

Abstract
Biological oxygen measurements by phosphorescence quenching make use of exogenous phosphorescent probes, which are introduced directly into the medium of interest (e.g. blood or interstitial fluid) where they serve as molecular sensors for oxygen. The byproduct of the quenching reaction is singlet oxygen, a highly reactive species capable of damaging biological tissue. Consequently, potential probe phototoxicity is a concern for biological applications. Herein, we compared the ability of polyethyleneglycol (PEG)-coated Pd tetrabenzoporphyrin (PdTBP)-based dendritic nanoprobes of three successive generations to sensitize singlet oxygen. It was found that the size of the dendrimer has practically no effect on the singlet oxygen sensitization efficiency in spite of the strong attenuation of the triplet quenching rate with an increase in the dendrimer generation. This unexpected result is due to the fact that the lifetime of the PdTBP triplet state in the absence of oxygen increases with dendritic generation, thus compensating for the concomitant decrease in the rate of quenching. Nevertheless, in spite of their ability to sensitize singlet oxygen, the phosphorescent probes were found to be non-phototoxic when compared with the commonly used photodynamic drug Photofrin in a standard cell-survival assay. The lack of phototoxicity is presumably due to the inability of PEGylated probes to associate with cell surfaces and/or penetrate cellular membranes. In contrast, conventional photosensitizers bind to cell components and act by generating singlet oxygen inside or in the immediate vicinity of cellular organelles. Therefore, PEGylated dendritic probes are safe to use for tissue oxygen measurements as long as the light doses are less than or equal to those commonly employed in photodynamic therapy.
Introduction
Oxygen-dependent quenching of phosphorescence1,2 is a widely used method for imaging of oxygen in biological systems, including tissue in vivo (for several recent examples, see ref. 3–7). The method makes use of the energy transfer between the molecules of exogenous phosphorescent probes in their excited triplet states (T1) and ground state triplet oxygen (X3Σg-) (Scheme 1).
Scheme 1.
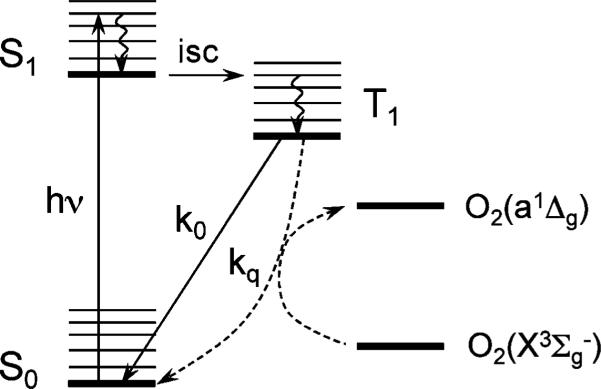
Diagram of photophysical processes occurring in phosphorescent molecules: hν – excitation, isc – intersystem crossing; k0 – rate constant of phosphorescence emission (k0 = 1/τ0 = kr + knr, where kr and knr are the rate constants of the radiate and non-radiative deactivation of the triplet state in the absence of oxygen); kq – rate constant of bimolecular quenching, which results in conversion of ground state oxygen O2(X3Σg-) into excited singlet state oxygen O2(a1Δg).
The dependence of the phosphorescence lifetime τ on the partial pressure of oxygen (p(O2)) throughout the physiological range of oxygen concentrations is described by the Stern–Volmer relationship:
| (1) |
where τ0 is the phosphorescence lifetime in the absence of oxygen and kq is the bimolecular collisional rate constant expressed in the units of pressure (mmHg-1 s-1).8
The phosphorescence quenching occurs presumably via the energy transfer by the triplet–triplet exchange mechanism, resulting in sensitization of singlet oxygen.9 The latter is a highly reactive species, implicated in a variety of toxic effects. In fact, the mechanism of the phosphorescence quenching is the same as that of photodynamic therapy (PDT),10 albeit the purpose of the latter is the generation of high doses of singlet oxygen (a1Δg) for killing undesirable cells, whereas in phosphorescence imaging singlet oxygen is merely an undesirable byproduct. Nevertheless, since phosphorescent probes are introduced directly into the medium of interest (e.g. blood or interstitial fluid), phototoxicity is a legitimate concern.11,12 This is particularly true if excitation intensities are high, as, for example, in microscopy applications, and/or if measurements occur over long time periods.
