

Abstract
β-Cell mass increases during pregnancy in adaptation to the insulin resistance of pregnancy. This increase is accompanied by an increase in β-cell proliferation, a process that requires intact prolactin receptor (Prlr) signalling. Previously, it was found that during pregnancy, heterozygous prolactin receptor-null (Prlr+/−) mice had lower number of β-cells, lower serum insulin and higher blood glucose levels than wild-type (Prlr+/+) mice. An unexpected observation was that the glucose homeostasis of the experimental mouse depends on the genotype of her mother, such that within the Prlr+/+ group, the Prlr+/+ offspring derived from Prlr+/+ mothers (Prlr+/+(+/+)) had higher β-cell mass and lower blood glucose than those derived from Prlr+/− mothers (Prlr+/+(+/−)). Pathways that are known to regulate β-cell proliferation during pregnancy include insulin receptor substrate-2, Akt, menin, the serotonin synthetic enzyme tryptophan hydroxylase-1, Forkhead box M1 and Forkhead box D3. The aim of the present study was to determine whether dysregulation in these signalling molecules in the islets could explain the maternal effect on the phenotype of the offspring. It was found that the pregnancy-induced increases in insulin receptor substrate-2 and Akt expression in the islets were attenuated in the Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice. The expression of Forkhead box D3, which plays a permissive role for β-cell proliferation during pregnancy, was also lower in the Prlr+/+(+/−) mice. In contrast, the pregnancy-induced increases in phospho-Jak2, tryptophan hydroxylase-1 and FoxM1, as well as the pregnancy-associated reduction in menin expression, were comparable between the two groups. There was also no difference in expression levels of genes that regulate insulin synthesis and secretion (i.e. glucose transporter 2, glucokinase and pancreatic and duodenal homeobox-1) between these two groups. Taken together, these results suggest that the in utero environment of the Prlr+/− mother confers long-term changes in the pancreatic islets of her offspring such that when the offspring themselves became pregnant, they cannot adapt to the increased insulin demands of their own pregnancy.
Key points
Heterozygous prolactin receptor-null mice (Prlr+/−) are glucose intolerant during pregnancy. Their wild-type offspring are at greater risk of developing glucose intolerance during their own pregnancy.
The impaired glucose tolerance of the offspring during her own pregnancy is associated with a lower β-cell mass, as well as a reduction in expression of insulin receptor substrate-2, Akt and Forkhead box D3 in the islets, in comparison to mice born to wild-type mothers.
Signalling molecules that mediate prolactin-specific signals, such as Jak/Stat, tryptophan hydroxylase-1, menin and Forkhead box M1, are expressed in comparable levels between wild-type offspring derived from either wild-type or heterozygous prolactin receptor-null mothers.
The glucose-stimulated insulin secretory response during pregnancy is minimally affected in mice derived from heterozygous prolactin receptor-null mothers.
Introduction
Insulin resistance develops during pregnancy as a physiological response to shunt nutrients to the fetus. In order to prevent the development of diabetes, pregnant females adapt by increasing their β-cell mass (Sorenson & Brelje, 1997). Previous studies from our laboratory and others have convincingly demonstrated that pregnancy hormones, such as prolactin and placental lactogens, acting through the prolactin receptor (Prlr), are required for the increase in β-cell proliferation and β-cell mass observed in normal pregnancy (Sorenson & Brelje, 1997, 2009; Huang et al. 2009). We found that in comparison to the wild-type (Prlr+/+) mice, mice with heterozygous deletion of the prolactin receptor (Prlr+/−) had a lower β-cell proliferation rate accompanied by a lower β-cell mass, which accounted for the lower serum insulin concentrations and higher blood glucose in the Prlr+/− mice, both during the non-fasting state and during an intraperitoneal glucose tolerance test (Huang et al. 2009). An interesting observation from that study was the impact of maternal genotype on the phenotype of her offspring during the offspring's own pregnancy. Wild-type mice derived from heterozygous Prlr-null mothers (Prlr+/+(+/−)) had a lower insulin level, a higher glucose level and a lower β-cell mass than wild-type mice derived from wild-type mothers (Prlr+/+(+/+)). In fact, the phenotype of the Prlr+/+(+/−) mice is intermediate between that of the Prlr+/+ and Prlr+/− mice (Huang et al. 2009). This observation suggests that exposure to the Prlr+/− environment in utero somehow compromises the ability of the pancreas of the offspring to adapt during her own pregnancy.
Both human (Kasperska-Czyzyk et al. 1996; Dabelea et al. 2000a,b) and animal studies (Bihoreau et al. 1986; Aerts et al. 1997; Han et al. 2007; Pinney & Simmons, 2010) have shown that exposure to gestational diabetes increases the risk of abnormal glucose homeostasis in the offspring. Early animal studies clearly demonstrated that in utero exposure to hyperglycaemia and/or hyperinsulinaemia were associated with impaired glucose tolerance in the offspring (Bihoreau et al. 1986; Gauguier et al. 1990; Aerts et al. 1997). Furthermore, some of these pups were born with altered pancreatic islet structure (Aerts & Van Assche, 1998), and these islets did not increase in size and function to accommodate for pregnancy-induced insulin resistance, resulting in gestational diabetes, leading to the vicious cycle of ‘gestational diabetes begets more gestational diabetes’ (Aerts et al. 1997). Mechanisms that determine how the intrauterine environment affects the ability of the pancreatic islets of the offspring to adapt to the increased metabolic demands during her own pregnancy are not known.
Significant efforts have been directed at deciphering the molecular mechanisms regulating β-cell proliferation during pregnancy. The insulin receptor substrate-2 (IRS-2)/Akt/p21cip (Kubota et al. 2000; Dickson & Rhodes, 2004; Cozar-Castellano et al. 2006a,b; Fatrai et al. 2006; Hughes & Huang, 2011), the Jak/Stat (Brelje et al. 2002, 2004) and the menin/p27/p18 pathways (Karnik et al. 2007), as well as the serotonin synthetic enzyme, tryptophan hydroxylase-1 (Tph1; Kim et al. 2010; Schraenen et al. 2010), and the transcription factors Forkhead box M1 (FoxM1; Zhang et al. 2009) and Forkhead box D3 (Foxd3; Plank et al. 2011), have all been shown to regulate β-cell proliferation during pregnancy. Foxd3 is unique in this context because its primary known function is to regulate embryonic stem cell self-renewal. It is expressed in pancreatic β-cells, and its expression drops during pregnancy. However, Fox3-null mice develop gestational diabetes, which is caused by a reduction in β-cell proliferation during pregnancy (Plank et al. 2011). Hence, it was proposed that Foxd3 plays a permissive role for β-cell proliferation during pregnancy. Here, it is reported that in comparison to the Prlr+/+(+/+) mice, the Prlr+/+(+/−) mice had lower IRS-2 and Akt expression in the islets during pregnancy, but expression of other signalling molecules was not perturbed. In addition, the expression of Foxd3 was reduced in the non-pregnant Prlr+/+(+/−) mice.
Methods
Ethical approval
All experimental procedures were approved by the Animal Use Review Committee at the University of Calgary in accordance with standards of the Canadian Council on Animal Care.
