

Abstract
The myogenic response of resistance arteries to intravascular pressure elevation is a fundamental physiological mechanism of crucial importance for blood pressure regulation and organ-specific control of blood flow. The importance of Ca2+ entry via voltage-gated Ca2+ channels leading to phosphorylation of the 20 kDa myosin regulatory light chains (LC20) in the myogenic response is well established. Recent studies, however, have suggested a role for Ca2+ sensitization via activation of the RhoA/Rho-associated kinase (ROK) pathway in the myogenic response. The possibility that enhanced actin polymerization is also involved in myogenic vasoconstriction has been suggested. Here, we have used pressurized resistance arteries from rat gracilis and cremaster skeletal muscles to assess the contribution to myogenic constriction of Ca2+ sensitization due to: (1) phosphorylation of the myosin targeting subunit of myosin light chain phosphatase (MYPT1) by ROK; (2) phosphorylation of the 17 kDa protein kinase C (PKC)-potentiated protein phosphatase 1 inhibitor protein (CPI-17) by PKC; and (3) dynamic reorganization of the actin cytoskeleton evoked by ROK and PKC. Arterial diameter, MYPT1, CPI-17 and LC20 phosphorylation, and G-actin content were determined at varied intraluminal pressures ± H1152, GF109203X or latrunculin B to suppress ROK, PKC and actin polymerization, respectively. The myogenic response was associated with an increase in MYPT1 and LC20 phosphorylation that was blocked by H1152. No change in phospho-CPI-17 content was detected although the PKC inhibitor, GF109203X, suppressed myogenic constriction. Basal LC20 phosphorylation at 10 mmHg was high at ∼40%, increased to a maximal level of ∼55% at 80 mmHg, and exhibited no additional change on further pressurization to 120 and 140 mmHg. Myogenic constriction at 80 mmHg was associated with a decline in G-actin content by ∼65% that was blocked by inhibition of ROK or PKC. Taken together, our findings indicate that two mechanisms of Ca2+ sensitization (ROK-mediated phosphorylation of MYPT1-T855 with augmentation of LC20 phosphorylation, and a ROK- and PKC-evoked increase in actin polymerization) contribute to force generation in the myogenic response of skeletal muscle arterioles.
Key points
Blood flow to our organs is maintained within a defined range to provide an adequate supply of nutrients and remove waste products by contraction and relaxation of smooth muscle cells of resistance arteries and arterioles.
The ability of these cells to contract in response to an increase in intravascular pressure, and to relax following a reduction in pressure (the ‘myogenic response’), is critical for appropriate control of blood flow, but our understanding of its mechanistic basis is incomplete.
Small arteries of skeletal muscles were used to test the hypothesis that myogenic constriction involves two enzymes, Rho-associated kinase and protein kinase C, which evoke vasoconstriction by activating the contractile protein, myosin, and by reorganizing the cytoskeleton.
Knowledge of the mechanisms involved in the myogenic response contributes to understanding of how blood flow is regulated and will help to identify the molecular basis of dysfunctional control of arterial diameter in disease.
Introduction
Resistance arteries and arterioles are mechanosensitive, existing in a state of partial constriction due to the presence of intravascular pressure, and constricting and dilating in response to pressure elevation and reduction, respectively (Bayliss 1902). This ability of resistance arteries and arterioles to react to intravascular pressure, known as the ‘myogenic response’, is an essential determinant of peripheral resistance, blood pressure regulation, regional blood flow control and protection of capillaries from damage due to a sudden increase in pressure (Olsen et al. 1981; Osol et al. 2002; Smeda 2003; Bidani et al. 2009). The myogenic response has been traced to cellular mechanisms inherent to vascular smooth muscle cells in the arterial wall, and is known to occur in the absence of endothelial or neuronal input (McCarron et al. 1989). Substantial progress has been made towards identification of the intrinsic mechanisms involved; however, several critical gaps remain in our understanding of molecular events underlying the myogenic response. Our focus here was to evaluate the contribution of Rho-associated kinase (ROK)- and protein kinase C (PKC)-dependent mechanisms of Ca2+ sensitization to the myogenic response of skeletal muscle resistance arteries.
Although Ca2+–calmodulin-dependent activation of myosin light chain kinase (MLCK) is a requisite step for myogenic constriction (Knot & Nelson, 1998), it is evident that additional mechanisms that increase force generation are also involved (see reviews by Schubert & Mulvany, 1999; Hill et al. 2001; Osol et al. 2002; Schubert et al. 2008; Cole & Welsh, 2011). The relationship between [Ca2+]i and diameter (or tone development) in the myogenic response indicates that pressure elevation also enhances sensitivity of the contractile process to Ca2+ (D’Angelo et al. 1997; Karibe et al. 1997; VanBavel et al. 1998, 2001; Wesselman et al. 2001; Lagaud et al. 2002; Schubert et al. 2002; Gokina et al. 2005). Three distinct mechanisms have been advanced as potential causes of increased force at constant [Ca2+]i in smooth muscle: (1) inhibition of myosin light chain phosphatase (MLCP) activity due to (a) ROK-mediated phosphorylation of the myosin targeting subunit (MYPT1) of MLCP (Kimura et al. 1996), or (b) direct interaction of the 17 kDa PKC-activated phosphatase inhibitor protein, CPI-17, with the catalytic PP1cδ subunit of MLCP following phosphorylation of CPI-17 by PKC (Eto et al. 1995); (2) dynamic remodelling of the actin cytoskeleton involving increased actin polymerization (Cipolla et al. 2002); and (3) suppression of thin filament regulation by PKC-mediated phosphorylation of caldesmon and/or calponin (Tanaka et al. 1990; Winder & Walsh, 1990).
Pharmacological inhibition of ROK or PKC activity has been shown to reduce myogenic constriction in several vessel types, implying that ROK- and PKC-dependent mechanisms of Ca2+ sensitization contribute to force generation in the myogenic response (Hill et al. 1990; Osol et al. 1991; Dessy et al. 2000; Schubert et al. 2002; Nakamura et al. 2003; Dubroca et al. 2005; Gokina et al. 2005). However, identification of the specific sensitization mechanism(s) activated by ROK and PKC signalling has been impeded by the small size of resistance vessels, making biochemical analysis challenging. This limitation has now been resolved with the development of highly sensitive western blotting techniques that permit accurate quantification of MYPT1, CPI-17 and LC20 phosphorylation in resistance arteries and arterioles (Takeya et al. 2008; Johnson et al. 2009). In our initial analysis, we detected increased phosphorylation of MYPT1 and LC20 in rat middle cerebral arteries (RMCAs) following pressurization from 10 to 60 and 100 mmHg, which was prevented by ROK inhibition (Johnson et al. 2009). In contrast, no change in CPI-17 phosphorylation was detected upon pressure elevation, and inhibition of the myogenic response by PKC inhibitor treatment was not associated with a change in MYPT1 or LC20 phosphorylation (Johnson et al. 2009). These findings led to the conclusion that ROK-mediated phosphorylation of MYPT1 and subsequent inhibition of MLCP activity contributes to Ca2+ sensitization in the myogenic response through suppression of LC20 dephosphorylation (Somlyo & Somlyo, 2003), but do not discount additional roles for ROK. On the other hand, the lack of a role for CPI-17 in the cerebral arterial myogenic response implies that an alternative mechanism(s) of Ca2+ sensitization, not involving MLCP inhibition and increased LC20 phosphorylation, must be evoked by PKC.
Accumulating evidence suggests that dynamic reorganization of the actin cytoskeleton plays an important role in smooth muscle contraction (Cipolla et al. 2002). Remodelling of the cytoskeleton involving increased actin polymerization is thought to enhance the efficiency of force transmission from the contractile apparatus to the cell membrane/extracellular matrix. Increased polymerization within the actin cytoskeleton can increase force generation in the absence of elevation in [Ca2+]i or LC20 phosphorylation, and may optimize the energetic cost of smooth muscle contraction (Jones et al. 1999). Agonist-induced or myogenic constriction of vascular smooth muscle is associated with actin remodelling, possibly in an isoform-specific manner (Cipolla & Osol, 1998; Cipolla et al. 2002; Gokina & Osol, 2002; Shaw et al. 2003; Flavahan et al. 2005; Ohanian et al. 2005; Kim et al. 2008; Gunst & Zhang, 2008). For example, the extent of myogenic constriction was affected by compounds that disrupt (cytochalasin D), prevent (latrunculin A), or enhance (jasplakinolide) actin polymerization, and confocal fluorescence microscopic measurements of phalloidin and DNAse 1 binding to filamentous (F)- and monomeric globular (G)-actin, respectively, indicate that there is an increase in the F- to G-actin ratio with pressure elevation (Cipolla & Osol, 1998; Cipolla et al. 2002; Gokina & Osol, 2002; Flavahan et al. 2005). However, the mechanism(s) responsible for the pressure-induced rise in F- to G-actin ratio has not been identified. In addition to regulating Ca2+ sensitization via inhibition of MLCP activity, ROK and PKC are established mediators of cytoskeletal remodelling in smooth muscle contraction (Gerthoffer, 2005; Gunst & Zhang, 2008). For example, ROK is known to phosphorylate and activate LIM kinase that inhibits actin depolymerization via phosphorylation of ADF/cofilin family proteins and promotes increased actin polymerization (Bernard, 2007).
