

Abstract
Although single-photon fluorescence lifetime imaging microscopy (FLIM) is widely used to image molecular processes using a wide range of excitation wavelengths, the captured emission of this technique is confined to the visible spectrum. Here, we explore the feasibility of utilizing near-infrared (NIR) fluorescent molecular probes with emission >700 nm for FLIM of live cells. The confocal microscope is equipped with a 785 nm laser diode, a red-enhanced photomultiplier tube, and a time-correlated single photon counting card. We demonstrate that our system reports the lifetime distributions of NIR fluorescent dyes, cypate and DTTCI, in cells. In cells labeled separately or jointly with these dyes, NIR FLIM successfully distinguishes their lifetimes, providing a method to sort different cell populations. In addition, lifetime distributions of cells co-incubated with these dyes allow estimate of the dyes’ relative concentrations in complex cellular microenvironments. With the heightened interest in fluorescence lifetime-based small animal imaging using NIR fluorophores, this technique further serves as a bridge between in vitro spectroscopic characterization of new fluorophore lifetimes and in vivo tissue imaging.
Keywords: Fluorescence lifetime imaging microscopy, live cell imaging, near infrared fluorescent molecular probes
I. Introduction
Fluorescence lifetime imaging microscopy (FLIM) is a promising technique for imaging molecular processes. It uses time-resolved measurements of fluorescence from cells and thin tissue sections to generate images of the characteristic fluorescence lifetimes (FLTs) within a pixel or voxel. The FLT is the average time a molecule resides in the excited state before returning to the ground state through fluorescence emission (Berezin and Achilefu 2010). Typically, FLIM is performed in the frequency or time domain. In the frequency domain, a sinusoidal modulated (0.1–1 GHz) light source illuminates the sample, and FLTs are measured by detecting and analyzing the amplitude and phase shift between the excitation light and the fluorescence emission (Gadella et al. 1993). In the time domain, pulsed light illuminates the sample, and the time-course of fluorescence emission is detected and analyzed for FLTs. Imaging systems use either time-gated wide field image intensifiers (Elson et al. 2002) or time-resolved laser scanning point detection (Morgan et al. 1995). Applications include imaging molecular signaling (Webb et al. 2008) and trafficking (Verveer et al. 2000), imaging spatial concentration of intracellular ions (Lahn et al. 2011), assessing intracellular environment (Kneen et al. 1998), characterizing tissue slices in vivo (Ushakov et al. 2011), and determining molecular interactions using fluorescence resonance energy transfer (Keese et al. 2010).
Conventional FLIM systems detect fluorescence at visible wavelengths. However, operation at these wavelengths limits the imaging performance due to high auto-fluorescence and light scattering (Yazdanfar et al. 2010). These issues become severe for low target concentrations, necessitating the use of strategies that reduce auto-fluorescence and improve the signal-to-noise ratio (SNR). Previous studies have partially addressed these issues by using two-photon (2P) excitations at near-infrared (NIR) wavelengths (Ushakov et al. 2011), as molecular imaging in the NIR region (>700 nm) minimizes auto-fluorescence and increases penetration depth. However, these 2P studies suffer from fluorescence detection in the visible region. Moreover, recent interests in using FLT techniques to image small animals and thick tissues have focused on the FLT properties of NIR fluorescent dyes (Bloch et al. 2005; Nothdurft et al. 2009; Raymond et al. 2010). Unfortunately, cellular level microscopic imaging of these in vivo NIR FLT measurements has not been reported.
To address these problems, we have built an NIR FLIM system consisting of a fiber-coupled laser diode with nominal 785 nm excitation; a confocal laser scanning microscope; a photomultiplier tube (PMT) (PMT Handbook, 3rd Ed., Hamamatsu Photonics, Japan) with sensitivity at NIR wavelengths; and a time-correlated single photon counting (TCSPC) card (TCSPC Handbook, 4th Ed., Becker-Hickl). We demonstrated the use of this system to determine the intracellular heterogeneous distribution of NIR fluorescent dyes, cypate (Achilefu et al. 2000) and 3,3-diethylthiatricarbocyanine iodine (DTTCI) (Sigma-Aldrich, St. Louis, MO) (Berezin et al. 2007). The approach offers a new analytical tool to characterize the trafficking and sequestration of NIR fluorescent probes in cells.