Mice
Heterozygous prolactin receptor-null (Prlr+/−) mice on a C57BL/6 background were purchased from Jackson Laboratories (West Grove, PA, USA), and working mouse stock was generated by crossing Prlr+/− with wild-type Prlr+/+ mice. The pups were genotyped as previously described (Bouchard et al. 1999). Mice were maintained on a 12 h light–12 h dark cycle, with liberal access to food and water. Mice were studied at 3–4 months of age. Mice were killed by cervical dislocation, followed by isolation of the pancreas or islets. Messenger RNA from islets of six to eight mice (from 5–7 separate litters) was used for real-time RT-PCR experiments, while total protein from islets of six to 22 mice (from 5–19 separate litters) was used for Western immunoblotting studies. Mice were used on days 0 and 15 of pregnancy. Day 15 of pregnancy was chosen for study because β-cell proliferation peaks on days 14–15 of pregnancy (Sorenson & Brelje, 1997). Non-fasted blood glucose was determined using a glucometer (OneTouch FastTake) (by Lifescan; Burnaby, BC, Canada) by sampling from the tail vein at 08.00 h and, at the same time, an additional 30 μl of serum was taken and stored at −80°C for later measurement of insulin by ensyme-linked immunosorbent assay (ELISA). An intraperitoneal glucose tolerance test (IPGTT) was performed after a 14–16 h overnight fast. Mice were injected with 2 g (kg body weight)-1 of glucose (20%d-glucose solution) intraperitoneally, followed by blood sampling from the tail vein for blood glucose at 0, 15, 30, 45, 60 and 120 min after the injection, using a glucometer (One Touch FastTake). An additional blood sample (30 μl) was obtained at 0 and 30 min for measurement of serum insulin, using an ELISA kit (catalogue no. 80-INSMSU-E01; Alpco (Salem, NH, USA)).
Materials
The protease inhibitor ‘COMPLETE’ Mini Tablet was from Roche USA (Indianapolis, IN, USA). Collagenase P (ref. no. 11 213 865 001) was from Roche Germany (Mannheim, Germany). Dextran (D-4751), trypsin 1 mg tablets (T7168) and all chemicals were from Sigma (Oakville, ON, Canada). Sources of antibodies are specified.
Immunohistochemistry
Mice were injected with bromodeoxyuridine (BrdU; 100 mg (kg body weight)−1) 4 h before isolation of the pancreas. The pancreas was isolated from pregnant and non-pregnant mice, weighed, then fixed in 4% paraformaldehyde–PBS solution at 4°C over night, embedded in paraffin blocks and cut into longitudinal serial sections 7 μm thick. Every 40th tissue section was stained for insulin to identify β-cells. This provides at least 280 μm distance between sections stained, minimizing the possibility of sampling the same islet twice. Antigen retrieval was achieved by incubating tissue sections with 1 mg ml−1 of trypsin (Sigma) at 37°C for 20 min. After 1 h of blocking with 1% goat serum–PBS at room temperature, tissues were incubated with primary antibody over night at 4°C (guinea-pig anti-insulin at 1:750 dilution from DAKO (Carpinteria, CA, USA); rabbit anti-glucagon at 1:500 dilution from Linco (St. Charles, MO, USA); and mouse anti-BrdU at 1:500 dilution from Sigma; all diluted in 1% goat serum–PBS). This was followed by 1 h incubation with fluorophore-conjugated secondary antibodies (Cy3-anti-guinea-pig from Jackson Laboratories; and Alexa-488 goat anti-rabbit from Molecular Probes (Eugene, OR, USA); both diluted in 1% goat serum–PBS at 1:300). Bis-benzimide H 33342 trihydrochloride (0.1 μg ml−1; Sigma) was added to the secondary antibody for nuclear staining. Stained sections were mounted using a fluorescent mounting medium from DakoCytomation (Carpinteria, CA, USA) and stored at 4°C.
Islet morphometry
For each pancreatic section, consecutive images of adjacent non-overlapping areas of the entire section were acquired using a Leica fluorescence microscope (Wetzlar, Germany), and captured with a CoolSnap digital camera (Tucson, AZ, USA) (Teta et al. 2005). Images were analysed by ImageJ software http://rsbweb.nih.gov/ij/download.html to measure the insulin-positive area and the area of the entire pancreatic section (identified by nuclear staining). β-Cell mass was calculated by multiplying the pancreatic weight by the β-cell fraction (i.e. the ratio of insulin-positive cell area to total pancreatic tissue area on the entire section; Freemark et al. 2002). Results represent the average of four or five tissue sections per animal from five or six animals of each genotype.
Islet isolation
Pancreatic islets were isolated from non-fasted adult pregnant female mice. Immediately postmortem, the pancreas was distended using collagenase P [0.66 mg ml−1 in Hank's Balanced Salt Solution (HBSS; Gibco, Burlington, ON, Canada); 2.5 ml/pancreas], excised and incubated at 37°C for 15 min with constant agitation. The digested pancreas was passed through a sterile 500 μm filter and then separated from exocrine tissue by centrifugation on a dextran gradient (1.100, 1.085, 1.075 and 1.045 g ml−1). Islets were picked off the 1.085–1.075 and 1.075–1.045 g ml−1 gradient interfaces, washed with HBSS (supplemented with 0.25% fraction V bovine serum albumin; Sigma), then lysed for protein determination in lysis buffer (Hughes & Huang, 2011).
Analysis of protein expression
Whole-cell protein extracts were obtained by disrupting the isolated islets in lysis buffer (2% SDS, 125 mm Tris, pH 7, 1 mm DTT in PBS, plus protease inhibitors, i.e. COMPLETE Mini Tablets, 50 mm NaF, 10 nm okadaic acid, 1 mm Na3VO4 and 1 mm phenylmethylsulfonyl fluoride; 200 islets (100 μl)-1), sonicated three times for 30 s, followed by determination of the protein concentration using the Bradford method. Protein was separated by SDS-PAGE (30 μg for menin and 10 μg for all other molecules) and transferred onto polyvinylidene difluoride filters, blocked in 3% bovine serum albumin at room temperature for 1 h, then incubation with primary antibody (rabbit anti-Akt antibody at 1:1000 dilution from Cell Signaling (Danvers, MA, USA); rabbit anti-phospho-Akt (Ser473)(D9E) antibody at 1:2000 dilution from Cell Signaling; rabbit anti-IRS-2 at 1:500 dilution from Millipore (Billerica, MA, USA); goat anti-menin antibody at 1:1000 dilution from Bethyl laboratories (Montgomery, TX, USA); rabbit anti-cyclin D2 at 1:1000 dilution from Santa Cruz (Santa Cruz, CA, USA); rabbit anti-p21 at 1:500 dilution from Santa Cruz; and rabbit anti-actin antibody at 1:1000 dilution from Santa Cruz) at 4°C overnight, followed by a 1 h incubation with horseradish peroxidase-conjugated secondary antibody (sheep anti-mouse antibody at 1:10,000 dilution from Amersham (Little Chalfont, Buckinghamshire, UK); donkey anti-rabbit antibody at 1:10,000 dilution from Amersham; and donkey anti-goat at 1:5000 to 1:10,000 dilution from Jackson Laboratories). Protein was visualized by the enhanced chemiluminescence method and scanned within the linear range using ImageJ software. The expression level of each protein was normalized to that of actin, which was used as a gel loading control. Each islet sample was assayed two or three times on separate SDS-PAGE gels, depending on the amount of protein available from each mouse, and the results for each mouse from the separate experiments were averaged. The ‘n’ value listed in the Results sections and figure legends represents the number of animals used.
