a week.
There is a complex interplay between cellular mediators, ROS, the complement system, cytokines/chemokines and other secreted factors that activate several parallel intracellular pathways that include transcription factors, nitric oxide synthase and haem oxygenase-1, all of which is beginning to be unravelled. Further laboratory work using knockout models and large animal studies of liver IRI will provide further mechanistic insights into this phenomenon and identify pharmacological agents that could be entered into clinical trials for reducing IRI.
Footnotes
P- Reviewers Jaeschke H, Mars WM S- Editor Gou SX L- Editor A E- Editor Zhang DN
References
- 1.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 3.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature. 1984;309:255–256. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- 5.Lin FL, Sperle K, Sternberg N. Recombination in mouse L cells between DNA introduced into cells and homologous chromosomal sequences. Proc Natl Acad Sci USA. 1985;82:1391–1395. doi: 10.1073/pnas.82.5.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature. 1985;317:230–234. doi: 10.1038/317230a0. [DOI] [PubMed] [Google Scholar]
- 7.Thomas KR, Folger KR, Capecchi MR. High frequency targeting of genes to specific sites in the mammalian genome. Cell. 1986;44:419–428. doi: 10.1016/0092-8674(86)90463-0. [DOI] [PubMed] [Google Scholar]
- 8.Thomas KR, Capecchi MR. Introduction of homologous DNA sequences into mammalian cells induces mutations in the cognate gene. Nature. 1986;324:34–38. doi: 10.1038/324034a0. [DOI] [PubMed] [Google Scholar]
- 9.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 10.Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol. 2006;291:H2067–H2074. doi: 10.1152/ajpheart.00272.2006. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am J Physiol. 1993;264:G801–G809. doi: 10.1152/ajpgi.1993.264.4.G801. [DOI] [PubMed] [Google Scholar]
- 12.Hanschen M, Zahler S, Krombach F, Khandoga A. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation. 2008;86:710–718. doi: 10.1097/TP.0b013e3181821aa7. [DOI] [PubMed] [Google Scholar]
- 13.Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC, Goldschmidt-Clermont PJ, Irani K. Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J. 2000;14:418–429. doi: 10.1096/fasebj.14.2.418. [DOI] [PubMed] [Google Scholar]
- 14.Theruvath TP, Czerny C, Ramshesh VK, Zhong Z, Chavin KD, Lemasters JJ. C-Jun N-terminal kinase 2 promotes graft injury via the mitochondrial permeability transition after mouse liver transplantation. Am J Transplant. 2008;8:1819–1828. doi: 10.1111/j.1600-6143.2008.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–1504. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frankenberg MV, Weimann J, Fritz S, Fiedler J, Mehrabi A, Büchler MW, Kraus TW. Gadolinium chloride-induced improvement of postischemic hepatic perfusion after warm ischemia is associated with reduced hepatic endothelin secretion. Transpl Int. 2005;18:429–436. doi: 10.1111/j.1432-2277.2004.00058.x. [DOI] [PubMed] [Google Scholar]
- 17.Uhlmann D, Gaebel G, Armann B, Ludwig S, Hess J, Pietsch UC, Fiedler M, Tannapfel A, Hauss J, Witzigmann H. Attenuation of proinflammatory gene expression and microcirculatory disturbances by endothelin A receptor blockade after orthotopic liver transplantation in pigs. Surgery. 2006;139:61–72. doi: 10.1016/j.surg.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Ari Z, Pappo O, Cheporko Y, Yasovich N, Offen D, Shainberg A, Leshem D, Sulkes J, Vidne BA, Hochhauser E. Bax ablation protects against hepatic ischemia/reperfusion injury in transgenic mice. Liver Transpl. 2007;13:1181–1188. doi: 10.1002/lt.21221. [DOI] [PubMed] [Google Scholar]