II. Materials and Methods
A. Near-infrared fluorescence lifetime microscopy system
We built an NIR FLIM system consisting of a fiber-coupled laser diode (BDL-785-SMC, Becker-Hickl, Gremany), a confocal laser scanning fluorescence microscope (FV1000, Olympus, Center Valley, PA), a thermoelectrically cooled, red-enhanced PMT (PMC-100-20, Becker-Hickl, Germany), and a TCSPC card (SPC-730, Becker-Hickl, Germany). The laser diode operates at TEM00 mode (Cheng 1989) and provides ~785 nm excitation light. Its pulsed wave (PW) duration is nominally 60–80 ps with frequency of 50 MHz. It also provides continuous wave (CW) illumination for single photon confocal microscopy. The PMT offers a minimum photon-count rate of 5 MHz. The TCSPC card has a transit-time spread of 180 ps and a dead time of 125 ns. The single photon counting operation employs equally spaced 256 time gates of duration 16.6 ns, with an initial delay of 1.4 ns for eliminating photons from the excitation pulse in the measurement.
To obtain FLIM images, the laser light was collimated, passed through the confocal system, and focused onto the sample using a 20×, 0.95-NA objective. Single photon fluorescence from the sample was collected through the same objective and directed by a dichroic mirror toward the confocal pinhole and detected by the PMT. Residual excitation light was removed using a bandstop filter with cutoff wavelengths of 765 nm and 805 nm before the PMT. Data acquisition was performed using the TCSPC card, triggered via a synchronization signal generated by the laser driver for each laser pulse. Images were acquired by unidirectional scanning with the excitation beam using a galvanometric mirror pair as embedded in the confocal microscope system.
B. Near-infrared contrast agents
The FLIM system was used to determine the heterogeneous cellular distribution of NIR dyes, cypate and DTTCI with absorption/emission peaks of 792/810 nm and 771/800 nm, respectively. Their absorption maxima overlap with the excitation spectrum of the laser used in our system. Cypate and DTTCI are heptamethine cyanine fluorescence dyes. They are selected because of their biocompatibility and their ability to absorb and emit light in the NIR wavelength window.
C. Cell culture
Live mouse leukaemic monocyte macrophage (Raw264.7, Tissue Culture Support Center, Washington University School of Medicine, St. Louis, MO) cells treated for 4 hours with 5 µM cypate and/or 2 µM DTTCI were imaged at room temperature. The different dye concentrations for treating cells were computed to normalize the relative brightness (molar extinction coefficient multiplied by fluorescence quantum yield) of these dye molecules in the cellular microenvironment using our previous spectroscopic study (Berezin et al. 2007). The cells were cultured in Dulbecco’s modified eagle medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin 100× (Invitrogen, Grand Island, NY). They were maintained before imaging at 37°C in a humidified atmosphere of 5% CO2.
D. Imaging
We performed NIR FLIM in four set of cells treated with (1) cypate alone, (2) DTTCI alone, (3) both cypate and DTTCI, and (4) a mixture of cells separately incubated either with cypate or with DTTCI. The FLIM was performed using 0.921 mW of PW laser power, with a pixel integration time of 7.3 ms.