Islet RNA isolation and quantitative real-time RT-PCR
Total islet RNA (100–200 islets per mouse) was extracted using the RNeasy Mini Kit (Qiagen; Mississauga, ON, Canada). The RNA concentration and integrity were assessed using the ND-1000 Spectrophotometer (NanoDrop by Thermo Scientific, Wilmington, DE, USA). Complementary DNA was synthesized using the Quantitect Reverse Transcription Kit (Qiagen). Reactions were carried out in triplicate with QuantiFast SYBR Green Master Mix (Qiagen) at an annealing temperature of 60°C. Data were collected using the DNA Engine Opticon2 Continuous Fluorescence Detection System and software (Bio-Rad; Philadelphia, PA, USA). The sequences for the primers were previously published (Zhang et al. 2009; Kim et al. 2010; Wang et al. 2010). The relative amount of RNA was determined by comparison with phosphoglycerate kinase 1 mRNA as a reference gene. Phosphoglycerate kinase 1 was chosen after testing >10 genes from Qiagen's reference gene panel; phosphoglycerate kinase 1 showed the least variability of all the genes tested (which included other commonly used reference genes, such as Hprt and cyclophilin A). The primer sequences used were as follows: Tph1, 5′-GATGGACAGCTGAGAGTCTTTGG-3′ (forward) and 5′-TGTCCAGAAAGTGCATGTTTGAG-3′ (reverse; Kim et al. 2010); pancreatic and duodenal homeobox-1 (Pdx1), 5′-AGGTCACCGCACAATCTTGCT-3′ (forward) and 5′- CTTTCCCGAATGGAACCGA-3′ (reverse; Xu et al. 2008); Htr2b, 5′-CCTCATCAATCATCCTCCTCG-3′ (forward) and 5′-CTGACCTGCTCTTCCGCTTT-3′ (reverse); and FoxM1, 5′-CACTTGGATTGAGGACCACTT-3′ (forward) and 5′-GTCGTTT CTGCTGTGATTCC -3′ (reverse; Zhang et al. 2009). Primers for glucokinase (QT00140007), glucose transporter 2 (GLUT2; QT00103537), and Foxd3 (QT01749951) were designed and supplied by Qiagen. Primers were validated by melt curve analysis (i.e. the dissociation curve contained a single peak with no shoulders) and that the amplified product contained a single gene product of the predicted amplicon length.
Statistical analysis
All statistics were performed using GraphPad Prism 4 software (by GraphPad software; San Diego, CA, USA). Two-tailed Student's unpaired t tests or two-way ANOVA with Bonferroni post hoc tests were performed where appropriate. Comparisons were made between the Prlr+/+(+/+) and the Prlr+/+(+/−) mice on day 0 or day 15 of pregnancy, as stated in the figure legends.
Results
Wild-type offspring of Prlr+/− mothers have higher serum glucose levels, lower serum insulin concentrations and reduced β-cell mass during their own pregnancy
Using mice with a heterozygous deletion of the prolactin receptor (Prlr+/−), we previously reported that the Prlr, which is a receptor for both prolactin and placental lactogens, is required for maintenance of normal glucose homeostasis during pregnancy (Huang et al. 2009). Interestingly, it was noted that the genotype of the mother has an impact on the phenotype of her offspring during the offspring's pregnancy, such that on day 15 of pregnancy Prlr+/+ mice derived from a Prlr+/+ mother (Prlr+/+(+/+)) had a lower non-fasted blood glucose level (7.71 ± 0.30 mmol l−1, n= 18, from 15 separate litters) than Prlr+/+ mice derived from a Prlr+/− mother (Prlr+/+(+/−); 8.66 ± 0.35 mmol l−1, n= 12, from 9 separate litters; P= 0.05; Fig. 1A). In the present study, day 15 of pregnancy was chosen for investigation because β-cell proliferation peaks on days 14–15 of pregnancy, as well to be consistent with the previous studies.
Figure 1. Effects of maternal genotype on the serum glucose and insulin concentrations of the offspring during the offspring's pregnancy.
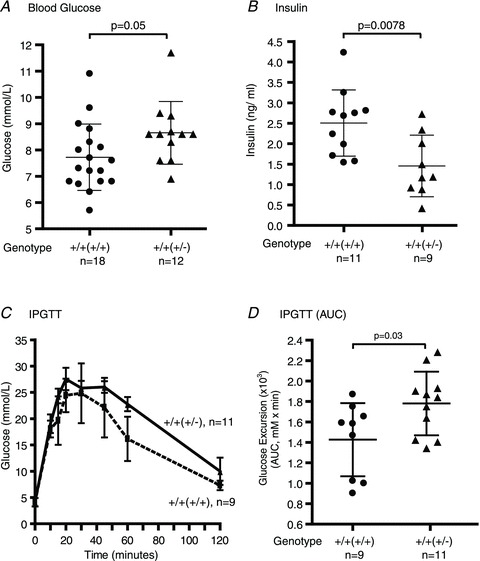
Non-fasted serum glucose [A; Prlr+/+(+/+), n= 18 (from 15 litters) and Prlr+/+(+/−), n= 12 (from 9 litters)] and insulin concentrations [B; Prlr+/+(+/+), n= 11 (from 9 litters) and Prlr+/+(+/−), n= 9 (from 8 litters)] were measured at 08.00 h on day 15 of pregnancy. Results are presented as means (±SD) and compared by Student's unpaired t test. C, an intraperitoneal glucose tolerance test (IPGTT) was performed after an overnight fast on 9–11 mice (from 5–9 litters). Results at each time point are expressed as the mean (±SD). Comparison between the two groups was made by two-way ANOVA. D, glucose excursion during an IPGTT is calculated as the area under the curve (AUC; millimolar glucose × time in minutes), and the results from 9–11 animals (from 5–9 litters) are presented as means (±SD). The genotype of the mother from which the experimental mouse was derived is in parentheses; ‘+/+(+/+)’indicates a wild-type mouse derived from wild-type mother and ‘+/+(+/−)’ indicates a wild-type mouse derived from heterozygous prolactin receptor-null mother.