Here, we have tested the hypothesis that mechanisms of ROK- and PKC-dependent Ca2+ sensitization involving phosphorylation of MYPT1 and CPI-17, and/or dynamic remodelling of the actin cytoskeleton, contribute to the myogenic response of rat gracilis (RGAs) and cremaster (RCrAs) skeletal muscle resistance arteries. ROK- and PKC-dependent phosphorylations of MYPT1 and CPI-17, respectively, were assessed in response to pressurization from 10 to between 80 and 140 mmHg, together with quantification of LC20 phosphorylation. The contribution of the actin cytoskeleton was investigated using latrunculin B that sequesters free G-actin and prevents F-actin assembly (Couéet al. 1987), and by quantification of G-actin content in the absence and presence of inhibitors of ROK (H1152) and PKC (GF109203X). Our findings provide evidence that mechanisms of Ca2+ sensitization involving ROK-dependent phosphorylation of MYPT1, and ROK- and PKC-evoked actin polymerization, contribute to the myogenic response of skeletal muscle resistance arteries.
Methods
Ethical approval
Male Sprague–Dawley rats (250–275 g; Charles River, Montreal, Quebec, Canada) were maintained and subsequently killed by halothane inhalation and exsanguination according to a protocol approved by the Animal Care Committee of the Faculty of Medicine, University of Calgary and conforming to the standards of the Canadian Council on Animal Care and The Journal of Physiology's ethical policies and regulations as outlined in Drummond (2009). A total of 110 rats were used.
Rat gracilis and cremaster artery pressure myography
The gracilis muscle was exposed by an incision of the skin, isolated from surrounding tissues, removed and placed in ice-cold Krebs' saline solution containing (in mmol l−1): NaCl 120, NaHCO3 25, KCl 4.8, NaH2PO4 1.2, MgSO4 1.2, glucose 11, CaCl2 2.5 (pH 7.4 when aerated with 95% air–5% CO2) as previously described (Sun et al. 1994). First- and second-order RGAs were removed, dissected free of the surrounding tissue and cut into 2 mm segments in preparation for arterial pressure myography. Cremaster muscles were surgically excised, as previously described (Meininger et al. 1991), and placed in a cooled dissection chamber filled with ice-cold Krebs' solution. First-order RCrAs were isolated and cut into 2 mm segments in preparation for arterial pressure myography.
Arterial segments were cannulated and mounted in a myograph chamber connected to a pressure controller (Living Systems, Burlington, VT, USA) and external arterial diameter measured by edge detection (IonOptix, Milton, MA, USA). Endothelial cells were removed from all arteries by briefly passing a stream of air through the vessel lumen and confirmed by the loss of vasodilatation to 10 μmol l−1 bradykinin. Arteries were allowed to warm to 37°C for 20 min in Krebs' solution, then pressurized to 60 mmHg and allowed to develop active tone over 30–45 min. All arteries were then subjected to three 5 min pressure steps from 20 to 80 mmHg to ensure the development of a stable level of pressure-dependent myogenic constriction. Preparations that exhibited leaks (indicated by a spontaneous, transient drop in intraluminal pressure) or a lack of stable myogenic constriction during these test steps were discarded. When a stable myogenic response was detected, pressure was reduced to 10 mmHg for 10 min before application of an appropriate pressure protocol.
Pressure protocol 1
To determine the effect of ROK and PKC inhibition on myogenic tone the vessels were subjected to a series of pressure steps from 10 to 120 mmHg in 20 mmHg increments. The first set of pressure steps was applied in normal Krebs' solution (control conditions). Pressure was then reduced to 10 mmHg and H1152 (0.5 μmol l−1; Sasaki et al. 2002) or GF109203X (3.0 μmol l−1; Toullec et al. 1991) added to the superfusate before a second series of steps. Pressure was then reduced to 10 mmHg again and superfusate replaced with zero Ca2+-containing Krebs' saline solution (i.e. no added Ca2+ and 2 mmol l−1 EGTA) before a third series of pressure steps to determine the passive diameter of the artery at each pressure. A second set of vessels was similarly pressurized to between 10 and 120 mmHg in two series of pressure steps in normal Krebs' solution to confirm the presence of an unaltered myogenic response with time (see online Supplemental Fig. 1). Active myogenic constriction was defined as the difference between arterial diameter in zero Ca2+ and normal Krebs' solution or saline containing drug.
Pressure protocol 2
Analysis of the phosphorylation of MYPT1, CPI-17 and LC20 was accomplished using vessels flash-frozen after a pressure protocol involving two 5 min steps to 80 mmHg and 10 min at 10 mmHg before a 10 min step to a test pressure of 10, 80, 120 or 140 mmHg. For some measurements of MYPT1 and LC20 phosphorylation, vessels were treated with H1152 (0.5 μmol l−1) after a stable arterial diameter was achieved at the test pressure. CPI-17 phosphorylation was also assessed by flash freezing vessels after 20 s at test pressures of 80 or 120 mmHg. Additional vessels were exposed to 1 μm phorbol 12,13-dibutyrate (PDBu) to serve as a positive control for CPI-17 phosphorylation. LC20 phosphorylation was also quantified in experiments in which RGAs were (1) maintained at 10 or 80 mmHg and exposed to ML-7 (10 μmol l−1) or latrunculin B (10 μmol l−1) until a stable level of vasodilatation was achieved, and (2) maintained at 10 mmHg and exposed to zero extracellular Ca2+ solution. For comparative purposes, we also quantified LC20 phosphorylation in RMCAs at 10 mmHg in the absence and presence of zero extracellular Ca2+ solution, as previously described (Johnson et al. 2009).
Pressure protocol 3
To determine the effect of latrunculin B (10 μmol l−1) on arterial diameter and G-actin content, arteries were maintained at 20 mmHg and treated with drug before or after pressure elevation from 20 to 80 mmHg. In a second set of experiments, vessels were maintained at 20 mmHg and exposed to two applications of phenylephrine (PE; 10 μmol l−1), with the second exposure performed in the presence of latrunculin B.
Protein extraction
Arteries were immersed in a wet ice-cold mixture of 10% trichloroacetic acid and dithiothreitol (DTT; 10 mmol l−1) in acetone. Segments were then washed in wet ice-cold acetone containing DTT, lyophilized overnight and stored at −80°C. The cannulated ends were dissected from each lyophilized vessel segment and discarded (to avoid including tissue that was not subjected to the test pressure) before protein extraction in 60 μl of sample buffer (4% SDS, 100 mmol l−1 DTT, 10% glycerol, 0.01% bromophenol blue, 60 mmol l−1 Tris-HCl, pH 6.8). Each sample contained one to two pooled RGAs or two to three pooled RCrAs for a single n value. Samples were heated at 95°C for 10 min and rotated overnight at 4°C before gel electrophoresis.
Phosphoprotein western blotting
A three-step western blotting protocol was used to quantify phospho-MYPT1 (at the ROK sites, T697 and T855), -CPI-17 and -LC20 content, as previously described in detail (Takeya et al. 2008; Johnson et al. 2009). Phospho-MYPT1-T697 and -MYPT1-T855 levels were determined by normalization to the level of Coomassie blue-stained actin in each sample. Phospho-CPI-17 and phospho-LC20 were separated by phosphate-affinity tag SDS-PAGE (Phos-Tag SDS-PAGE) and quantified as a percentage of total CPI-17 and LC20, respectively, as previously described (Takeya et al. 2008; Johnson et al. 2009).