Recently we described a general strategy for construction of “protected” soluble molecular probes for biological imaging of oxygen.13 These probes consist of phosphorescent Pt and Pd porphyrins or π-extended porphyrins (tetrabenzoporphyrins and tetranaphthoporphyrins) encapsulated inside poly-arylglycine (AG) dendrimers. Dendrimers14,15 are branched tree-like macromolecules that exhibit a defined structure and a high degree of order. From a topological viewpoint, dendrimers contain three distinct regions: core, branches and surfaces. In the oxygen probes, phosphorescent metalloporphyrins play the role of cores, while the surfaces are made of polyethyleneglycol (PEG) residues. PEGylated dendritic jackets regulate the sensitivity and dynamic range of measurements via control of oxygen quenching constant kq (eqn (1)). In addition, they insure probes’ aqueous solubility and prevent interaction of the probes with biological systems. The latter property is very important, since association of probes with proteins and other macromolecular objects in biological environments may significantly affect the accessibility of the triplet cores to oxygen, unpredictably changing the rate of quenching. PEGylation of the dendrimer periphery makes probes insensitive to the presence of proteins and other biological solutes. At the same time, PEG residues themselves only slightly affect the probe's accessibility to oxygen, making it possible to change the probe's size without significantly altering oxygen quenching parameters.
While several approaches to the construction of biological oxygen sensors are currently under scrutiny,16–22 dendritic porphyrins23–27 are perhaps the most widely used for in vivo oxygen measurements. This approach offers tunable optical properties and flexible chemical design, which can be adapted to the needs of different types of in vivo imaging applications. Consequently, the question of their phototoxicity is becoming increasingly important.
Comprehensive evaluation of phototoxicity is a complex task, requiring specially designed experiments for each type of tissue and/or each mode of imaging application, e.g. wide field vs. scanning microscopy vs. near-infrared tomography. Nevertheless, generally useful information can be obtained by comparing phosphorescent probes with standard PDT sensitizers, whose primary function is to generate singlet oxygen and for which a wealth of biological information is already available. Comparative tests can be carried out using standard in vitro cell-survival assays, which directly measure cytotoxicity of a treatment under selected exposure conditions.
In this paper, we evaluated oxygen quenching properties of Pd meso-tetra(3,5-dicarboxyphenyl)tetrabenzoporphyrin (PdTBP, also termed G0 dendrimer) and PdTBP-based PEGylated AG-dendrimers of three successive generations (G1–G3). Relative singlet oxygen sensitization efficiencies were estimated by measuring singlet oxygen phosphorescence in C2H5OH–CH3OH mixture and in D2O. Dendrimer PdTBP-(AG2-PEG)8 (G2) was tested in a cell survival assay against Photofrin, a clinically approved photosensitizer. The tests revealed negligible phototoxicity of G2 compared to that of Photofrin, suggesting that the oxygen probes can be safely used under illumination conditions commonly employed in tissue oxygen measurements.
Experimental
All solvents and reagents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on aluminium-supported silica gel plates (Aldrich). Column chromatography was performed on Selecto silica gel (Fisher). Preparative size exclusion chromatography (SEC) was performed on S-X1 beads (Bio-Rad), using THF as a mobile phase. 1H and 13C NMR spectra were recorded on a Brucker DPX-400 spectrometer. The mass spectra were obtained on a MALDI-TOF Voyager-DE™ RP BioSpectrometry workstation, using α-cyano-4-hydroxycinnamic acid as the matrix.
Quartz fluorometric cells (Starna Cells, 1 cm optical path length) were used in both UV-Vis and fluorescence experiments. Optical absorption spectra were recorded on Perkin Elmer Lambda 40 or Perkin Elmer Lambda 650 UV-Vis spectrophotometers. Emission measurements were performed on a Perkin Elmer LS-55 fluorometer, equipped with a Hamamatsu R928-P PMT and a Xenon flash lamp or on an Edinburgh Instruments FS920 fluorometer, equipped with a Hamamatsu R2658P PMT. Singlet oxygen emission was measured using a liquid nitrogen cooled ultrapure germanium crystal as a detector. Time-resolved phosphorescence measurements were performed using either the Perkin Elmer LS-55 instrument or an in-house constructed phosphorometer,28 adapted for time domain operation. Photophysical experiments were performed using either air-equilibrated solutions or solutions degassed by four pump-freeze-thaw cycles. Oxygen titrations were performed using a previously described system.24
Electrochemical measurements were carried out using argon-purged CH3CN (Romil Hi-Dry™) solutions at room temperature with an EcoChemie Autolab 30 multipurpose instrument interfaced to a PC. The working electrode was a glassy carbon electrode (0.08 cm2, Amel) and the counter electrode was a Pt wire. A silver wire was employed as a quasi-reference electrode (QRE). Tetraethylammonium hexafluorophosphate (TEAPF6) (0.1 M) was used as a supporting electrolyte. The potentials are reported relative to SCE. These were measured with AgQRE (quasi-reference electrode) using ferrocene as an internal standard (+0.395 V vs. SCE). The concentrations of the compounds examined were ca 10-3 M. Cyclic voltammograms were obtained at scan rates in the range 0.2–5 V s-1. The experimental error for the potential values was estimated to be ±10 mV.