When the mice were challenged with a glucose load during the IPGTT, there was a trend for higher glucose excursion in the Prlr+/+(+/−) mice (P= 0.0597 by two-way ANOVA). Glucose excursion, measured as integrated area under the curve (AUC) across the time points, was higher in the Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice (Fig. 1D; 1731 ± 93 vs. 1427 ± 119 min × mm; P= 0.03 by Student's unpaired t test; n= 9–11 from 5–9 separate litters). The Prlr+/+(+/−) mice also had a lower non-fasted serum insulin level (1.44 ± 0.25 ng ml−1, n= 9, from 8 separate litters) than the Prlr+/+(+/+) mice (2.50 ± 0.24 ng ml−1, n= 11, from 9 separate litters; P= 0.0078; Fig. 1B). To determine the cause of this difference, the β-cell mass was measured. It was found that in non-pregnant mice, there is no difference in β-cell mass between the Prlr+/+(+/+) and Prlr+/+(+/−) mice. During pregnancy, however, the Prlr+/+(+/−) mice had a lower β-cell mass (2.81 ± 0.32 mg, n= 5, from 4 separate litters) in comparison to the Prlr+/+(+/+) mice (3.96 ± 0.47 mg, n= 5, from 3 separate litters; P= 0.025; Fig. 2A). The reduced β-cell mass in the Prlr+/+(+/−) mice was accompanied by a lower β-cell proliferation rate, as measured by the percentage of β-cells that incorporated BrdU, in comparison to that of the Prlr+/+(+/+) mice (0.14 ± 0.03 vs. 0.24 ± 0.03%, n= 5–6, from 4 separate litters; P= 0.045; Fig. 2B). It is important to note that the Prlr+/+(+/+) and the Prlr+/+(+/−) mice had similar insulin sensitivities, because both dropped their glucose levels by 40% in response to insulin during an insulin tolerance test, as we reported in an earlier study (Huang et al. 2009).
Figure 2. β-Cell mass and proliferation rate on day 15 of pregnancy.
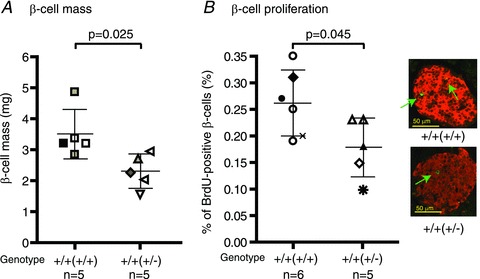
A, β-cell mass was calculated as the β-cell fraction (defined as the percentage area of the pancreas section stained positive for insulin) multiplied by the weight of the pancreas. n= 5 mice. B, β-cell proliferation was determined by counting the percentage of β-cells that also stained positive for bromodeoxyuridine (BrdU). n= 5–6 mice. For both A and B, each symbol represents a single mouse from a different litter, and results are summarized as means (±SD), and compared by Student's unpaired t test. The genotype of the mother from which the experimental mouse was derived is in parentheses. A representative image of insulin (red) and BrdU (green) co-staining is also presented. Green arrows point to β-cells stained positive for BrdU.
Mice born to Prlr+/− mothers had a lower level of Akt and IRS-2 in their islets during pregnancy
In order to understand the mechanism underlying the difference in serum glucose, insulin concentration and β-cell mass between the Prlr+/+(+/−) and Prlr+/+(+/+) mice, the expression levels of signalling molecules that regulate β-cell mass and function in isolated islets were examined. Previously, we reported that the IRS-2/Akt, Jak/Stat and the menin signalling pathways participated in a prolactin-mediated increase in β-cell mass during pregnancy (Hughes & Huang, 2011). Here, it was found that during pregnancy, there was a 2.1-fold increase in IRS-2 expression in the Prlr+/+(+/+) mice, whereas in the Prlr+/+(+/−) mice this was blunted, at 1.48-fold (Fig. 3A). Downstream of IRS-2, Akt expression increased by 2.1-fold in the Prlr+/+(+/+) mice during pregnancy, a response that was blunted in the Prlr+/+(+/−) mice, at 1.24-fold (Fig. 3B). When Akt activation (as measured by an increase in phospho-Akt expression) was examined during pregnancy, it was found that the pregnancy-induced increase in phospho-Akt expression was reduced in the Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice (Fig. 3C).
Figure 3. Effects of maternal genotype on insulin receptor substrate-2 (IRS-2) and Akt expression in the islets of the offspring.

Protein expression levels of IRS-2 (A), Akt (B) and phospho-Akt (C) were quantified by Western immunoblotting. Each lane represents an individual mouse. The optical density of the IRS-2/Akt/phospho-Akt bands was normalized to that of actin, which was used as the loading control. Results are presented as means (±SEM) of all animals within each group, and comparison was made between Prlr+/+(+/+) and Prlr+/+(+/−) mice on day 15 of pregnancy by Student's unpaired t test. For IRS-2, 10–13 mice were used (as indicated in the figure), and they came from 8or 9 litters within each group. For Akt, samples from 20–23 mice were used, and they came from 12–20 litters (except for the group ‘Prlr+/+(+/−) G0’, which included 11 mice from 11 litters). For phospho-Akt, samples from 20–22 mice were used, and they came from 11–18 litters (except for the group ‘Prlr+/+(+/−) G0’, which included 10 mice from 6 litters). Abbreviations: G0, non-pregnant; and G15, day 15 of pregnancy.
Prolactin-responsive genes that participate in upregulation of β-cell proliferation during pregnancy are minimally affected by maternal genotype
Next, the expression levels of signalling molecules that regulate β-cell mass during pregnancy or are regulated by placental hormones were examined, i.e. Jak2, menin, p21cip, the cyclins, Tph1, FoxM1 and Foxd3.
First, no difference in phospho-Jak2 expression was detected in the pancreatic islets between Prlr+/+(+/+) and Prlr+/+(+/−) mice on days 0 and 15 of pregnancy (Fig. 4A). Next, it was found that the pregnancy-induced reduction in menin expression in the islets was comparable between the Prlr+/+(+/+) and the Prlr+/+(+/−) mice (Fig. 4B). When the expression levels of cell cycle proteins was examined, it was found that cyclin D2 increased slightly in both Prlr+/+(+/−) and Prlr+/+(+/+) mice during pregnancy (Fig. 4D). Likewise, the pregnancy-associated increase in p21cip expression was not significantly different between the Prlr+/+(+/+) and the Prlr+/+(+/−) mice (Fig. 4C).
Figure 4. Effects of maternal genotype on phospho-Jak2, menin, p21 and cyclin D2 expression in the islets of the offspring.
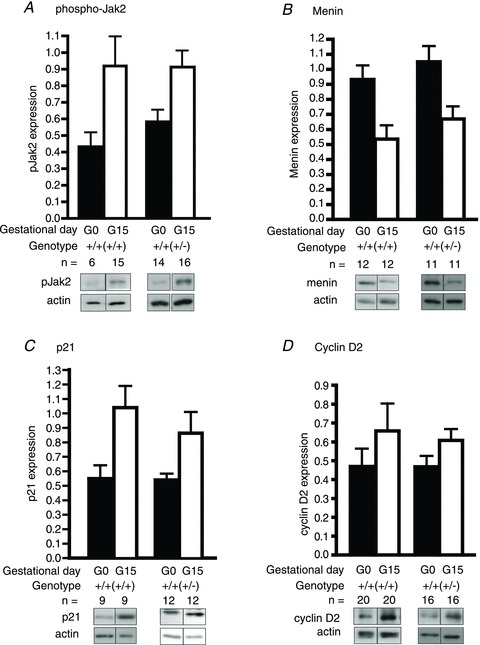
Protein expression levels of phospho-Jak2 (A; n= 6–16 from 5–12 litters), menin (B; n= 11–12 from 8–9 litters), p21 (C; n= 9–12 from 7–9 litters) and cyclin D2 (D; n= 16–20 from 10–14 litters) were determined by Western immunoblotting. Each lane represents an individual mouse. The optical density of the phospho-Jak2/menin/p21/cyclin D2 bands was normalized to that of actin. Results are presented as means (±SEM) of all animals within each group on days 0 or 15 (G0 and G15) of pregnancy, and comparison was made between Prlr+/+(+/+) and Prlr+/+(+/−) mice on G15 using Student's unpaired t test. The genotype of the mother from which the experimental mouse was derived is in parentheses.