G-actin determination
G-actin content was determined as previously described (Corteling et al. 2007; Luykenaar et al. 2009; Walsh et al. 2011) for individual RGA segments pressurized to 10 or 80 mmHg ± latrunculin B, H1152 or GF109203X. Each vessel was transferred to F-actin stabilization buffer (Cytoskeleton, Denver, CO, USA) containing: 50 mmol l−1 PIPES (pH 6.9), 50 mmol l−1 KCl, 5 mmol l−1 MgCl2, 5 mmol l−1 EGTA, 5% v/v glycerol, 0.1% Nonidet P40, 0.1% Triton X-100, 0.1% Tween 20, 0.1% 2-mercaptoethanol, 0.001% anti-foamC, and then homogenized in 100 μl of stabilizing buffer at room temperature. The homogenate was centrifuged at 100,000 g for 1 h at 22°C to separate G- and F-actin; 30 μl of the high-speed supernatant containing G-actin was carefully removed and added to 30 μl of 2 × sample buffer. Samples were then heated at 95°C for 10 min and stored at −20°C before SDS-PAGE and western blotting. SDS-PAGE was carried out in 1.5 mm thick minigels (10% acrylamide in the resolving gel with 4.5% acrylamide stacking gel) at 30 mA for 1.5 h in a Mini Protean Cell (Bio-Rad Laboratories, Mississauga, ON, Canada). Following electrophoresis, proteins were transferred on to a 0.2 μm nitrocellulose membrane at 100 V for 90 min at 4°C, in transfer buffer containing 25 mmol l−1 Tris-HCl, 192 mmol l−1 glycine and 20% methanol. Membranes were then washed in PBS for 5 min, incubated in 0.5% glutaraldehyde in PBS for 15 min to fix proteins on the membrane and washed (2 × 5 min) with TBST (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.05% Tween 20). Membranes were blocked with 5% non-fat dried milk in TBS containing 0.1% Tween 20 (0.1% TBST) for 1 h and then cut at the 35 kDa molecular mass marker. The high molecular mass proteins were incubated with rabbit polyclonal actin antibody (1:1000 dilution; Cytoskeleton) while the low molecular mass proteins were incubated with goat polyclonal SM-22α antibody (1:2000 dilution; Novus Biologicals, Oakville, ON, Canada). Both antibody incubations were performed overnight at 4°C in 1% dry milk in 0.1% TBST. A standard two-step western blotting protocol was sufficient to detect G-actin and SM-22 in individual RGAs. Membranes were washed (4 × 5 min) in TBST and incubated for 1 h in 1% dry milk in 0.1% TBST containing antirabbit IgG–horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution) or antigoat IgG–horseradish peroxidase-conjugated secondary antibody (1:5000 dilution; Millipore, Bellerica, MA, USA), respectively. After incubation with secondary antibodies, the membranes were washed (4 × 5 min) with TBST and (1 × 5 min) with TBS before chemiluminescence signal detection using Amersham ECL advanced western blotting detection kit (GE Healthcare, Mississauga, ON, Canada). The emitted light was detected and quantified with a chemiluminescence imaging analyser (LAS3000 Mini; Fujifilm Canada, Mississauga, ON, Canada) and images were analysed with MultiGauge v3.0 software (Fujifilm).
Chemicals
All chemicals were purchased from Sigma (Oakville, ON, Canada) unless indicated otherwise. H1152 and ML-7 were obtained from Calbiochem (San Diego, CA, USA), and latrunculin B and GF109203X were from Enzo Life Sciences (Plymouth Meeting, PA, USA). Tween 20, Coomassie brilliant blue-R250, TEMED, PVDF and nitrocellulose membranes were from Bio-Rad Laboratories. Rabbit polyclonal antibodies specific for MYPT1 phosphorylated at T697 (anti-MYPT1-T697) or T855 (anti-MYPT1-T855) and anti-CPI-17 were from Upstate USA (Charlottesville, VA, USA). Polyclonal rabbit anti-LC20 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Biotin-conjugated goat anti-rabbit; secondary antibody was from Chemicon International and horseradish peroxidase-conjugated streptavidin was from Pierce Biotechnology (Rockford, CT, USA). Phos-tag acrylamide was obtained from NARD Institute Ltd (Amajasaki City, Hyogo Prefecture, Japan).
Statistical analysis
All values are presented as means ±s.e.m., with n values indicative of the number of vessels studied for each treatment. In general, vessels from one rat were employed to minimize variability between control and treatment groups. Statistical difference was determined using unpaired Student's t tests, repeated measures ANOVA followed by Bonferroni's post hoc test, or ANOVA followed by Dunnett's multiple comparisons test as indicated. P < 0.05 was considered statistically significant.
Results
Inhibition of Rho-associated kinase attenuates myogenic constriction of skeletal muscle resistance arteries
RGA and RCrA skeletal muscle resistance arteries exhibited an intraluminal pressure-dependent myogenic vasoconstriction between 40 and 80 mmHg (Fig. 1). Active constriction at 80 mmHg (i.e. the difference in diameter ± external Ca2+) was 48 ± 4 μm (n= 9) and 55 ± 5 μm (n= 9) for RGAs and RCrAs, respectively, consistent with previous studies (Sun et al. 1994; Zou et al. 1995). To evaluate the contribution of ROK in the myogenic response, vessels were subjected to three sequential series of pressure steps to between 20 and 120 mmHg in increments of 20 mmHg in: (1) control solution; (2) solution containing the ROK inhibitor, H1152 (0.5 μmol l−1); and (3) zero external Ca2+ saline. Representative recordings of arterial diameter and mean data of Fig. 1 show that the myogenic response of RGAs and RCrAs was markedly reduced by H1152 (see online Supplemental Fig. 1 for time-control data).
Figure 1. Inhibition of Rho-associated kinase suppresses the myogenic response of rat gracilis arteries (RGAs) and rat cremaster arteries (RCrAs).
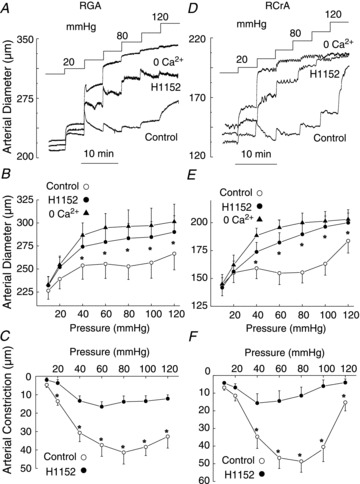
Effect of H1152 on RGA (A–C; n= 6) and RCrA (D–F; n= 6) myogenic response. Representative pressure-induced changes in arterial diameter (A and D) and mean diameter (±s.e.m.)–pressure relations (B and E) for 10–120 mmHg in normal Krebs' saline (control), 0.5 μmol l−1 H1152, and zero Ca2+ saline (0 Ca2+), as well as mean active constriction–pressure relations ± H1152 (C and F). *Significantly different (P < 0.05) from control value.
Pressure elevation evokes a Rho-associated kinase-mediated increase in MYPT1 and LC20 phosphorylation
RGAs and RCrAs segments exhibiting stable diameter at 10 min following pressure steps from 10 mmHg to 80, 120 or 140 mmHg ± H1152 (0.5 μmol l−1) were flash-frozen using pressure protocol 2 (see Methods section). These pressures were selected as 10 mmHg represents the predominantly relaxed condition, 80 mmHg was near, or at the maximal level of myogenic constriction (Fig. 1) and consistent with the physiological intravascular pressures for this calibre of skeletal muscle vessel (Fronek & Zweifach, 1975), and 120 and 140 mmHg were at and above the physiological range. The latter were associated with a loss of myogenic control of arterial diameter and forced dilation (Osol et al. 2002).
Figures 2 and 3 show representative western blots of phospho-MYPT1-T697 and -MYPT1-T855 and the corresponding Coomassie blue-stained actin content of each sample, as well as mean normalized protein phosphorylation levels as a function of pressure ± H1152 for RGAs and RCrAs, respectively. Basal levels of MYPT1-T697 and MYPT1-T855 phosphorylation were detected at 10 mmHg in both vessels (Figs 2A and B and 3A and B). The level of phospho-MYPT1-T855 was significantly increased at 80, 120 and 140 compared to 10 mmHg (Figs 2B and 3B), but MYPT1-T697 phosphorylation was unaltered even at 140 mmHg (Figs 2A and 3A). The pressure-induced increase in MYPT1-T855 phosphorylation was significantly reduced by H1152, but phospho-MYPT1-T697 content was unaffected by the ROK inhibitor over the entire pressure range of 10–140 mmHg.
Figure 2. Pressure elevation evokes Rho-associated kinase-mediated increase in MYPT1-T855 phosphorylation in rat gracilis arteries.
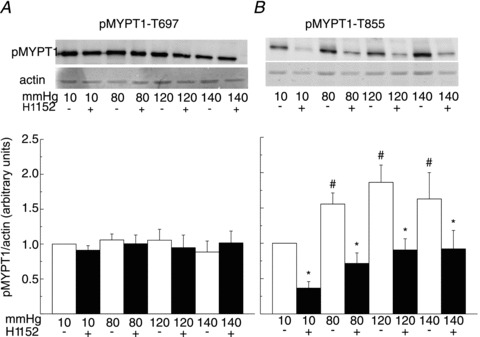
A and B, representative blots of MYPT1-T697 and MYPT1-T855 phosphorylation and corresponding levels of actin in each lane (upper) and means ±s.e.m. of phosphoprotein content normalized to actin expressed as a fraction of the value at 10 mmHg (lower) in samples derived from rat gracilis arteries pressurized to 10, 80, 120 and 140 mmHg ± H1152 (0.5 μmol l−1) (n= 7 blots with 1–2 arterial segments per sample). #,*Significant difference (P < 0.05) from value at 10 mmHg and in control solution at each pressure, respectively. pMYPT1, MYPT1 phosphorylation.