- 19.Sawaya DE, Zibari GB, Minardi A, Bilton B, Burney D, Granger DN, McDonald JC, Brown M. P-selectin contributes to the initial recruitment of rolling and adherent leukocytes in hepatic venules after ischemia/reperfusion. Shock. 1999;12:227–232. doi: 10.1097/00024382-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Singh I, Zibari GB, Brown MF, Granger DN, Eppihimer M, Zizzi H, Cruz L, Meyer K, Gonzales E, McDonald JC. Role of P-selectin expression in hepatic ischemia and reperfusion injury. Clin Transplant. 1999;13:76–82. doi: 10.1034/j.1399-0012.1999.130103.x. [DOI] [PubMed] [Google Scholar]
- 21.Young CS, Palma JM, Mosher BD, Harkema J, Naylor DF, Dean RE, Crockett E. Hepatic ischemia/reperfusion injury in P-selectin and intercellular adhesion molecule-1 double-mutant mice. Am Surg. 2001;67:737–744. [PubMed] [Google Scholar]
- 22.Khandoga A, Biberthaler P, Enders G, Axmann S, Hutter J, Messmer K, Krombach F. Platelet adhesion mediated by fibrinogen-intercelllular adhesion molecule-1 binding induces tissue injury in the postischemic liver in vivo. Transplantation. 2002;74:681–688. doi: 10.1097/00007890-200209150-00016. [DOI] [PubMed] [Google Scholar]
- 23.Hines IN, Kawachi S, Harada H, Pavlick KP, Hoffman JM, Bharwani S, Wolf RE, Grisham MB. Role of nitric oxide in liver ischemia and reperfusion injury. Mol Cell Biochem. 2002;234-235:229–237. [PubMed] [Google Scholar]
- 24.Kawachi S, Hines IN, Laroux FS, Hoffman J, Bharwani S, Gray L, Leffer D, Grisham MB. Nitric oxide synthase and postischemic liver injury. Biochem Biophys Res Commun. 2000;276:851–854. doi: 10.1006/bbrc.2000.3559. [DOI] [PubMed] [Google Scholar]
- 25.Shimamura K, Kawamura H, Nagura T, Kato T, Naito T, Kameyama H, Hatakeyama K, Abo T. Association of NKT cells and granulocytes with liver injury after reperfusion of the portal vein. Cell Immunol. 2005;234:31–38. doi: 10.1016/j.cellimm.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am J Pathol. 2009;174:2265–2277. doi: 10.2353/ajpath.2009.080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Edwards MJ, Lentsch AB. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology. 2008;48:1213–1223. doi: 10.1002/hep.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caldwell CC, Okaya T, Martignoni A, Husted T, Schuster R, Lentsch AB. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2005;289:G969–G976. doi: 10.1152/ajpgi.00223.2005. [DOI] [PubMed] [Google Scholar]
- 29.Kuboki S, Sakai N, Tschöp J, Edwards MJ, Lentsch AB, Caldwell CC. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1054–G1059. doi: 10.1152/ajpgi.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203:2639–2648. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y, Rabb H, Womer KL. Ischemia-reperfusion and immediate T cell responses. Cell Immunol. 2007;248:4–11. doi: 10.1016/j.cellimm.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen XD, Ke B, Zhai Y, Gao F, Tsuchihashi S, Lassman CR, Busuttil RW, Kupiec-Weglinski JW. Absence of toll-like receptor 4 (TLR4) signaling in the donor organ reduces ischemia and reperfusion injury in a murine liver transplantation model. Liver Transpl. 2007;13:1435–1443. doi: 10.1002/lt.21251. [DOI] [PubMed] [Google Scholar]
- 33.Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, Busuttil RW, Kupiec-Weglinski JW. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 34.Camargo CA, Madden JF, Gao W, Selvan RS, Clavien PA. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology. 1997;26:1513–1520. doi: 10.1002/hep.510260619. [DOI] [PubMed] [Google Scholar]
- 35.Okaya T, Lentsch AB. Peroxisome proliferator-activated receptor-alpha regulates postischemic liver injury. Am J Physiol Gastrointest Liver Physiol. 2004;286:G606–G612. doi: 10.1152/ajpgi.00191.2003. [DOI] [PubMed] [Google Scholar]