Tryptophan hydroxylase-1, the serotonin synthetic enzyme, is one of the highest expressing genes in the islets in response to pregnancy (Kim et al. 2010), and it was found that indeed, its expression increased sharply during pregnancy in both the Prlr+/+(+/+) and the Prlr+/+(+/−) mice to a comparable level (Fig. 5A). Expression of FoxM1, another transcription factor that is important for regulation of β-cell mass during pregnancy (Zhang et al. 2009), was also comparable between the Prlr+/+(+/+) and the Prlr+/+(+/−) mice during pregnancy (Fig. 5B).
Figure 5. Effects of maternal genotype on tryptophan hydroxylase-1 (Tph1), FoxM1 and Forkhead box D3 (Foxd3) expression in the islets of the offspring.
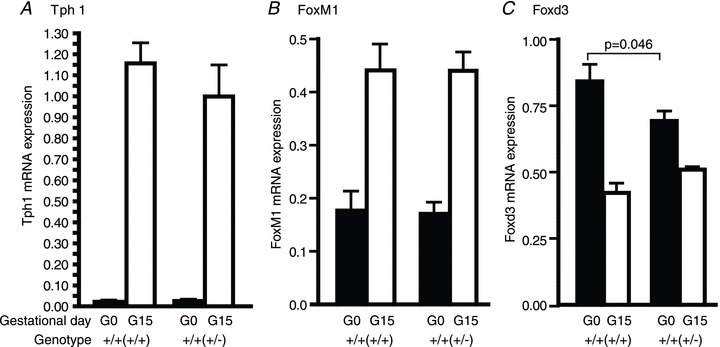
Messenger RNA expressions of Tph1 (A), FoxM1 (B) and Foxd3 (C)in islets isolated from non-pregnant and pregnant (day 15) Prlr+/+(+/+) and Prlr+/+(+/−) mice were determined by quantitative RT-PCR. The relative amount of RNA was determined by comparison with phosphoglycerate kinase (Pkg1) mRNA as a reference gene. Results are presented as means (±SEM) of 8–10 mice (from 5–6 litters), and comparisons are made between Prlr+/+(+/+) and Prlr+/+(+/−) mice on days 0 or 15 of pregnancy. The genotype of the mother from which the experimental mouse was derived is in parentheses.
A newly identified regulator of β-cell mass in pregnancy is the transcription factor Foxd3, which plays a permissive role and allows β-cell proliferation during pregnancy (Plank et al. 2011). Here, it was found that in comparison to the Prlr+/+(+/+) mice, the Prlr+/+(+/−) mice had a lower level of Foxd3 in the non-pregnant state, which may contribute in part to the lower β-cell proliferation rate in the Prlr+/+(+/−) mice (Fig. 5C).
Glucose-stimulated insulin secretion is minimally affected by maternal genotype
To determine whether the lower serum insulin concentration of the Prlr+/+(+/−) mice is solely explained by the lower β-cell mass, or if there is a defect in insulin secretion, insulin release in response to glucose was measured in vivo. The Prlr+/+(+/−) mice had a lower non-fasting (Fig. 1B) and fasting insulin concentration in comparison to the Prlr+/+(+/+) mice on day 15 of pregnancy (Table 1). During an IPGTT, insulin concentrations increased 2-fold in both the Prlr+/+(+/+) and the Prlr+/+(+/−) mice. There was a significant difference in insulin concentration between 0 and 30 min within each group, and at both time points the Prlr+/+(+/−) mice had lower insulin concentrations than the Prlr+/+(+/+) mice; however, there was no interaction between these two variables and it was not possible to detect a statistically significant difference in the change in insulin concentration in response to the glucose load (Table 1). To investigate the possibility that the lower serum insulin concentration in the Prlr+/+(+/−) mice was due to a decreased ability to sense glucose, the expression of GLUT2 was investigated. It was found that GLUT2 expressions were comparable between the Prlr+/+(+/+) and Prlr+/+(+/−) mice, both before and during pregnancy (Fig. 6A). Glucokinase, the rate-limiting enzyme for glucose metabolism, was also expressed at similar levels in the Prlr+/+(+/+) and the Prlr+/+(+/−) mice on day 15 of pregnancy (Fig. 6B). Likewise, pancreatic and duodenal homeobox-1 (Pdx-1), which is important for maintaining β-cell mass (Stoffers et al. 1997; Ackermann & Gannon, 2007) and for regulating insulin gene transcription (Iype et al. 2005), was expressed at similar levels in the pregnant Prlr+/+(+/+) and Prlr+/+(+/−) mice. Interestingly, a modest (but statistically significant) reduction was observed in glucokinase and Pdx-1 expression in the virgin Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice, although this difference no longer existed during pregnancy (Fig. 6B and C).
Table 1.
Effects of maternal genotype on insulin secretion
| Insulin concentration (ng ml-1) | |||
|---|---|---|---|
| Genotype | 0 (min) | 30 (min) | Change (30 min – 0 min) |
| Prlr+/+(+/+) (n= 9) | 0.51 ± 0.09 | 1.00 ± 0.13 | 0.49 ± 0.08 |
| Prlr+/+(+/−) (n= 10) | 0.22 ± 0.04 | 0.45 ± 0.07 | 0.24 ± 0.04 |
| P value | <0.05 | <0.01 | n.s. |
Insulin concentrations were measured at 0 and 30 min of an intraperitoneal glucose tolerance test (2 g of glucose (kg body weight)−1) after an overnight fast. Results are presented as means ± SEM and analysed by two-way ANOVA.
Figure 6. Effects of maternal genotype on mRNA expressions of glucose transporter 2 (GLUT2; A), glucokinase (B) and pancreatic and duodenal homeobox-1 (Pdx-1; C) in islets isolated from non-pregnant and pregnant (day 15) Prlr+/+(+/+) and Prlr+/+(+/−) mice were determined by quantitative RT-PCR.
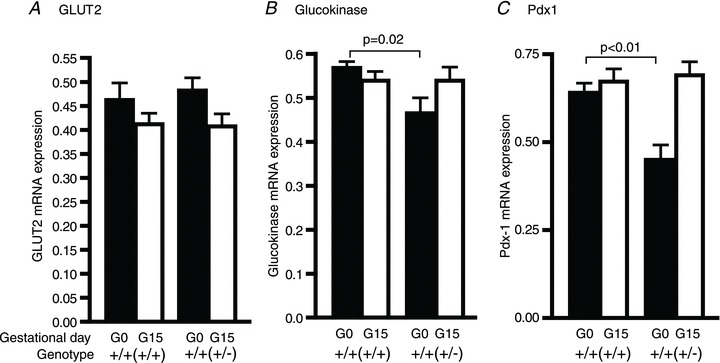
The relative amount of RNA was determined by comparison with phosphoglycerate kinase (Pkg1) mRNA as a reference gene. Results represent means (±SEM) of 8–10 mice (from 5–6 litters), and comparisons are made between Prlr+/+(+/+) and Prlr+/+(+/−) mice on days 0 or 15 of pregnancy. The genotype of the mother from which the experimental mouse was derived is in parentheses.