Figure 3. Pressure elevation evokes Rho-associated kinase-mediated increase in MYPT1-T855 phosphorylation in rat cremaster arteries.
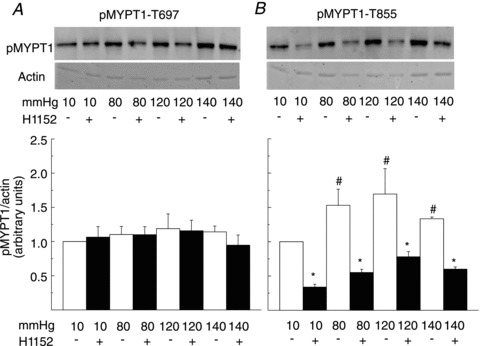
A and B, representative western blots of MYPT1-T697 and MYPT1-T855 phosphorylation and corresponding levels of actin in each lane (upper) and means ±s.e.m. of phosphoprotein content normalized to actin expressed as a fraction of the value at 10 mmHg (lower) in samples derived from rat cremaster arteries pressurized to 10, 80, 120 and 140 mmHg ± H1152 (0.5 μmol l−1) (n= 6 blots with 3 arterial segments per sample). #,*Significant difference (P < 0.05) from value at 10 mmHg and in control solution at each pressure, respectively. pMYPT1, MYPT1 phosphorylation.
If ROK-mediated MYPT1-T855 phosphorylation contributes to Ca2+ sensitization in RGAs and RCrAs, then the pressure-dependent increase in phosphorylation would be expected to evoke a concomitant H1152-sensitive increase in LC20 phosphorylation at 80, 120 and 140 mmHg. Figures 4 and 5 show representative western blots of LC20 and mean phospho-LC20 content as a percentage of total LC20 at 10, 80, 120 and 140 mmHg ± H1152 in RGAs and RCrAs, respectively. Pressure elevation to 80, 120 or 140 mmHg in the presence of extracellular Ca2+ was associated with an increase in LC20 phosphorylation from 43 ± 5% at 10 mmHg to 53 ± 4, 58 ± 5 and 56 ± 10% at 80, 120 and 140 mmHg, respectively, in RGAs, and from 38 ± 2% at 10 mmHg to 51 ± 3, 54 ± 7 and 53 ± 7% at 80, 120 and 140 mmHg, respectively, in RCrAs. Inhibition of ROK with H1152 significantly reduced basal LC20 phosphorylation in RGAs and RCrAs, as well as the increase in phosphoprotein content detected following pressure elevation (Figs 4 and 5).
Figure 4. Pressure elevation evokes Rho-associated kinase-mediated increase in LC20 phosphorylation in rat gracilis arteries.
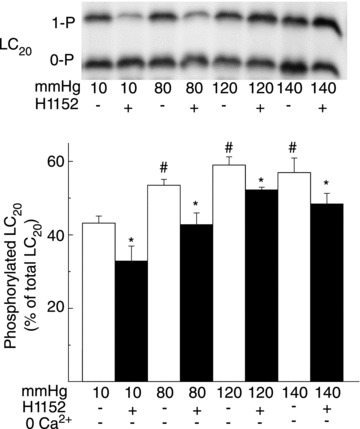
Representative western blot (upper) and means ±s.e.m. of LC20 phosphorylation (lower) as % of total LC20 at 10 (±0 Ca2+), 80, 120 and 140 mmHg ± Rho-associated kinase inhibition with H1152 (n= 7 with 1–2 arterial segments per sample) with monophosphorylated and unphosphorylated LC20 separated by Phos-tag SDS-PAGE. #,*Significant difference (P < 0.05) from value at 10 mmHg and in control solution at each pressure, respectively.
Figure 5. Pressure elevation evokes Rho-associated kinase-mediated increase in LC20 phosphorylation in rat cremaster arteries.
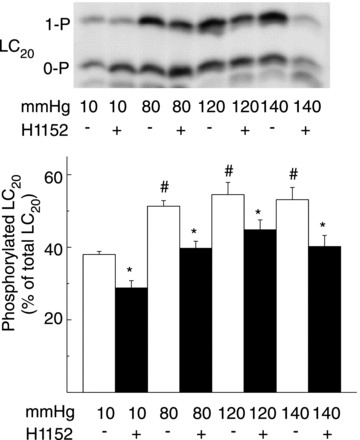
Representative western blot (upper) and means ±s.e.m. of LC20 phosphorylation (lower) as % of total LC20 at 10, 80, 120 and 140 mmHg ± Rho-associated kinase inhibition with H1152 (n= 6 with 3 arterial segments per sample) with unphosphorylated and monophosphorylated LC20 separated by Phos-tag SDS-PAGE. #,*Significant difference (P < 0.05) from value at 10 mmHg and in control solution at each pressure, respectively.
Lack of pressure-induced CPI-17 phosphorylation
The PKC inhibitor, GF109203X (3 μmol l−1) produced a similar inhibition of myogenic constriction in RGAs (Fig. 6) as observed for H1152 (Fig. 1). Therefore, the contribution of a PKC-mediated Ca2+ sensitization mechanism to the myogenic response of RGAs was examined by measuring CPI-17 phosphorylation in vessel segments pressurized at 10, 80 or 120 mmHg. In the positive control, PDBu (2 μmol l−1) significantly increased CPI-17 phosphorylation (Fig. 6C). However, no CPI-17 phosphorylation was observed at 10, 80 or 120 mmHg (Fig. 6C). CPI-17 phosphorylation was also analysed at an additional time point of 20 s after pressure steps to 80 or 120 mmHg to permit detection of a rapid change in phosphorylation, as was reported for agonist-evoked contraction of conduit arteries (maximal change at 15 s; Dimopoulos et al. 2007). However, no CPI-17 phosphorylation was detected even at this early sampling time (Fig. 6C).
Figure 6. Pressure elevation is not accompanied by an increase in CPI-17 phosphorylation content in rat gracilis arteries.
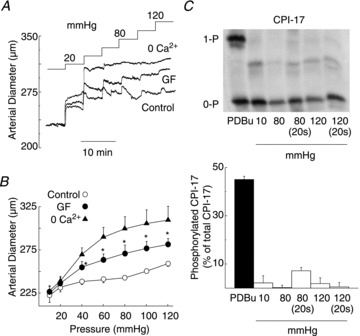
A, representative pressure-induced changes in arterial diameter and (B) mean diameter (±s.e.m.)–pressure relations (n= 3) for 10–120 mmHg in normal Krebs' saline (control), 3 μmol l−1 GF109203X (GF), and zero Ca2+ saline (0 Ca2+). C, representative western blot and mean CPI-17 phosphorylation level ±s.e.m. expressed as a percentage of total CPI-17 in rat gracilis arteries treated with PDBu (2 μmol l−1) or pressurized to 10, 80, 80 (sampled at 20 s), 120 and 120 (sampled at 20 s) mmHg (n= 3 blots with 4 arterial segments in each sample). *Significantly different (P < 0.05) from control value. PDBu, phorbol 12,13-dibutyrate.
Dynamic cytoskeletal reorganization contributes to the myogenic response of skeletal muscle resistance arteries
The high basal level of LC20 phosphorylation, limited increase in phosphorylation between 10 and 80 mmHg, lack of additional increase following pressurization to ≥80 mmHg and absence of a role for PKC-mediated CPI-17 phosphorylation suggested that additional mechanisms of force generation due to Ca2+ sensitization possibly contributed to the myogenic response of skeletal muscle arteries. We therefore examined the effect on myogenic constriction of the G-actin-binding, cytoskeleton-disrupting compound latrunculin B (Couéet al. 1987). Figure 7 shows representative recordings and mean diameter of RGAs and RCrAs sequentially exposed to latrunculin B (10 μmol l−1) and zero Ca2+ superfusate after a stable myogenic response was achieved at 80 mmHg. Latrunculin B treatment evoked dilation to the passive diameter observed in the absence of active, Ca2+-dependent constriction in both vessels. Figure 8A and B show a representative recording and mean diameter during a step increase in pressure from 20 to 80 mmHg ± pretreatment with latrunculin B, prior to zero Ca2+ superfusate. Latrunculin B did not affect basal tone development at 20 mmHg, as no change in diameter was detected but RGAs exhibited an immediate dilation to the passive diameter when pressure was elevated to 80 mmHg, i.e. latrunculin B completely prevented maintenance of diameter with pressure elevation. PE (10 μmol l−1) was applied ± latrunculin B at 20 mmHg to determine whether sequestration of G-actin disrupted the existing actin cytoskeleton before the pressure step, thereby preventing subsequent myogenic constriction. Figure 8C and D show representative recordings and mean diameters before and during PE ± latrunculin B treatment. PE evoked a sustained constriction of 53 ± 5 μm at 20 mmHg, but in the presence of latrunculin B, the constriction to agonist was transient (see representative data in Fig. 8C and E). The peak of the PE-induced constriction was not significantly altered by latrunculin B at 46 ± 5 μm, and time-to-peak constriction was not affected (86 ± 15 s before and 83 ± 14 s after latrunculin B). However, the PE-evoked constriction was not sustained in the presence of latrunculin B and final diameter was not different from that observed before addition of PE, indicating a lack of maintenance of agonist-induced force generation (Fig. 8D).