E. Spectroscopic fluorescence lifetime measurement
Spectroscopic FLTs of mixtures of cypate and DTTCI diluted in cell culture media were measured using a TCSPC technique (Horiba, Japan) with excitation source NanoLed at 773 nm (Horiba, Japan) and impulse repetition rate of 1 MHz at 90° to a R928P detector (Hamamatsu Photonics, Japan), as described previously (Berezin et al. 2007). The detector was set to 820 nm with a 20 nm band pass filter. The electrical signal was amplified by a TB-02 pulse amplifier (Horiba, Japan), and the amplified signal was fed to the constant fraction discriminator (Philips, The Netherlands). The first detected photon represented the start signal by the time-to-amplitude converter (TAC), and the excitation pulse triggered the stop signal. The TAC measures the time of a detected photon from the pulse at the detector input to the next pulse at the reference input. The multichannel analyzer recorded repetitive start-stop signals from the TAC and generated a histogram of photons as a function of time-calibrated channels (6.88 ps/channel) until the peak signal reached 10,000 counts. The lifetime was recorded on a 50 ns scale. The instrument response function was obtained by using Rayleigh scatter of Ludox-40 (Sigma-Aldrich, St. Louis, MO) (0.03% in Millipore quality water) in a quartz cuvette at 773 nm emission. DAS6 v6.1 decay analysis software (Horiba, Japan) was used for lifetime calculations. The goodness of fit was judged by Chi-r-squared () values and Durbin-Watson parameters and visual observations of fitted line, residuals, and autocorrelation function.
F. Image analysis
Collected time traces captured from the NIR FLIM system were analyzed in the SPCImage software (Becker-Hickl, Germany). It replaces time traces per pixel with the cumulative time traces computed using the target pixel and its neighboring eight pixels to increase the SNR. Maximum SNR is defined by the square root of the number of photons acquired in the peak channel of the 256 time gates used to perform TCSPC acquisition (Kollner and Wolfrum 1992). The earliest time points allow fitting of the instrument response function, while a single-exponential curve was sufficient to fit the subsequent falloff with time. Pixels with total counts <8200 photons in the approximated time traces were considered to be noise and discarded (see Section III.A for more explanation). A fitness test determined the validity of the fit, providing values <1.5 for all pixels. To visualize the data, the collected photon-count image and the corresponding FLIM image of sizes 128×128 were interpolated using the SPCImage software to produce final set of 512×512 images.
For other computations, FLIM images of original sizes 128×128 were exported to MATLAB (Mathworks, Natick, MA). Here noise from the data was discarded as stated above. Empirical distribution of the corresponding non-zero FLTs in cells was derived for the respective experiment described in Section II.D.
To integrate spectroscopic FLT measurements of the mixtures of cypate and DTTCI with the FLIM image of cells co-incubated with both cypate and DTTCI, a simple MATLAB code was implemented. This code first generates a calibration curve using smoothing spline based interpolation to fit the relationship between the spectroscopic FLT measurements of the mixtures of cypate and DTTCI and their molar ratios. Then it transfers the FLIM image of the cells co-incubated with both of these dyes to a ratiometric image using the calibration curve, exhibiting an estimate of the molar ratio between cypate and DTTCI at each pixel in the ratiometric image. This code also computes the distribution of these ratios using the non-zero components of this image.
III. Results and Discussion
A. Near-infrared fluorescence lifetime microscopy system
Fig. 1 summarizes the instrument design. Positive material dispersion of optical components in the fiber leads to pulse broadening and reduces excitation efficiency at the sample. These losses can be recovered if the pulses arriving at the sample are transform-limited by pre-compensating for material dispersion in the system (Kim et al. 2008). In the present scheme, the length of the fiber (~0.5 m) is optimally designed to pre-compensate any material dispersion. Moreover, single TEM00 mode operation of the laser provides an additional pre-compensation to such dispersion effect.
Figure 1.