Discussion
Previous work from this laboratory has shown that the prolactin receptor is required for β-cell proliferation and maintenance of normal glucose homeostasis during pregnancy (Huang et al. 2009). An unexpected observation was that the phenotype of the wild-type mouse depends on the genotype of her mother. In comparison to wild-type (Prlr+/+) mice derived from Prlr+/+ mothers (i.e. Prlr+/+(+/+) mice), Prlr+/+ mice derived from Prlr+/− mothers (i.e. Prlr+/+(+/−) mice) have significantly higher blood glucose with lower serum insulin concentrations during pregnancy, which is associated with a reduction in pancreatic islet mass and β-cell proliferation. Therefore, the goal of the present study was to decipher the potential mechanisms underlying this transgenerational effect of the maternal genotype on β-cell mass and function of her offspring. It is important to note that the experimental mice were derived from many separate litters, because each Prlr mouse typically has only six to eight pups per litter, and most litters give rise to only one or two wild-type female mice for these experiments.
The insulin signalling pathway (insulin receptor → insulin receptor substrate → phosphoinositol-3-kinase → Akt) is required for normal β-cell function and glucose-stimulated insulin secretion (Withers et al. 1999; Dickson & Rhodes, 2004; Taniguchi et al. 2006). Here, it is reported that the pregnancy-associated increase in IRS-2 and Akt expression was decreased in the Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice (Fig. 3). A potential cause of this blunted Akt expression is the transcription factor, Foxd3. Forkhead box D3 regulates embryonic stem cell self-renewal, and its expression is normally downregulated in the pancreatic β-cell during pregnancy (Plank et al. 2011). β-Cell-specific knockdown of Foxd3 has no impact on glucose homeostasis in basal physiological conditions, but during pregnancy the Foxd3-null mice have glucose intolerance, reduced β-cell mass, blunted β-cell proliferation and lower serum insulin concentration in comparison to the wild-type mice (Plank et al. 2011). Given that the Foxd3-null mice have glucose intolerance during pregnancy despite the normal downregulation of Foxd3 expression during pregnancy, the study by Plank et al. (2011) suggested that Foxd3 plays a permissive role in the non-pregnant state to allow for β-cell proliferation during pregnancy. One of the downregulated genes in the Foxd3-null mice was Akt. In the present study, it was found that the non-pregnant Prlr+/+(+/−) mice had a lower Foxd3 expression than the Prlr+/+(+/+) mice, while Foxd3 expression was reduced to similar levels during pregnancy in both Prlr+/+(+/+) and Prlr+/+(+/−) mice (Fig. 5C). It is possible that the blunted Akt expression observed in the Prlr+/+(+/−) mice is explained in part by the lower Foxd3 expression level in these mice, and contributes to the observed reduction in β-cell mass. This reduced Foxd3 level is unlikely to explain the reduction in IRS-2, because the Foxd3-null mice had normal IRS-2 expression levels.
Other genes that are known to regulate β-cell mass in response to prolactin/placental hormones include the genes for FoxM1, the tumor suppressor menin, cyclin inhibitory protein p21cip and the serotonin synthetic enzyme, Tph1. Transgenic mice with reduced FoxM1 expression developed gestational diabetes accompanied by reduced β-cell proliferation and β-cell mass (Zhang et al. 2009). Interestingly, β-cells of FoxM1-null mice had higher menin levels, and overexpression of menin has been shown to cause gestational diabetes (Karnik et al. 2007). Indeed, during pregnancy, menin expression in islets is reduced, allowing for β-cell proliferation. In the present study, it was found that the expression levels of FoxM1 and menin are comparable between the Prlr+/+(+/−) and Prlr+/+(+/+) mice in the pancreatic islets on day 15 of pregnancy. The role of cell cycle protein has also been examined in the context of prolactin/placental lactogen exposure. In transgenic mice with islet-specific overexpression of placental lactogen, the cyclin inhibitory protein p21cip was the only cell cycle protein that was upregulated, while expression levels of cyclins, other cyclin inhibitory proteins or inhibitory kinases were not affected (Cozar-Castellano et al. 2006b). In the present study, it was found that the pregnancy-associated increase in p21cip is preserved in the Prlr+/+(+/−) mice. Likewise, no significant difference was detected in cyclin D2 expression between the Prlr+/+(+/+) and Prlr+/+(+/−) mice. These observations are not entirely surprising given that both genotypes have intact prolactin receptor expression; we would therefore expect that the Prlr+/+(+/−) mice would maintain a robust response in at least some of the Prlr-regulated pathways.
There are emerging data to support a role for the serotonin synthetic enzyme, Tph1, in regulation of β-cell mass during pregnancy. Kim et al. (2010) reported that Tph1 inhibition causes gestational diabetes due to a reduction in β-cell proliferation and mass. In contrast, Schraenen et al. (2010)) found that despite very strong induction of Tph1 in a subpopulation of β-cells during pregnancy, they could not detect a difference in β-cell proliferation between the wild-type and Tph1-null mice. Furthermore, they could not detect the Tph1 receptors, Htr1d and Htr2b, in islets of pregnant mice, casting some doubt on the biological importance of Tph1 (and serotonin) in β-cells during pregnancy (Schraenen et al. 2010). In the present study, it was found that both Prlr+/+(+/+) and Prlr+/+(+/−) mice expressed a similar amount of Tph1 on day 15 of pregnancy. However, in agreement with the study of Schraenen et al. (2010), very low expression levels of the Tph1 receptor, Htr2b, were detected, and the levels detected were comparable between the two genotypes (data not shown). In view of the discrepant results from the studies of Kim et al. (2010) and Schraenen et al. (2010) regarding the requirement of Tph1 for β-cell proliferation during pregnancy, the precise role of Tph1 in regulation of β-cell proliferation during pregnancy requires further study.
Interestingly, we found no significant difference between the Prlr+/+(+/+) and Prlr+/+(+/−) mice in either the incremental increase in insulin secretion or the expression of genes that regulate insulin synthesis and secretion (i.e. GLUT2, glucokinase and Pdx1) on day 15 of pregnancy. Of note, expressions of both glucokinase and Pdx1 were significantly lower in the non-pregnant Prlr+/+(+/−) mice in comparison to the Prlr+/+(+/+) mice. This may contribute to the lower insulin level detected in the non-pregnant Prlr+/+(+/−) mice. However, expression of both glucokinase and Pdx1 were restored to the levels observed in the Prlr+/+(+/+) mice by day 15 of pregnancy and, as there is no statistically significant difference in glucose-stimulated insulin response between the Prlr+/+(+/+) and the Prlr+/+(+/−) mice, it appears that the lower prepregnant glucokinase and Pdx1 levels did not translate into functional differences between the two groups during pregnancy. The cause of this reduction in glucokinase and Pdx1 expression is unclear. Although hyperglycaemia can cause a reduction in Pdx1 expression (Zangen et al. 1997), it is unlikely to be the sole explanation because hyperglycaemia in the Prlr+/+(+/−) mice persists until day 15 of pregnancy, a time point when the Pdx1 expression level has returned to the level observed in the Prlr+/+(+/+) mice. Taken together, our results suggest that the lower non-fasted insulin and higher glucose levels observed in the Prlr+/+(+/−) mice on day 15 of pregnancy are due to their lower β-cell mass and not due to an insulin secretory defect.