Figure 7. Disruption of dynamic cytoskeletal reorganization with latrunculin B (LatB) is accompanied by full dilation of rat gracilis arteries (RGAs) and rat cremaster arteries (RCrAs) at 80 mmHg.
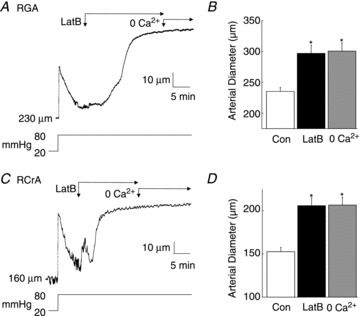
A and C, representative recordings of RGA and RCrA diameter during a pressure step from 20 to 80 mmHg followed by LatB (10 μmol l−1) and then zero Ca2+ saline. B and D, mean diameter ±s.e.m. of RGAs and RCrAs (n= 6 and 3, respectively) at 80 mmHg in control Krebs' (Con) and after treatment with LatB and zero Ca2+ saline. *Significantly different (P < 0.05) from control value.
Figure 8. Pretreatment with latrunculin B (LatB) abolishes the myogenic response and maintenance of phenylephrine (PE)-induced contraction in rat gracilis arteries (RGAs).
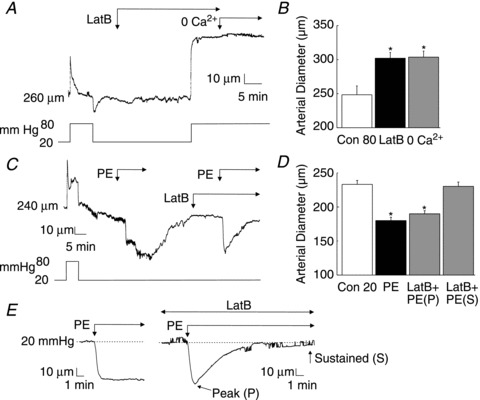
A, representative recording of RGA diameter during treatment with LatB (10 μmol l−1) before a pressure step from 20 to 80 mmHg and subsequent exposure to zero Ca2+ saline. B, mean diameter ±s.e.m. of RGAs at 80 mmHg in the absence and presence of LatB or zero Ca2+ saline (n= 3). C, representative recording of RGA diameter during two applications of PE (10 μmol l−1) at 20 mmHg, with the second treatment in the presence of LatB. D, mean diameter ±s.e.m. before and in PE in the absence and presence of LatB, with the latter expressed as diameter at maximal (peak or P) constriction and final, maintained diameter (sustained or S) (n= 5). E, representative traces of the recordings obtained in (C), showing the sustained constriction in response to PE alone and the dilation back to original diameter when PE was applied in the presence of LatB. *Significantly different (P < 0.05) from control value.
Two sets of experiments were performed to exclude the possibility that the dilation evoked by latrunculin B treatment was due to changes in MLCK or MLCP activity secondary to cytoskeletal disruption, i.e. (1) latrunculin B was applied in the presence of the MLCK inhibitor, ML-7 (10 μmol l−1) at 80 mmHg, and (2) the effect of ML-7 and latrunculin B on LC20 phosphorylation was determined. ML-7 caused RGAs to dilate to ∼45% of the level in zero Ca2+ saline, and subsequent exposure to latrunculin B produced full dilation to the passive diameter (Fig. 9A). ML-7 treatment reduced phospho-LC20 levels at 10 and 80 mmHg to below the level detected in untreated vessels at 10 mmHg (Fig. 9B). In contrast, latrunculin B had no effect on LC20 phosphorylation compared to untreated vessels at 80 mmHg (Fig. 9B). These data support the conclusion that the dilation evoked by latrunculin B was independent of a change in the balance of MLCK and MLCP activities.
Figure 9. Latrunculin B (LatB) evokes LC20-independent inhibition of myogenic constriction in rat gracilis arteries (RGAs).
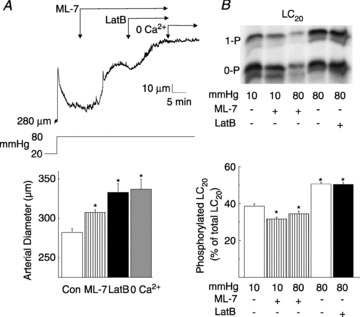
A, representative recording (upper) and means ±s.e.m. (lower) of RGA diameter owing to a pressure step from 20 to 80 mmHg followed by sequential exposure to ML-7 (10 μmol l−1), LatB (10 μmol l−1) and zero Ca2+ saline (n= 5). B, representative western blot (upper) and means ±s.e.m. of LC20 phosphorylation (lower) as a percentage of total LC20 in RGAs at 10 mmHg ± ML-7 (10 μmol l−1), and at 80 mmHg ± ML-7 (10 μmol l−1) or ± LatB (10 μmol l−1) (n= 3 blots with 1 arterial segment per sample) with monophosphorylated and unphosphorylated LC20 separated by Phos-tag SDS-PAGE. *Significantly different (P < 0.05) from control value.
If dynamic reorganization of actin cytoskeleton involving increased actin polymerization contributes to force generation in the myogenic response of skeletal muscle resistance arteries, G-actin content would be expected to decrease following an acute increase in pressure. Therefore, G-actin content was quantified in RGAs at 80 mmHg ± latrunculin B, normalized to the level of SM-22, which is recovered exclusively in the supernatant following high-speed centrifugation (Luykenaar et al. 2009; Walsh et al. 2011), and expressed as a fraction of the level detected at 10 mmHg (Fig. 10A). G-actin content was reduced by ∼60% in response to a pressure step from 10 to 80 mmHg. No decline was detected in the presence of latrunculin B at 80 mmHg, rather G-actin content was increased relative to that observed at 10 mmHg in control conditions (Fig. 10A).
Figure 10. Pressure elevation reduces G-actin content in rat gracilis arteries (RGAs) via Rho-associated kinase and protein kinase C-dependent signalling.
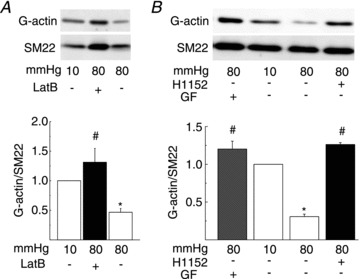
A, representative western blots of G-actin and SM-22 (upper) and means ±s.e.m. of G-actin normalized to SM-22 (lower) and expressed as a fraction of the value at 10 mmHg (n= 6 blots with 1 arterial segment per sample) for RGAs at 10 mmHg and 80 mmHg ± LatB (10 μmol l−1). B, representative western blots of G-actin and SM-22 (upper) and means ±s.e.m. of G-actin normalized to SM-22 (lower) and expressed as a fraction of the value at 10 mmHg (n= 4 blots with 1 arterial segment per sample) for RGAs at 10 mmHg and 80 mmHg ± H1152 (0.5 μmol l−1) or GF109203X (GF; 3 μmol l−1). *Significantly different (P < 0.05) from control value. LatB, latrunculin B.
Finally, we tested the possibility that the change in G-actin content was dependent on ROK and/or PKC activity by quantifying G-actin levels at 80 mmHg in the absence or presence of H1152 or GF109203X. Figure 10B shows that the decline in G-actin content evoked by pressure elevation from 10 to 80 mmHg was prevented following inhibition of ROK or PKC.
Discussion
This study examined the contribution of Ca2+ sensitization mechanisms evoked by ROK and PKC to pressure-induced, myogenic constriction of rat skeletal muscle resistance arteries. We identified a ROK-dependent increase in phosphorylation of MYPT1 and LC20 with pressure elevation, as well as a pressure-dependent decrease in G-actin content that is dependent on signalling pathways involving ROK and PKC. These novel findings have important implications for our understanding of the molecular basis of force generation in myogenic control of skeletal muscle resistance arterial diameter in response to changes in intravascular pressure.
Our findings support the view that Ca2+ sensitization because of MLCP inhibition is a fundamental mechanism contributing to force generation in the myogenic response of resistance arteries. Here, we show that the myogenic response of skeletal muscle resistance arteries was accompanied by a ROK-mediated increase in MYPT1 phosphorylation at T855 and an increase in LC20 phosphorylation. In the presence of ROK inhibition with H1152, the levels of phospho-MYPT1-T855 and phospho-LC20 at 80 to 140 mmHg were not different from the basal levels at 10 mmHg. Ca2+ sensitization induced by ROK-mediated phosphorylation of MYPT1 and reduction in MLCP activity is therefore essential to permit the rise in LC20 phosphorylation associated with the myogenic response of rat skeletal muscle arteries, consistent with previous findings for cerebral resistance arteries (Johnson et al. 2009; El-Yazbi et al. 2010). Interestingly, increased MYPT1 phosphorylation at T855 and T697 was detected in RMCAs at 140 mmHg (El-Yazbi et al. 2010), but T855 was the exclusive site of increased phosphorylation in RGAs and RCrAs over the entire range of pressure analysed here. The physiological significance of this difference in phosphorylation at MYPT1-T697 is not evident at this time.