- 36.Teoh N, Field J, Sutton J, Farrell G. Dual role of tumor necrosis factor-alpha in hepatic ischemia-reperfusion injury: studies in tumor necrosis factor-alpha gene knockout mice. Hepatology. 2004;39:412–421. doi: 10.1002/hep.20035. [DOI] [PubMed] [Google Scholar]
- 37.Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, Chan MH, Jeyabalan G, Yim JH, Geller DA. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1261–G1268. doi: 10.1152/ajpgi.00460.2005. [DOI] [PubMed] [Google Scholar]
- 38.Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–1800. doi: 10.1111/j.1600-6143.2005.00932.x. [DOI] [PubMed] [Google Scholar]
- 39.Rüdiger HA, Clavien PA. Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology. 2002;122:202–210. doi: 10.1053/gast.2002.30304. [DOI] [PubMed] [Google Scholar]
- 40.Tian Y, Jochum W, Georgiev P, Moritz W, Graf R, Clavien PA. Kupffer cell-dependent TNF-alpha signaling mediates injury in the arterialized small-for-size liver transplantation in the mouse. Proc Natl Acad Sci USA. 2006;103:4598–4603. doi: 10.1073/pnas.0600499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, Kupiec-Weglinski JW. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 42.Hamada T, Fondevila C, Busuttil RW, Coito AJ. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47:186–198. doi: 10.1002/hep.21922. [DOI] [PubMed] [Google Scholar]
- 43.Tsuchihashi S, Livhits M, Zhai Y, Busuttil RW, Araujo JA, Kupiec-Weglinski JW. Basal rather than induced heme oxygenase-1 levels are crucial in the antioxidant cytoprotection. J Immunol. 2006;177:4749–4757. doi: 10.4049/jimmunol.177.7.4749. [DOI] [PubMed] [Google Scholar]
- 44.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, Luster AD, Busuttil RW, Kupiec-Weglinski JW. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 45.Diepenhorst GM, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg. 2009;249:889–899. doi: 10.1097/SLA.0b013e3181a38f45. [DOI] [PubMed] [Google Scholar]
- 46.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Austen WG, Kyriakides C, Favuzza J, Wang Y, Kobzik L, Moore FD, Hechtman HB. Intestinal ischemia-reperfusion injury is mediated by the membrane attack complex. Surgery. 1999;126:343–348. [PubMed] [Google Scholar]
- 48.Stahl GL, Xu Y, Hao L, Miller M, Buras JA, Fung M, Zhao H. Role for the alternative complement pathway in ischemia/reperfusion injury. Am J Pathol. 2003;162:449–455. doi: 10.1016/S0002-9440(10)63839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams JP, Pechet TT, Weiser MR, Reid R, Kobzik L, Moore FD, Carroll MC, Hechtman HB. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol. 1999;86:938–942. doi: 10.1152/jappl.1999.86.3.938. [DOI] [PubMed] [Google Scholar]
- 50.Xu DZ, Zaets SB, Chen R, Lu Q, Rajan H, Yang X, Zhang J, Feketova E, Bogdan N, Deitch EA, et al. Elimination of C5aR prevents intestinal mucosal damage and attenuates neutrophil infiltration in local and remote organs. Shock. 2009;31:493–499. doi: 10.1097/SHK.0b013e318188b3cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 52.Møller-Kristensen M, Wang W, Ruseva M, Thiel S, Nielsen S, Takahashi K, Shi L, Ezekowitz A, Jensenius JC, Gadjeva M. Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol. 2005;61:426–434. doi: 10.1111/j.1365-3083.2005.01591.x. [DOI] [PubMed] [Google Scholar]
- 53.Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest. 2000;105:1363–1371. doi: 10.1172/JCI8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh MC, Bourcier T, Takahashi K, Shi L, Busche MN, Rother RP, Solomon SD, Ezekowitz RA, Stahl GL. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol. 2005;175:541–546. doi: 10.4049/jimmunol.175.1.541. [DOI] [PubMed] [Google Scholar]