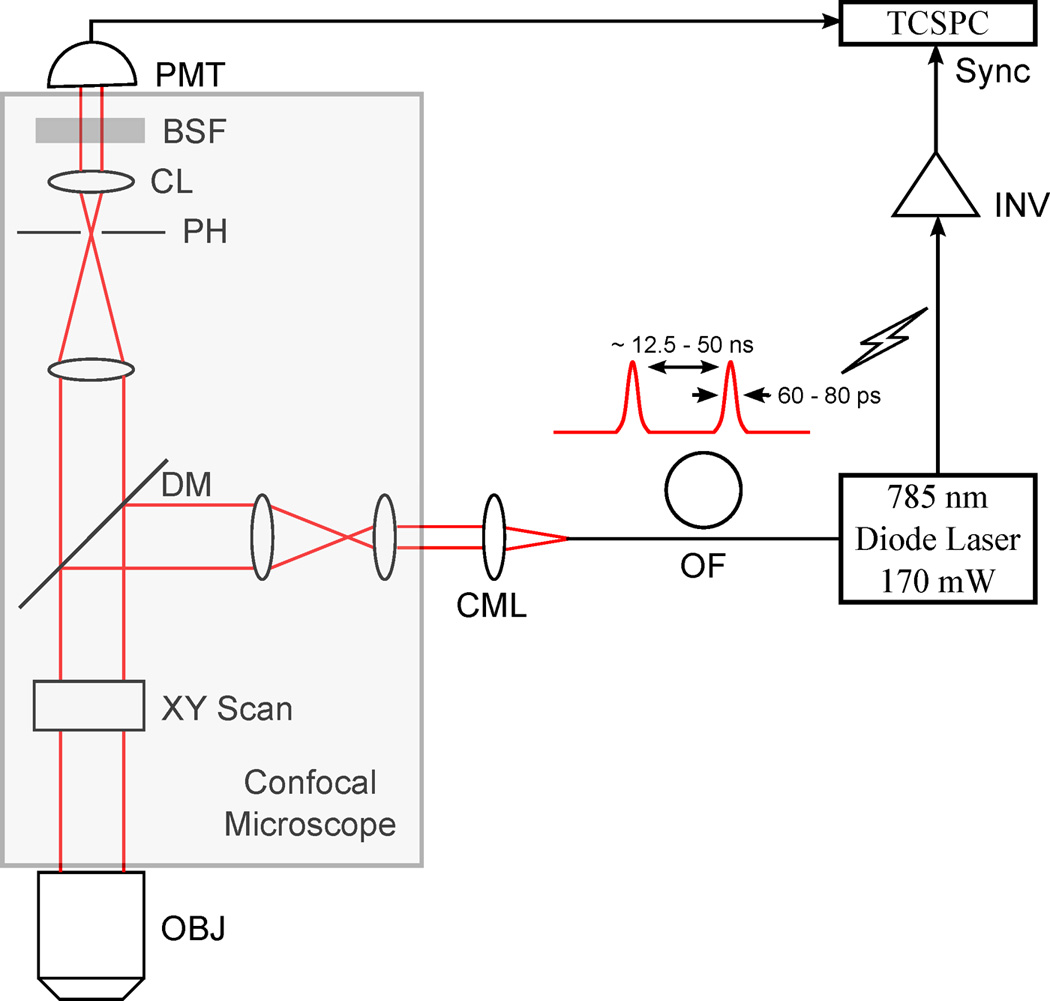
Schematic of the FLIM system. OF, optical fiber; CML, collimator lens; OBJ, objective; DM, dichroic mirror; PH, pinhole; CL, collection lens; BSF, bandstop filter; and INV, inverter.
The minimum count rate for the developed system is computed to be 8200 photons/pixel (Gerritsen et al. 2002). This count rate efficiently estimates lifetimes of organic NIR dyes, whose FLTs typically vary from 500 ps to 2 ns. This rate requires a minimum collection time of 27 sec for capturing a 128×128 image.