Considerable heterogeneity with respect to the presence of a myogenic response, level of myogenic responsiveness, time course of myogenic constriction and relationship between membrane potential, [Ca2+]i and myogenic constriction, has been reported for different vascular beds, and resistance vessels of decreasing diameter within individual beds (Johnson, 1986; Davis, 1993; Davis & Hill, 1999; Schubert & Mulvany, 1999; Hill et al. 2001; Brekke et al. 2002; Kotecha & Hill, 2005). As noted by Dora (2005), it is logical that the distinct, vessel-specific features of the myogenic response contribute to the varied physiological behaviour of different vascular beds, such as vasculature supplying skeletal muscle and the brain. Comparison of present and previous findings for skeletal muscle and cerebral arteries, respectively, provides evidence for heterogeneity in the magnitude of change in LC20 and MYPT1 phosphorylation associated with myogenic constriction. Specifically,
The increase in LC20 phosphorylation associated with pressurization to 80 mmHg or greater was significantly smaller in RGAs and RCrAs compared to RMCAs (Johnson et al. 2009; El-Yazbi et al. 2010). This may be attributed to a higher basal, but identical maximal stoichiometry of phosphorylation of 50–55% of total LC20 at ≥80 mmHg in the skeletal muscle compared to cerebral vessels (Johnson et al. 2009; El-Yazbi et al. 2010). Basal LC20 phosphorylation at 10 mmHg was 0.43 ± 0.05 and 0.38 ± 0.02 mol Pi/mol LC20 in RGAs and RCrAs, respectively, compared to ∼0.25 mol Pi/mol LC20 in RMCAs (Johnson et al. 2009; El-Yazbi et al. 2010). We confirmed this difference in the current study by quantifying LC20 phosphorylation levels in RGAs and RMCAs in the same experiments run side-by-side. To validate the high basal level of phospho-LC20 detected here using skeletal muscle vessels, RMCAs were pressurized to 10 mmHg ± external Ca2+ (n= 4 each), frozen and then prepared in parallel and run on the same Phos-tag gels as samples from RGAs; online Supplemental Fig. 2 shows that the phospho-LC20 content of RMCAs was 26 ± 2% and significantly lower than that in RGAs (P < 0.05%). In contrast, the level of phosphorylation in RGAs and RMCAs in the absence of external Ca2+ was similar at 8 ± 2 and 10 ± 1%, respectively (online Supplemental Fig. 2).
The relative change in MYTP1-T855 phosphorylation in skeletal muscle resistance arteries was smaller at ∼55% (based on the difference between values at 10 mmHg and 80–140 mmHg that were not statistically different) compared to cerebral arteries at >∼110% (at 10 versus 100 mmHg; Johnson et al. 2009).
The decline in basal MYPT1-T855 and LC20 phosphorylation at 10 mmHg in the presence of ROK inhibition was greater in skeletal muscle vessels at ∼70% compared to ∼50% in RMCAs (a similar value of ∼50% was detected at >80 mmHg in both vessel types; Johnson et al. 2009). This implies a greater basal level of ROK-mediated phosphorylation of MYPT1-T855, consistent with the difference in phospho-LC20 content, in skeletal muscle compared to cerebral resistance arteries. These differences in phosphoprotein content complement previous finding that the relative change in membrane potential in the myogenic response of RCrAs is smaller, and occurs over a narrower pressure range, than in posterior cerebral arteries (Knot & Nelson, 1998; Knot et al. 1998; Kotecha & Hill, 2005), possibly due to reduced large conductance Ca2+-activated K+ channel subunit expression and activity (Yang et al. 2009).
Although the PKC inhibitor, GF109203X, blocked myogenic constriction of RGAs, our data do not support the view that Ca2+ sensitization via PKC-mediated phosphorylation of CPI-17, with inhibition of MLCP activity, contributes to the myogenic response. Previous studies on skeletal muscle resistance arteries have claimed a role for PKC-induced Ca2+ sensitization (Hill et al. 1990; Liu et al. 1994; Karibe et al. 1997; Massett et al. 2002) and the ability of PKC inhibitors and activators to suppress and enhance myogenic constriction in these vessels, respectively, is consistent with findings for RMCAs and other vessels (Gokina et al. 1999; Lagaud et al. 2002; Yeon et al. 2002; Jarajapu & Knot, 2005; Schubert et al. 2008; Johnson et al. 2009). However, no change in CPI-17 phosphorylation was detected following step increases in pressure from 10 to 80 or 120 mmHg, even when RGAs were flash-frozen at 20 s, i.e. within the period of elevated phospho-CPI-17 associated with agonist treatment of conduit arterial smooth muscle tissues (Dimopoulos et al. 2007). A similar lack of CPI-17 phosphorylation was noted for RMCAs (Johnson et al. 2009). On the other hand, our data do indicate a role for PKC in evoking reorganization of the actin cytoskeleton.
Our findings indicate that a second mechanism of increased force generation at constant [Ca2+]i also contributes to myogenic constriction in skeletal muscle resistance arteries. The role of an additional mechanism(s) of Ca2+ sensitization was considered here based on the lack of change in LC20 phosphorylation at pressures of >80 mmHg, and the sensitivity of the myogenic response to PKC inhibition in the absence of a role for CPI-17. Our data suggest that a dynamic reorganization of the cytoskeleton involving increased actin polymerization, and possibly disruption (severing) and depolymerization of existing cytoskeletal connections is triggered in parallel to the activation of MLCK and inhibition of MLCP during the myogenic response of skeletal muscle resistance arteries. Latrunculin B blocked and completely reversed myogenic constriction of RGAs when applied before and after pressure elevation from 20 to 80 mmHg, respectively. This is consistent with findings of previous studies in which cytochalasin D and/or latrunculin B were used to inhibit myogenic constriction (Cipolla & Osol, 1998; Cipolla et al. 2002; Gokina & Osol, 2002; Flavahan et al. 2005). We can exclude an effect of the actin cytoskeleton on myogenic control of membrane potential and [Ca2+]i, as well as MLCK and MLCP activities in the myogenic response, as no change in LC20 phosphorylation was detected in the presence of latrunculin B. On the other hand, pressurization to 80 mmHg was associated with a latrunculin B-sensitive reduction in G-actin by ∼60% compared to the level detected at 10 mmHg. This finding is consistent with previous studies using a confocal fluorescence microscopic approach to detect increased F-actin and/or decreased G-actin levels associated with myogenic constriction of rat cerebral and murine tail arteries (Cipolla & Osol, 1998; Cipolla et al. 2002; Flavahan et al. 2005). Here, we show that the decline in G-actin with pressure elevation in RGAs was completely blocked by H1152 or GF109203X. This result indicates that pressure-evoked ROK signalling elicits two mechanisms of Ca2+ sensitization, MLCP inhibition via MYPT1-T855 phosphorylation as described above, and cytoskeletal reorganization involving increased actin polymerization. The lack of change in G-actin level associated with PKC inhibition detected here provides an explanation for the sensitivity of the myogenic response to PKC inhibition in the absence of a role for CPI-17. Whether an additional mechanism of PKC-dependent suppression of thin filament regulation contributes to myogenic force generation in skeletal muscle resistance arteries remains to be determined; however, we did not detect a change in caldesmon or calponin phosphorylation levels following pressure elevation in RMCAs (unpublished observation). Cellular signalling pathways involving ROK and PKC are known to play an important role in regulating actin cytoskeletal dynamics in smooth muscle and other cell types and both enzymes phosphorylate intermediates that directly or indirectly regulate actin polymerization/depolymerization (Gerthoffer, 2005; Gunst & Zhang, 2008). Further analysis is required to elucidate the specific signalling intermediates involved in the reorganization evoked by ROK and PKC activities.