- 55.Chung KY, Park JJ, Han KH. Pig to canine auxiliary hepatic xenotransplantation model: prevention of hyperacute rejection via Kupffer cell blockade and complement regulation. Transplant Proc. 2008;40:2755–2759. doi: 10.1016/j.transproceed.2008.08.056. [DOI] [PubMed] [Google Scholar]
- 56.Fondevila C, Shen XD, Tsuchihashi S, Uchida Y, Freitas MC, Ke B, Busuttil RW, Kupiec-Weglinski JW. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury. Liver Transpl. 2008;14:1133–1141. doi: 10.1002/lt.21496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inderbitzin D, Beldi G, Avital I, Vinci G, Candinas D. Local and remote ischemia-reperfusion injury is mitigated in mice overexpressing human C1 inhibitor. Eur Surg Res. 2004;36:142–147. doi: 10.1159/000077255. [DOI] [PubMed] [Google Scholar]
- 58.Lee VG, Johnson ML, Baust J, Laubach VE, Watkins SC, Billiar TR. The roles of iNOS in liver ischemia-reperfusion injury. Shock. 2001;16:355–360. doi: 10.1097/00024382-200116050-00006. [DOI] [PubMed] [Google Scholar]
- 59.Theruvath TP, Zhong Z, Currin RT, Ramshesh VK, Lemasters JJ. Endothelial nitric oxide synthase protects transplanted mouse livers against storage/reperfusion injury: Role of vasodilatory and innate immunity pathways. Transplant Proc. 2006;38:3351–3357. doi: 10.1016/j.transproceed.2006.10.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hines IN, Harada H, Bharwani S, Pavlick KP, Hoffman JM, Grisham MB. Enhanced post-ischemic liver injury in iNOS-deficient mice: a cautionary note. Biochem Biophys Res Commun. 2001;284:972–976. doi: 10.1006/bbrc.2001.5069. [DOI] [PubMed] [Google Scholar]
- 61.Khandoga A, Enders G, Biberthaler P, Krombach F. Poly(ADP-ribose) polymerase triggers the microvascular mechanisms of hepatic ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2002;283:G553–G560. doi: 10.1152/ajpgi.00085.2002. [DOI] [PubMed] [Google Scholar]
- 62.Kimura H, Katsuramaki T, Isobe M, Nagayama M, Meguro M, Kukita K, Nui A, Hirata K. Role of inducible nitric oxide synthase in pig liver transplantation. J Surg Res. 2003;111:28–37. doi: 10.1016/s0022-4804(03)00036-2. [DOI] [PubMed] [Google Scholar]
- 63.Meguro M, Katsuramaki T, Nagayama M, Kimura H, Isobe M, Kimura Y, Matsuno T, Nui A, Hirata K. A novel inhibitor of inducible nitric oxide synthase (ONO-1714) prevents critical warm ischemia-reperfusion injury in the pig liver. Transplantation. 2002;73:1439–1446. doi: 10.1097/00007890-200205150-00013. [DOI] [PubMed] [Google Scholar]
- 64.Meguro M, Katsuramaki T, Kimura H, Isobe M, Nagayama M, Kukita K, Nui A, Hirata K. Apoptosis and necrosis after warm ischemia-reperfusion injury of the pig liver and their inhibition by ONO-1714. Transplantation. 2003;75:703–710. doi: 10.1097/01.TP.0000053400.42842.5C. [DOI] [PubMed] [Google Scholar]
- 65.Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 66.Lv X, Yang L, Tao K, Liu Y, Yang T, Chen G, Yu W, Lv H, Wu F. Isoflurane preconditioning at clinically relevant doses induce protective effects of heme oxygenase-1 on hepatic ischemia reperfusion in rats. BMC Gastroenterol. 2011;11:31. doi: 10.1186/1471-230X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katori M, Anselmo DM, Busuttil RW, Kupiec-Weglinski JW. A novel strategy against ischemia and reperfusion injury: cytoprotection with heme oxygenase system. Transpl Immunol. 2002;9:227–233. doi: 10.1016/s0966-3274(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 68.Su H, van Dam GM, Buis CI, Visser DS, Hesselink JW, Schuurs TA, Leuvenink HG, Contag CH, Porte RJ. Spatiotemporal expression of heme oxygenase-1 detected by in vivo bioluminescence after hepatic ischemia in HO-1/Luc mice. Liver Transpl. 2006;12:1634–1639. doi: 10.1002/lt.20852. [DOI] [PubMed] [Google Scholar]