B. Near-infrared fluorescence lifetime microscopy imaging
To demonstrate the versatility of the FLIM, we compared FLT distributions in four set of cells treated with (1) cypate alone, (2) DTTCI alone, (3) both cypate and DTTCI, and (4) a mixture of cells separately incubated either with cypate or with DTTCI. Figs. 2(A-D) show the photon count images (top row), the FLIM images (middle row), and the FLT distributions (bottom row) for subsets of cells from these four experiments. During the FLIM, a total of ~107 photons were captured in each image. Analysis of the time traces for all pixels with sufficient signal (Nothdurft et al. 2009) (>8200 photon counts) were used to generate the FLT images. The FLIM images of cells with cypate alone (Fig. 2A) or DTTCI alone (Fig. 2B) show a remarkable difference. Although the differential localization of cypate and DTTCI is not possible from the intensity images, the FLIM images decipher cells treated with each dye, with cells exhibiting higher or lower lifetime regions predominantly possessing DTTCI or Cypate, respectively. Expectedly, when cells separately incubated with either cypate or DTTCI were mixed, the FLT distribution was distinctly bimodal, representing the individual cells labeled with each dye (Fig. 2C). However, for the cells co-incubated with both cypate and DTTCI, while the FLT distribution still appears bi-modal, the peaks are shifted towards the center from the original peak positions of the separate cell images (Fig. 2D). This is likely due to interaction between the dyes in being co-localized within the same cells.
Figure 2.
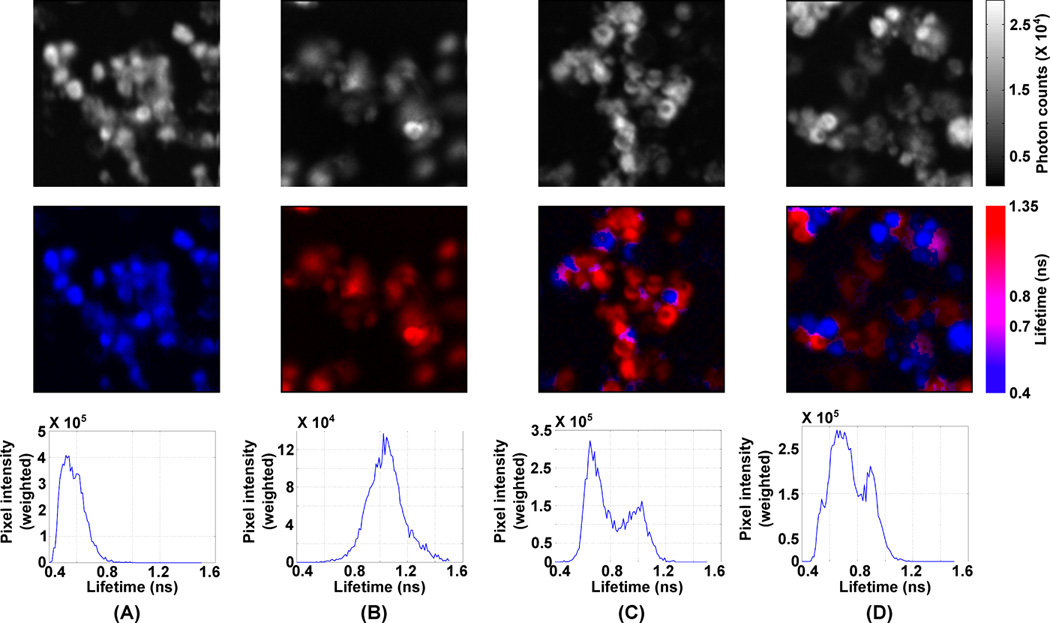
(A-D) Intensity images (top row), FLIM images (middle row), and FLT distributions (bottom row) of cells treated with cypate alone (A), with DTTCI alone (B), either with cypate or DTTCI (C), and with both cypate and DTTCI (D).
C. Integrating spectroscopic and microscopic fluorescence lifetime data
To estimate the relative concentrations of cypate and DTTCI in cells co-incubated with both of these dyes, we used information from spectroscopic FLT measurements of their mixtures diluted in culture media using TCSPC technique. The concentration of cypate was varied from 2 µM to 10 µM at a fixed concentration of DTTCI (2 µM). For consistency with our cell studies, we used single-exponential fitting to estimate lifetimes from the fluorometer measurements. The measured lifetime vs. the ratio between the concentrations of these dyes was used to generate a calibration curve (Fig. 3A). This curve was used to estimate the relative concentrations of cypate and DTTCI in the cells. The resulting image of the estimated concentration ratios between DTTCI and cypate in the cells and the estimated distribution of these ratios are shown in Figs. 3B and 3C, respectively. Interestingly, the ratio distribution plot suggests two distinct regions exhibiting high concentration of either cypate or DTTCI. Note that the separation between the peaks is greater in the distribution of concentration ratios (Fig. 3C) than in the lifetimes (Fig 2D, bottom row).