Both human and animal studies have shown that gestational diabetes causes β-cell dysfunction in the offspring. In the absence of severe hyperglycaemia, exposure to mild hyperglycaemia in utero also has deleterious effects on pancreatic function of the offspring. In pregnant rats, Gauguier et al. (1991) infused glucose to induce mild hyperglycaemia (6.5–8 mm) during the last week of gestation, and found that the offspring had impaired glucose tolerance starting at 3 months of age. The cause of glucose intolerance in that model was a defect in neuroregulation of insulin secretion in vivo, because ex vivo glucose-stimulated insulin secretion was intact (Gauguier et al. 1991). In a separate study, the same group reported that offspring exposed to hyperglycaemia in utero also had glucose intolerance during their own pregnancy (Gauguier et al. 1990). Unfortunately, neither of these studies reported on β-cell mass, so the extent of the β-cell defect in the offspring was unknown. In our model, the Prlr+/− dams have significantly higher blood glucose by day 9 of pregnancy (10.1 ± 0.4 mmol l−1, n= 7) in comparison to the pregnant Prlr+/+ dams (9.1 ± 0.3 mmol l−1, n= 8; P= 0.03), a difference that persisted until day 15 of pregnancy (9.0 ± 0.2 mmol l−1 in Prlr+/− dams and 8.1 ± 0.3 mmol l−1 in Prlr+/+ dams). The mild hyperglycaemia experienced by the Prlr+/+(+/−) mice in utero may contribute to their blunted IRS-2 and Akt responses during pregnancy, because prolonged exposure to high glucose has been shown to downregulate IRS-2 expression in β-cells in vitro, although the glucose levels used in those studies were in the range of 15–20 mm (Briaud et al. 2005; Lingohr et al. 2006). The potential metabolic effects of prolactin also need to be considered. The Prlr+/− dams have significantly elevated prolactin levels (unpublished data; E Hughes and C Huang), but prolactin does not cross the placenta (Ben-Jonathan et al. 2008), so the elevated prolactin in the pregnant Prlr+/− dams is unlikely to have a direct effect on her offspring. However, the placenta is a metabolically active organ and produces many hormones, including adiponectin and leptin. A recent study in humans showed that mild maternal hyperglycaemia in the second and third trimester is associated with lower DNA methylation on the fetal side of the placenta, which in turn is associated with a higher maternal circulation level of adiponectin during pregnancy (Bouchard et al. 2012). Whether a similar scenario is operating in our model is unknown, but it is highly plausible that placentae of the Prlr+/− dams have a very different metabolic and hormonal profile from that of the Prlr+/+ dams. This requires further investigation.
In summary, we found that maternal genotype has a significant impact on the ability of her wild-type offspring to adapt to the increased demand for insulin during the offspring's own pregnancy. When we examined the molecular mechanisms, it appeared that signalling molecules that regulate β-cell mass, i.e. IRS-2 and Akt, are downregulated in the Prlr+/+(+/−) mice, but molecules that mediate prolactin/placental hormone-specific signals, i.e. Jak2, menin, Tph1, FoxM1 and p21CIP, were unaffected, which is not surprising given that these mice have a normal complement of Prlr. Furthermore, expression levels of molecules that regulate insulin synthesis and secretion (GLUT2, glucokinase and Pdx-1) were unaffected in the offspring. The physiological consequences of these changes are that the Prlr+/+(+/−) mice had higher blood glucose and lower insulin concentrations during pregnancy, which is likely to reflect their reduced β-cell mass, while maintaining glucose-stimulated insulin release capacity (Fig. 7). These data suggest that normal prolactin activity during pregnancy is important for normal glucose homeostasis in both the present pregnancy and pregnancies in the future generations. Identification of a therapeutic strategy that can break this cycle of ‘gestational diabetes begets gestational diabetes’ will be of upmost interest.
Figure 7. Working model.

Wild-type experimental mice (F2) derived from Prlr+/− mothers (F1) have lower IRS-2 and Akt expression in the islets, resulting in lower β-cell mass during pregnancy. Genes that regulate prolactin/placental hormone-specific responses (i.e. Jak2, Tph1, FoxM1 and menin) and genes that regulate insulin synthesis and secretion, such as GLUT2, glucokinase (GCK) and Pdx1, were not significantly affected by the maternal genotype.
Translational perspective
The results of this study are consistent with previous human and animal studies showing that prenatal exposure to gestational diabetes increases the risk of gestational diabetes in the next generation. In the present model, the molecular mechanism appears to involve downregulation of insulin receptor substrate-2, Akt and Forkhead box D3, leading to a reduction in β-cell mass and low insulin concentrations. Therefore, finding small-molecule therapeutic agents that can specifically target these signalling molecules and restore their expression levels can potentially rescue the β-cell defect and break the cycle of ‘gestational diabetes begets gestational diabetes’. As the obesity epidemic continues, which increases the risk of gestational diabetes, discovery of effective therapeutic interventions for treatment of the affected offspring will have a significant positive impact on the health of the population.
Acknowledgments
Thanks to Francis Snider, Martha Hughes and Sheila Li for technical assistance and to Dr Jay Cross for discussions of results on the project. This work was supported by grants from the Canadian Diabetes Association (Grant OG-3-07-2410-CH) and Alberta Children's Hospital Foundation to C.H.
Glossary
- AUC
area under the curve
- BrdU
bromodeoxyuridine
- Foxd3
Forkhead box D3
- GLUT2
glucose transporter 2
- IPGTT
intraperitoneal glucose tolerance test
- IRS-2
insulin receptor substrate-2
- Pdx-1
pancreatic and duodenal homeobox-1
- Prlr
prolactin receptor
- Tph1
tryptophan hydroxylase-1
- FoxM1
Forkhead box M1
- Htr2b
5-hydroxytryptamine receptor 2B
- Htr1d
5-hydroxytryptamine receptor 1d
- p21cip
cyclin inhibitory protein p21
Author contributions
The author (C.H.) is responsible for conception and design of the experiments, performance of 30% of the experiments, analysis and interpretation of all data, and drafting and revising the manuscript.