The present study shows that the myogenic response of skeletal muscle resistance arteries is associated with increased levels of LC20 and MYPT1 phosphorylation. This observation is consistent with previous experiments on RCrAs (Zou et al. 1995) and RMCAs (Johnson et al. 2009), but differences are also apparent. For example, the levels of phospho-LC20 detected in RGAs and RCrAs at pressures associated with myogenic constriction are greater than those previously reported for RCrAs (Zou et al. 1995). We detected a maximal stoichiometry of phosphorylation of ∼0.55 mol Pi/mol LC20 in segments of RGAs and RCrAs pressurized to 80, 120 and 140 mmHg, but Zou et al. (1995) reported levels of ∼0.27 at 70 mmHg and ∼0.40 mol Pi/mol LC20 at 120 and 150 mmHg in RCrAs. We attribute this difference to the method employed to prepare the vessels for biochemical analysis. Here, RGAs and RCrAs were immersed in TCA–acetone–DTT on wet ice before washing in wet ice-cold acetone–DTT and lyophilization. Zou et al. (1995) clamped vessels between acetone–dry ice cooled tongs before transfer to acetone–dry ice solution, slow warming to room temperature, and homogenization in a urea–DTT–Triton X-100 buffer, used 2-D gel electrophoresis and pooled vessels. A comprehensive comparison of different methods of preparing smooth muscle tissues for biochemical analysis of LC20 phosphorylation was recently performed by Walsh et al. (2011). This analysis revealed that lower and more variable levels of LC20 phosphorylation are detected using dry ice cooled tongs or solutions compared to the method used here. Specifically, it was suggested that dry ice cooled tongs or solutions may prevent effective quenching within the core of tissue samples and/or the slow warming step may enable residual tissue phosphatase activity and dephosphorylation of LC20 before enzyme denaturation in homogenization buffer or by boiling (Walsh et al. 2011).
In summary, this study indicates that Ca2+ sensitization due to MLCP inhibition by ROK-mediated phosphorylation of MYPT1-T855, and dynamic cytoskeletal reorganization involving increased actin polymerization evoked by ROK- and PKC-mediated signalling pathways contribute to the myogenic response of skeletal muscle resistance arteries. The fact that the myogenic response is blocked by the inhibition of MLCK activity following removal of extracellular Ca2+, suppression of ROK-mediated MLCP inhibition by H1152, or suppression of actin polymerization with H1152 or GF109203X, implies that the appropriate activation of all three pathways for control of MLCK, MLCP and cytoskeletal reorganization is essential for physiological regulation of skeletal muscle resistance arterial diameter (Fig. 11). Differences in pressure dependence and extent of regulation owing to these mechanisms may contribute to vessel-specific heterogeneity in the myogenic response and permit varied patterns of physiological behaviour in different vascular beds.
Figure 11. Schematic diagram depicting the involvement of ROK and PKC in Ca2+ sensitization during the myogenic response of skeletal muscle arterioles.
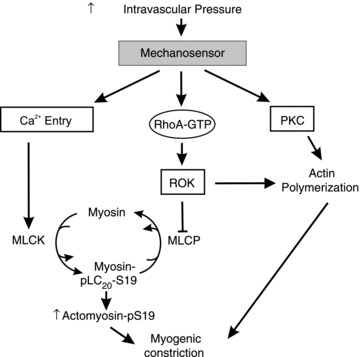
An increase in intraluminal pressure is detected by the mechanosensor, leading to depolarization, Ca2+ influx and activation of MLCK, and the activation of ROK and PKC. ROK, through phosphorylation of MYPT1 (the myosin binding regulatory subunit of MLCP) at T855, inhibits MLCP activity to increase pLC20 and enhance constriction. Both ROK and PKC induce actin polymerization to strengthen connections between the contractile machinery, plasma membrane and extracellular matrix, thereby enhancing force transmission and myogenic constriction. Thus, the mechanisms of ROK-mediated MLCP inhibition, and ROK- and PKC-mediated actin polymerization together result in increased force at constant cytosolic free Ca2+ concentration, i.e. Ca2+ sensitization. MLCP, myosin light chain phosphatase; MLCK, myosin light chain kinase; pLC20, LC20 phosphorylation; PKC, protein kinase C; ROK, Rho-associated kinase.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (MOP-97988). A.M.D., O.C. and A.E.-Y. were supported by the Kertland Family Postdoctoral Fellowship in Vascular Biology, the Spanish Ministry of Education, and the Alberta Heritage Foundation for Medical Research and Canadian Institutes of Health Research, respectively. W.C.C. is the Andrew Family Professor in Cardiovascular Research and M.P.W. is an Alberta Heritage Foundation for Medical Research Scientist and recipient of a Canada Research Chair (Tier 1) in Vascular Smooth Muscle Research. A.E.-Y. is a Lecturer in Pharmacology at the Faculty of Pharmacy at Alexandria University. The authors have no conflicts of interest to disclose.
Glossary
- [Ca2+]i
cytosolic free Ca2+ concentration
- CPI-17
17 kDa PKC-potentiated protein phosphatase 1 inhibitor protein
- DTT
dithiothreitol
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- LC20
20 kDa myosin regulatory light chain subunit
- MYPT1
myosin targeting subunit of myosin light chain phosphatase
- PDBu
phorbol 12,13-dibutyrate
- PE
phenylephrine
- Phos-Tag SDS-PAGE
phosphate-affinity tag SDS-PAGE
- PKC
protein kinase C
- RCrAs
rat cremaster arteries
- RGAs
rat gracilis arteries
- RMCAs
rat middle cerebral arteries
- ROK
Rho-associated kinase
Author contributions
All authors were responsible for the conception and design of experiments, and drafting and critical revision of the manuscript. Data collection and analysis were conducted by A.M.-D., O.C., E.J.W. and A.E.-Y. All authors contributed to the interpretation of the findings and have approved the submitted version of this manuscript. The work was conducted in the laboratory of W.C.C. in the Smooth Muscle Research Group at the University of Calgary.
Supplementary material
Supplemental Figure 1
Supplemental Figure 2
References
- Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Bidani AK, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension. 2009;54:393–398. doi: 10.1161/HYPERTENSIONAHA.109.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke JF, Gokina NI, Osol G. Vascular smooth muscle cell stress as a determinant of cerebral artery myogenic tone. Am J Physiol Heart Circ Physiol. 2002;283:H2210–H2216. doi: 10.1152/ajpheart.00633.2002. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Osol G. Vascular smooth muscle actin cytoskeleton in cerebral artery forced dilatation. Stroke. 1998;29:1223–1228. doi: 10.1161/01.str.29.6.1223. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- Cole WC, Welsh DG. Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys. 2011;510:160–173. doi: 10.1016/j.abb.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Corteling RL, Brett SE, Yin H, Zheng XL, Walsh MP, Welsh DG. The functional consequence of RhoA knockdown by RNA interference in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;293:H440–H447. doi: 10.1152/ajpheart.01374.2006. [DOI] [PubMed] [Google Scholar]
- Coué M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- D’Angelo G, Davis MJ, Meininger GA. Calcium and mechanotransduction of the myogenic response. Am J Physiol Heart Circ Physiol. 1997;273:H175–H182. doi: 10.1152/ajpheart.1997.273.1.H175. [DOI] [PubMed] [Google Scholar]
- Davis MJ. Myogenic response gradient in an arteriolar network. Am J Physiol Heart Circ Physiol. 1993;264:H2168–H2179. doi: 10.1152/ajpheart.1993.264.6.H2168. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- Dessy C, Matsuda N, Hulvershorn J, Sougnez CL, Sellke FW, Morgan KG. Evidence for involvement of the PKC-alpha isoform in myogenic contractions of the coronary microcirculation. Am J Physiol Heart Circ Physiol. 2000;279:H916–H923. doi: 10.1152/ajpheart.2000.279.3.H916. [DOI] [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res. 2007;100:121–129. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA. Does arterial myogenic tone determine blood flow distribution in vivo. Am J Physiol Heart Circ Physiol. 2005;289:H1323–H1325. doi: 10.1152/ajpheart.00513.2005. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubroca C, You D, Levy BI, Loufrani L, Henrion D. Involvement of RhoA/Rho kinase pathway in myogenic tone in the rabbit facial vein. Hypertension. 2005;45:974–979. doi: 10.1161/01.HYP.0000164582.63421.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol. 2010;588:1747–1762. doi: 10.1113/jphysiol.2010.187146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem (Tokyo) 1995;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol. 2005;288:H660–H669. doi: 10.1152/ajpheart.00608.2004. [DOI] [PubMed] [Google Scholar]