Figure 3.
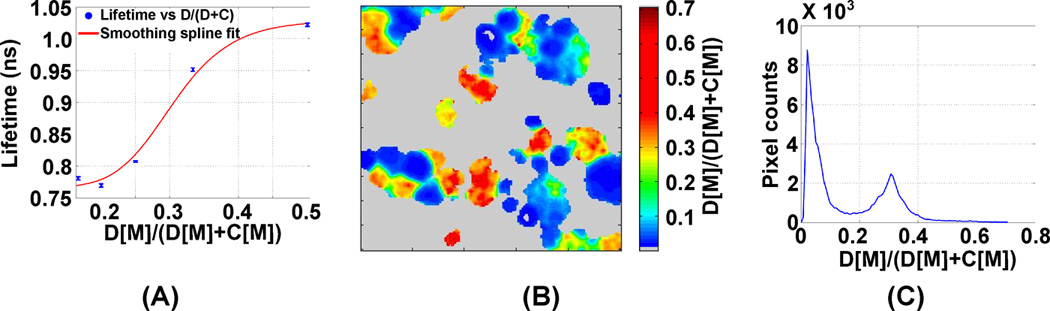
The plot between measured lifetime and concentration ratios of DTTCI and cypate, and a smoothing spline fit to this plot (A), estimated ratios between cypate and DTTCI concentrations in cellular microenvironment (B), and the distribution of these ratios (C).
IV. Conclusion
Previous studies have focused on in vitro spectroscopic characterizations or the tissue mapping of FLT properties of NIR fluorescent probes, but did not address their cellular FLT profiles. Our study bridges the gap between in vitro and in vivo FLT properties of NIR dyes for FLT imaging of probes with emission above 700 nm. The FLT data obtained using our system can complement the fluorescence intensity information obtained in both the visible and the NIR wavelengths, facilitating the co-registration of these images to understand complex cellular pathways. The NIR wavelengths of this system have reduced auto-fluorescence, as demonstrated in a recent report (Yazdanfar et al. 2010).
The major limitation of the current system is the need to capture the images at low resolution because most of the current organic NIR dyes are not highly photostable. Hence, capturing high resolution images would increase the image integration time, which would in turn result in photobleaching of the associated dyes. Note that lower resolution images hinder us from observing cellular details using FLT data. In future, we will therefore focus on improving the image acquisition speed of the current NIR FLIM instrument, which will allow us to capture higher resolution images and thus, improve our capacity to associate the heterogeneous intracellular lifetime map of NIR dyes with subcellular features.
Acknowledgements
This study is based upon work supported by the NIH grant R01 EB008111, and in part by the following NIH grants: R01 EB007276, R01 EB008111, R33 CA123537, U54 CA136398 – Network for Translational Research, and HHSN268201000046C – NHLBI Program of Excellence in Nanotechnology. We thank Mrs. Kexian Liang for preparing cypate.