- 69.Wyllie S, Seu P, Gao FQ, Gros P, Goss JA. Disruption of the Nramp1 (also known as Slc11a1) gene in Kupffer cells attenuates early-phase, warm ischemia-reperfusion injury in the mouse liver. J Leukoc Biol. 2002;72:885–897. [PubMed] [Google Scholar]
- 70.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 71.Stephanou A. Role of STAT-1 and STAT-3 in ischaemia/reperfusion injury. J Cell Mol Med. 2004;8:519–525. doi: 10.1111/j.1582-4934.2004.tb00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato A, Yoshidome H, Edwards MJ, Lentsch AB. Reduced hepatic ischemia/reperfusion injury by IL-4: potential anti-inflammatory role of STAT6. Inflamm Res. 2000;49:275–279. doi: 10.1007/PL00000207. [DOI] [PubMed] [Google Scholar]
- 73.Kuboki S, Schuster R, Blanchard J, Pritts TA, Wong HR, Lentsch AB. Role of heat shock protein 70 in hepatic ischemia-reperfusion injury in mice. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1141–G1149. doi: 10.1152/ajpgi.00491.2006. [DOI] [PubMed] [Google Scholar]
- 74.Beraza N, Lüdde T, Assmus U, Roskams T, Vander Borght S, Trautwein C. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132:2504–2517. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 75.Ban K, Cooper AJ, Samuel S, Bhatti A, Patel M, Izumo S, Penninger JM, Backx PH, Oudit GY, Tsushima RG. Phosphatidylinositol 3-kinase gamma is a critical mediator of myocardial ischemic and adenosine-mediated preconditioning. Circ Res. 2008;103:643–653. doi: 10.1161/CIRCRESAHA.108.175018. [DOI] [PubMed] [Google Scholar]
- 76.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, Bolli R. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation. 2005;112:1971–1978. doi: 10.1161/CIRCULATIONAHA.105.561522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cursio R, Mari B, Louis K, Rostagno P, Saint-Paul MC, Giudicelli J, Bottero V, Anglard P, Yiotakis A, Dive V, et al. Rat liver injury after normothermic ischemia is prevented by a phosphinic matrix metalloproteinase inhibitor. FASEB J. 2002;16:93–95. doi: 10.1096/fj.01-0279fje. [DOI] [PubMed] [Google Scholar]
- 78.Ricciardi R, Meyers WC, Schaffer BK, Kim RD, Shah SA, Wheeler SM, Donohue SE, Sheth KR, Callery MP, Chari RS. Protein kinase C inhibition abrogates hepatic ischemic preconditioning responses. J Surg Res. 2001;97:144–149. doi: 10.1006/jsre.2001.6139. [DOI] [PubMed] [Google Scholar]
- 79.Yanagida H, Kaibori M, Yamada M, Habara K, Yokoigawa N, Kwon AH, Kamiyama Y, Okumura T. Induction of inducible nitric oxide synthase in hepatocytes isolated from rats with ischemia-reperfusion injury. Transplant Proc. 2004;36:1962–1964. doi: 10.1016/j.transproceed.2004.08.055. [DOI] [PubMed] [Google Scholar]
- 80.Shen XD, Ke B, Zhai Y, Gao F, Anselmo D, Lassman CR, Busuttil RW, Kupiec-Weglinski JW. Stat4 and Stat6 signaling in hepatic ischemia/reperfusion injury in mice: HO-1 dependence of Stat4 disruption-mediated cytoprotection. Hepatology. 2003;37:296–303. doi: 10.1053/jhep.2003.50066. [DOI] [PubMed] [Google Scholar]
- 81.Lagoa CE, Vodovotz Y, Stolz DB, Lhuillier F, McCloskey C, Gallo D, Yang R, Ustinova E, Fink MP, Billiar TR, et al. The role of hepatic type 1 plasminogen activator inhibitor (PAI-1) during murine hemorrhagic shock. Hepatology. 2005;42:390–399. doi: 10.1002/hep.20797. [DOI] [PubMed] [Google Scholar]
- 82.Kato A, Edwards MJ, Lentsch AB. Gene deletion of NF-kappa B p50 does not alter the hepatic inflammatory response to ischemia/reperfusion. J Hepatol. 2002;37:48–55. doi: 10.1016/s0168-8278(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 83.Kato A, Graul-Layman A, Edwards MJ, Lentsch AB. Promotion of hepatic ischemia/reperfusion injury by IL-12 is independent of STAT4. Transplantation. 2002;73:1142–1145. doi: 10.1097/00007890-200204150-00023. [DOI] [PubMed] [Google Scholar]