References
- Ackermann AM, Gannon M. Molecular regulation of pancreaticβ-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206. doi: 10.1677/JME-06-0053. [DOI] [PubMed] [Google Scholar]
- Aerts L, Vercruysse L, Van Assche FA. The endocrine pancreas in virgin and pregnant offspring of diabetic pregnant rats. Diabetes Res Clin Pract. 1997;38:9–19. doi: 10.1016/s0168-8227(97)00080-6. [DOI] [PubMed] [Google Scholar]
- Aerts L, Van Assche FA. Ultrastructural evaluation ofβ-cell recruitment in virgin and pregnant offspring of diabetic mothers. Diabetes Res Clin Pract. 1998;41:9–14. doi: 10.1016/s0168-8227(98)00063-1. [DOI] [PubMed] [Google Scholar]
- Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans. Endocr Rev. 2008;29:1–41. doi: 10.1210/er.2007-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihoreau MT, Ktorza A, Kinebanyan MF, Picon L. Impaired glucose homeostasis in adult rats from hyperglycemic mothers. Diabetes. 1986;35:979–984. doi: 10.2337/diab.35.9.979. [DOI] [PubMed] [Google Scholar]
- Bouchard B, Ormandy CJ, Di Santo JP, Kelly PA. Immune system development and function in prolactin receptor-deficient mice. J Immunol. 1999;163:576–582. [PubMed] [Google Scholar]
- Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes. 2012;61:1272–1280. doi: 10.2337/db11-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelje TC, Stout LE, Bhagroo NV, Sorenson RL. Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of Langerhans. Endocrinology. 2004;145:4162–4175. doi: 10.1210/en.2004-0201. [DOI] [PubMed] [Google Scholar]
- Brelje TC, Svensson AM, Stout LE, Bhagroo NV, Sorenson RL. An immunohistochemical approach to monitor the prolactin-induced activation of the JAK2/STAT5 pathway in pancreatic islets of Langerhans. J Histochem Cytochem. 2002;50:365–383. doi: 10.1177/002215540205000308. [DOI] [PubMed] [Google Scholar]
- Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in β-cells. J Biol Chem. 2005;280:2282–2293. doi: 10.1074/jbc.M412179200. [DOI] [PubMed] [Google Scholar]
- Cozar-Castellano I, Haught M, Stewart AF. The cell cycle inhibitory protein p21cip is not essential for maintaining β-cell cycle arrest or β-cell function in vivo. Diabetes. 2006a;55:3271–3278. doi: 10.2337/db06-0627. [DOI] [PubMed] [Google Scholar]
- Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of β-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes. 2006b;55:70–77. [PubMed] [Google Scholar]
- Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH, Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000a;49:2208–2211. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- Dabelea D, Knowler WC, Pettitt DJ. Effect of diabetes in pregnancy on offspring: follow-up research in the Pima Indians. J Matern Fetal Med. 2000b;9:83–88. doi: 10.1002/(SICI)1520-6661(200001/02)9:1<83::AID-MFM17>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Dickson LM, Rhodes CJ. Pancreatic β-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt. Am J Physiol Endocrinol Metab. 2004;287:E192–E198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- Fatrai S, Elghazi L, Balcazar N, Cras-Méneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N, Breant B, Kelly PA. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology. 2002;143:1378–1385. doi: 10.1210/endo.143.4.8722. [DOI] [PubMed] [Google Scholar]
- Gauguier D, Bihoreau MT, Ktorza A, Berthault MF, Picon L. Inheritance of diabetes mellitus as consequence of gestational hyperglycemia in rats. Diabetes. 1990;39:734–739. doi: 10.2337/diab.39.6.734. [DOI] [PubMed] [Google Scholar]
- Gauguier D, Bihoreau MT, Picon L, Ktorza A. Insulin secretion in adult rats after intrauterine exposure to mild hyperglycemia during late gestation. Diabetes. 1991;40:109–114. doi: 10.2337/diab.40.2.s109. [DOI] [PubMed] [Google Scholar]
- Han J, Xu J, Long YS, Epstein PN, Liu YQ. Rat maternal diabetes impairs pancreatic β-cell function in the offspring. Am J Physiol Endocrinol Metab. 2007;293:E228–E236. doi: 10.1152/ajpendo.00479.2006. [DOI] [PubMed] [Google Scholar]
- Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology. 2009;150:1618–1626. doi: 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy-induced β-cell proliferation. Endocrinology. 2011;152:847–855. doi: 10.1210/en.2010-1250. [DOI] [PubMed] [Google Scholar]
- Iype T, Francis J, Garmey JC, Schisler JC, Nesher R, Weir GC, Becker TC, Newgard CB, Griffen SC, Mirmira RG. Mechanism of insulin gene regulation by the pancreatic transcription factor Pdx-1: application of pre-mRNA analysis and chromatin immunoprecipitation to assess formation of functional transcriptional complexes. J Biol Chem. 2005;280:16798–16807. doi: 10.1074/jbc.M414381200. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, Fontaine M, Yen MH, Kim SK. Menin controls growth of pancreatic β-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–809. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- Kasperska-Czyzyk T, Jedynasty K, Bowsher RR, Holloway DL, Stradowska I, Stepień K, Nowaczyk R, Szymczak W, Czyzyk A. Difference in the influence of maternal and paternal NIDDM on pancreatic beta-cell activity and blood lipids in normoglycaemic non-diabetic adult offspring. Diabetologia. 1996;39:831–837. doi: 10.1007/s001250050517. [DOI] [PubMed] [Google Scholar]
- Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, Yang K, Honig G, van der Hart M, Kishimoto N, Wang J, Yagihashi S, Tecott LH, Watada H, German MS. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota N, Tobe K, Terauchi Y, Eto K, Yamauchi T, Suzuki R, Tsubamoto Y, Komeda K, Nakano R, Miki H, Satoh S, Sekihara H, Sciacchitano S, Lesniak M, Aizawa S, Nagai R, Kimura S, Akanuma Y, Taylor SI, Kadowaki T. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory β-cell hyperplasia. Diabetes. 2000;49:1880–1889. doi: 10.2337/diabetes.49.11.1880. [DOI] [PubMed] [Google Scholar]
- Lingohr MK, Briaud I, Dickson LM, McCuaig JF, Alárcon C, Wicksteed BL, Rhodes CJ. Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet β-cells. J Biol Chem. 2006;281:15884–15892. doi: 10.1074/jbc.M600356200. [DOI] [PubMed] [Google Scholar]
- Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab. 2010;21:223–229. doi: 10.1016/j.tem.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank JL, Frist AY, Legrone AW, Magnuson MA, Labosky PA. Loss of Foxd3 results in decreased β-cell proliferation and glucose intolerance during pregnancy. Endocrinology. 2011;152:4589–4600. doi: 10.1210/en.2010-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraenen A, Lemaire K, de Faudeur G, Hendrickx N, Granvik M, Van Lommel L, Mallet J, Vodjdani G, Gilon P, Binart N, in't Veld P, Schuit F. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia. 2010;53:2589–2599. doi: 10.1007/s00125-010-1913-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: β-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- Sorenson RL, Brelje TC. Prolactin receptors are critical to the adaptation of islets to pregnancy. Endocrinology. 2009;150:1566–1569. doi: 10.1210/en.2008-1710. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Wang S, Yan J, Anderson DA, Xu Y, Kanal MC, Cao Z, Wright CV, Gu G. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol. 2010;339:26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated β-cell development and peripheral insulin signalling. Nat Genet. 1999;23:32–40. doi: 10.1038/12631. [DOI] [PubMed] [Google Scholar]
- Xu X, D’Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. β Cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Zangen DH, Bonner-Weir S, Lee CH, Latimer JB, Miller CP, Habener JF, Weir GC. Reduced insulin, GLUT2, and IDX-1 in β-cells after partial pancreatectomy. Diabetes. 1997;46:258–264. doi: 10.2337/diab.46.2.258. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. Gestational diabetes mellitus resulting from impaired β-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. 2009;59:143–152. doi: 10.2337/db09-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]