- Fronek K, Zweifach BW. Microvascular pressure distribution in skeletal muscle and the effect of vasodilation. Am J Physiol. 1975;228:791–796. doi: 10.1152/ajplegacy.1975.228.3.791. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT. Actin cytoskeletal dynamics in smooth muscle contraction. Can J Physiol Pharmacol. 2005;83:851–856. doi: 10.1139/y05-088. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Osol G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am J Physiol Heart Circ Physiol. 2002;282:H1410–H1420. doi: 10.1152/ajpheart.00441.2001. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Knot HJ, Nelson MT, Osol G. Increased Ca2+ sensitivity as a key mechanism of PKC-induced constriction in pressurized cerebral arteries. Am J Physiol Heart Circ Physiol. 1999;277:H1178–H1188. doi: 10.1152/ajpheart.1999.277.3.H1178. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Park KM, McElroy-Yaggy K, Osol G. Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J Appl Physiol. 2005;98:1940–1948. doi: 10.1152/japplphysiol.01104.2004. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol. 2008;295:C576–C587. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Falcone JC, Meininger GA. Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am J Physiol Heart Circ Physiol. 1990;259:H1586–H1594. doi: 10.1152/ajpheart.1990.259.5.H1586. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- Jarajapu YP, Knot HJ. Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am J Physiol Heart Circ Physiol. 2005;289:H1917–H1922. doi: 10.1152/ajpheart.01012.2004. [DOI] [PubMed] [Google Scholar]
- Johnson PC. Autoregulation of blood flow. Circ Res. 1986;59:483–495. doi: 10.1161/01.res.59.5.483. [DOI] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009;587:2537–2553. doi: 10.1113/jphysiol.2008.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Perkins WJ, Lorenz RR, Prakash YS, Sieck GC, Warner DO. F-actin stabilization increases tension cost during contraction of permeabilized airway smooth muscle in dogs. J Physiol. 1999;519:527–538. doi: 10.1111/j.1469-7793.1999.0527m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karibe A, Watanabe J, Horiguchi S, Takeuchi M, Suzuki S, Funakoshi M, Katoh H, Keitoku M, Satoh S, Shirato K. Role of cytosolic Ca2+ and protein kinase C in developing myogenic contraction in isolated rat small arteries. Am J Physiol Heart Circ Physiol. 1997;272:H1165–H1172. doi: 10.1152/ajpheart.1997.272.3.H1165. [DOI] [PubMed] [Google Scholar]
- Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008;295:C768–C778. doi: 10.1152/ajpcell.00174.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1326–H1334. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- Lagaud G, Gaudreault N, Moore ED, Van Breemen C, Laher I. Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. Am J Physiol Heart Circ Physiol. 2002;283:H2187–H2195. doi: 10.1152/ajpheart.00554.2002. [DOI] [PubMed] [Google Scholar]
- Liu J, Hill MA, Meininger GA. Mechanisms of myogenic enhancement by norepinephrine. Am J Physiol Heart Circ Physiol. 1994;266:H440–H446. doi: 10.1152/ajpheart.1994.266.2.H440. [DOI] [PubMed] [Google Scholar]
- Luykenaar KD, El-Rahman RA, Walsh MP, Welsh DG. Rho-kinase-mediated suppression of KDR current in cerebral arteries requires an intact actin cytoskeleton. Am J Physiol Heart Circ Physiol. 2009;296:H917–H926. doi: 10.1152/ajpheart.01206.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massett MP, Ungvari Z, Csiszar A, Kaley G, Koller A. Different roles of PKC and MAP kinases in arteriolar constrictions to pressure and agonists. Am J Physiol Heart Circ Physiol. 2002;283:H2282–H2287. doi: 10.1152/ajpheart.00544.2002. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Osol G, Halpern W. Myogenic responses are independent of the endothelium in rat pressurized posterior cerebral arteries. Blood Vessels. 1989;26:315–319. doi: 10.1159/000158780. [DOI] [PubMed] [Google Scholar]
- Meininger GA, Zawieja DC, Falcone JC, Hill MA, Davey JP. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am J Physiol Heart Circ Physiol. 1991;261:H950–959. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Hayashi K, Ozawa Y, Fujiwara K, Okubo K, Kanda T, Wakino S, Saruta T. Vessel- and vasoconstrictor-dependent role of Rho/Rho-kinase in renal microvascular tone. J Vasc Res. 2003;40:244–251. doi: 10.1159/000071888. [DOI] [PubMed] [Google Scholar]
- Ohanian V, Gatfield K, Ohanian J. Role of the actin cytoskeleton in G-protein-coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension. 2005;46:93–99. doi: 10.1161/01.HYP.0000167990.82235.3c. [DOI] [PubMed] [Google Scholar]
- Olsen TS, Larsen B, Skriver EB, Herning M, Enevoldsen E, Lassen NA. Focal cerebral hyperemia in acute stroke. Incidence, pathophysiology and clinical significance. Stroke. 1981;12:598–607. doi: 10.1161/01.str.12.5.598. [DOI] [PubMed] [Google Scholar]
- Osol G, Laher I, Cipolla M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ Res. 1991;68:359–367. doi: 10.1161/01.res.68.2.359. [DOI] [PubMed] [Google Scholar]
- Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol. 2002;283:H2260–H2267. doi: 10.1152/ajpheart.00634.2002. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Suzuki M, Hidaka H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol Ther. 2002;93:225–232. doi: 10.1016/s0163-7258(02)00191-2. [DOI] [PubMed] [Google Scholar]
- Schubert R, Mulvany MJ. The myogenic response: established facts and attractive hypotheses. Clin Sci (Lond) 1999;96:313–326. [PubMed] [Google Scholar]
- Schubert R, Kalentchuk VU, Krien U. Rho kinase inhibition partly weakens myogenic reactivity in rat small arteries by changing calcium sensitivity. Am J Physiol Heart Circ Physiol. 2002;283:H2288–H2295. doi: 10.1152/ajpheart.00549.2002. [DOI] [PubMed] [Google Scholar]
- Schubert R, Lidington D, Bolz SS. The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc Res. 2008;77:8–18. doi: 10.1016/j.cardiores.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Shaw L, Ahmed S, Austin C, Taggart M. Inhibitors of actin filament polymerisation attenuate force but not global intracellular calcium in isolated pressurised resistance arteries. J Vasc Res. 2003;40:1–10. doi: 10.1159/000068940. [DOI] [PubMed] [Google Scholar]
- Smeda JS. Stroke development in stroke-prone spontaneously hypertensive rats alters the ability of cerebrovascular muscle to utilize internal Ca2+ to elicit constriction. Stroke. 2003;34:1491–1496. doi: 10.1161/01.STR.0000073797.91891.9E. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sun D, Kaley G, Koller A. Characteristics and origin of myogenic response in isolated gracilis muscle arterioles. Am J Physiol Heart Circ Physiol. 1994;266:H1177–1183. doi: 10.1152/ajpheart.1994.266.3.H1177. [DOI] [PubMed] [Google Scholar]
- Takeya K, Loutzenhiser K, Shiraishi M, Loutzenhiser R, Walsh MP. A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol. 2008;294:F1487–F1492. doi: 10.1152/ajprenal.00060.2008. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Ohta H, Kanda K, Tanaka T, Hidaka H, Sobue K. Phosphorylation of high-Mr caldesmon by protein kinase-C modulates the regulatory function of this protein on the interaction between actin and myosin. Eur J Biochem. 1990;188:495–500. doi: 10.1111/j.1432-1033.1990.tb15427.x. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamell L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–81. [PubMed] [Google Scholar]
- VanBavel E, Wesselman JP, Spaan JA. Myogenic activation and calcium sensitivity of cannulated rat mesenteric small arteries. Circ Res. 1998;82:210–220. doi: 10.1161/01.res.82.2.210. [DOI] [PubMed] [Google Scholar]
- VanBavel E, Van der Meulen ET, Spaan JA. Role of Rho-associated protein kinase in tone and calcium sensitivity of cannulated rat mesenteric small arteries. Exp Physiol. 2001;86:585–592. doi: 10.1113/eph8602217. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Thornbury K, Cole WC, Sergeant G, Hollywood M, McHale N. Rho-associated kinase plays a role in rabbit urethral smooth muscle contraction, but not via enhanced myosin light chain phosphorylation. Am J Physiol Renal Physiol. 2011;300:F73–F85. doi: 10.1152/ajprenal.00011.2010. [DOI] [PubMed] [Google Scholar]
- Wesselman JP, Spaan JA, van der Meulen ET, VanBavel E. Role of protein kinase C in myogenic calcium-contraction coupling of rat cannulated mesenteric small arteries. Clin Exp Pharmacol Physiol. 2001;28:848–855. doi: 10.1046/j.1440-1681.2001.03534.x. [DOI] [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. Smooth muscle calponin: inhibition of actomyosin MgATPase and regulation by phosphorylation. J Biol Chem. 1990;265:10148–10155. [PubMed] [Google Scholar]
- Yang Y, Murphy TV, Ella SR, Grayson TH, Haddock R, Hwang YT, Braun AP, Peichun G, Korthuis RJ, Davis MJ, Hill MA. Heterogeneity in function of small artery smooth muscle BKCa: involvement of the beta1-subunit. J Physiol. 2009;587:3025–3044. doi: 10.1113/jphysiol.2009.169920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeon DS, Kim JS, Ahn DS, Kwon SC, Kang BS, Morgan KG, Lee YH. Role of protein kinase C- or RhoA-induced Ca2+ sensitization in stretch-induced myogenic tone. Cardiovasc Res. 2002;53:431–438. doi: 10.1016/s0008-6363(01)00496-5. [DOI] [PubMed] [Google Scholar]
- Zou H, Ratz PH, Hill MA. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am J Physiol Heart Circ Physiol. 1995;269:H1590–H1596. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.