References
- Achilefu S, Dorshow RB, et al. Novel receptor-targeted fluorescent contrast agents for in vivo tumor imaging. Investigative radiology. 2000;35(8):479–485. doi: 10.1097/00004424-200008000-00004. [DOI] [PubMed] [Google Scholar]
- Berezin MY, Achilefu S. Fluorescence lifetime measurements and biological imaging. Chemical reviews. 2010;110(5):2641–2684. doi: 10.1021/cr900343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezin MY, Lee H, et al. Near infrared dyes as lifetime solvatochromic probes for micropolarity measurements of biological systems. Biophysical journal. 2007;93(8):2892–2899. doi: 10.1529/biophysj.107.111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch S, Lesage F, et al. Whole-body fluorescence lifetime imaging of a tumor-targeted near-infrared molecular probe in mice. Journal of biomedical optics. 2005;10(5):054003. doi: 10.1117/1.2070148. [DOI] [PubMed] [Google Scholar]
- Cheng DK. Field and Wave Electromagnetics. Boston: Addison-Wesley; 1989. [Google Scholar]
- Elson DS, Siegel J, et al. Wide-field fluorescence lifetime imaging with optical sectioning and spectral resolution applied to biological samples. Journal of Modern Optics. 2002;49(5–6):985–995. [Google Scholar]
- Gadella TWJ, Jovin TM, et al. Fluorescence Lifetime Imaging Microscopy (Flim) - Spatial-Resolution of Microstructures on the Nanosecond Time-Scale. Biophysical Chemistry. 1993;48(2):221–239. [Google Scholar]
- Gerritsen HC, Asselbergs MA, et al. Fluorescence lifetime imaging in scanning microscopes: acquisition speed, photon economy and lifetime resolution. Journal of microscopy. 2002;206(Pt 3):218–224. doi: 10.1046/j.1365-2818.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Keese M, Yagublu V, et al. Fluorescence lifetime imaging microscopy of chemotherapy-induced apoptosis resistance in a syngenic mouse tumor model. International journal of cancer. Journal international du cancer. 2010;126(1):104–113. doi: 10.1002/ijc.24730. [DOI] [PubMed] [Google Scholar]
- Kim D, Choi H, et al. Ultrafast optical pulse delivery with fibers for nonlinear microscopy. Microscopy research and technique. 2008;71(12):887–896. doi: 10.1002/jemt.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneen M, Farinas J, et al. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophysical journal. 1998;74(3):1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollner M, Wolfrum J. How Many Photons Are Necessary for Fluorescence-Lifetime Measurements. Chemical Physics Letters. 1992;200(1–2):199–204. [Google Scholar]
- Lahn M, Dosche C, et al. Two-photon microscopy and fluorescence lifetime imaging reveal stimulus-induced intracellular Na+ and Cl- changes in cockroach salivary acinar cells. American journal of physiology. Cell physiology. 2011;300(6):C1323–C1336. doi: 10.1152/ajpcell.00320.2010. [DOI] [PubMed] [Google Scholar]
- Morgan CG, Murray JG, et al. Photon-Correlation System for Fluorescence Lifetime Measurements. Review of Scientific Instruments. 1995;66:3744–3749. [Google Scholar]
- Nothdurft RE, Patwardhan SV, et al. In vivo fluorescence lifetime tomography. Journal of biomedical optics. 2009;14(2):024004. doi: 10.1117/1.3086607. [DOI] [PubMed] [Google Scholar]
- Raymond SB, Boas DA, et al. Lifetime-based tomographic multiplexing. Journal of biomedical optics. 2010;15(4):046011. doi: 10.1117/1.3469797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushakov DS, Caorsi V, et al. Response of rigor cross-bridges to stretch detected by fluorescence lifetime imaging microscopy of myosin essential light chain in skeletal muscle fibers. The Journal of biological chemistry. 2011;286(1):842–850. doi: 10.1074/jbc.M110.149526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verveer PJ, Wouters FS, et al. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 2000;290(5496):1567–1570. doi: 10.1126/science.290.5496.1567. [DOI] [PubMed] [Google Scholar]
- Webb SED, Roberts SK, et al. Single-Molecule Imaging and Fluorescence Lifetime Imaging Microscopy Show Different Structures for High- and Low-Affinity Epidermal Growth Factor Receptors in A431 Cells. Biophysical journal. 2008;94(3):803–819. doi: 10.1529/biophysj.107.112623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanfar S, Joo C, et al. Multiphoton microscopy with near infrared contrast agents. Journal of biomedical optics. 2010;15(3):030505. doi: 10.1117/1.3420209. [DOI] [PMC free article] [PubMed] [Google Scholar]