Materials
The synthesis of PdTBP-core (G0) and generation-two dendrimer PdTBP-(AG2-PEG)8 (G2) followed the earlier reported protocol.13 Compounds PdTBP-(AG1-PEG)8 (G1) and PdTBP-(AG3-PEG)8 (G3) were synthesized analogously to the corresponding Pt porphyrin-based dendrimers,13 using octacarboxylic acid G0 as a core. The synthesis involved coupling of dendrons (H2N-AG1-OBu or H2N-AG3-OBu)13 to G0 with formation of poly-esters PdTBP-(AG1-OBu)8 and PdTBP-(AG3-OBu)8. The subsequent hydrolysis produced poly-acids PdTBP-(AG1-OH)8 and PdTBP-(AG3-OH)8, which were converted into the target compounds G1 and G3 by esterification using PEG350 (polyethyleneglycol monomethyl ether, Av. MW 350).
PdTBP-(AG1-OBu)8
The synthesis in general followed the protocol for compound Pt-1-(AG1-OBu)8 in ref. 13. Yield: 82%. 1H NMR (DMSO-d6) δ 10.62 (s, 8H), 9.46 (s, broad, 8H), 9.23 (s, broad, 4H), 9.08 (s, broad, 8H), 8.48 (s, broad, 16H), 8.10 (s, broad, 8H), 7.44 (m, 8H), 7.11 (m, 8H), 4.24 (t, J = 6.5 Hz, 32H), 4.19 (s, broad, 16H), 1.64 (m, 32H), 1.36 (m, 32H), 0.87 (t, J = 7.4 Hz, 48H). 13C NMR (DMSO-d6) δ 168.9, 167.0, 166.3, 165.4, 141.8, 140.5, 138.4, 137.9, 135.8, 135.1, 131.6, 131.3, 129.1, 127.1, 124.6, 124.4, 124.1, 65.5, 44.3, 30.8, 19.4, 14.2. MALDI-TOF (m/z): calcd. for C212H228N20O48Pd: 3930.6, found: 3931.5 [M + H+].
PdTBP-(AG1OH)8
The synthesis in general followed the protocol for compound Pt-1-(AG1-OH)8 in ref. 13. Yield: 92%. 1H NMR (DMSO-d6) δ 10.34 (s, broad, 8H), 9.21 (s, broad, 12H), 9.00 (s, broad, 8H), 8.44 (s, broad, 16H), 8.15 (s, broad, 8H), 7.41 (s, broad, 8H), 7.12 (s, broad, 8H), 4.18 (s, broad, 16H). 13C NMR (DMSO-d6) δ 168.6, 166.8, 166.3, 164.3, 141.6, 139.9, 138.2, 137.9, 135.9, 134.9, 134.8, 132.4, 128.6, 126.8, 125.1, 124.4, 44.3. MALDI-TOF (m/z): calcd. for C148H100N20O48Pd: 3032.9, found: 3033.7 (M + H+); 3056.6 (M + Na+).
PdTBP-(AG1PEG)8
The synthesis in general followed the protocol for compound Pt-1-(AG1-PEG)8 in ref. 13. Yield: ~60%. 1H NMR (DMSO-d6) δ 10.63 (s, 8H), 9.43 (s, 8H), 9.20 (s, 4H), 9.04 (s, 8H), 8.50 (s, 8H), 8.13 (m, 8H), 7.43 (s, 8H), 7.08 (m, 8H), 4.5-3.0 (multiple peaks, ~620H). MALDI-TOF spectrum shows a bell-shaped peak centred around 7915 Da. Calcd Av. MW. 8.2 kDa.
PdTBP-(AG3-OBu)8
The synthesis followed the protocol for compound Pt-1-(AG3-OBu)8 in ref. 13. Yield: 69%. MALDI-TOF (m/z): calcd. for C884H996N116O240Pd: 17180.83, found: 17204 [M + Na+] + 15215 [PdTBP-(AG3-OBu)7 + Na+] + 13226 [PdTBP-(AG3-OBu)6 + Na+].
PdTBP-(AG3-OH)8
The synthesis followed the protocol for compound Pt-1-(AG3-OH)8 in ref. 13. Yield: 60% (for coupling and hydrolysis combined). Because of the high sample viscosity and low mobility of the dendritic branches, it was impossible to obtain a high resolution NMR spectrum even at elevated temperature. 1H NMR (DMSO-d6) δ 10.45 (bs, 52H), 9.5–7.9 (series of overlapped peaks, 228 H), 4.1–3.8 (series of overlapped peaks, 112 H). MALDI-TOF (m/z): calcd. for C628H484N116O240Pd: 13592.8, found: bell-shaped peak centred at 12.9 kDa.
PdTBP-(AG3-PEG)8
The synthesis followed the protocol for compound Pt-1-(AG3-OPEG)8 in ref. 13. Yield: ~55%. 1H NMR (DMSO-d6) δ 10.7-10.3 (set of broad overlapping peaks, 52H), 9.5–7.9 (set of broad overlapping peaks, 244H), 4.6–3.0 (set of broad overlapping peaks, ~2400H). MALDI-TOF spectrum showed a broad peak centred at 27.5 kDa. Calculated Av. MW. 32 kDa.
Cellular clonogenic assays
All tissue culture solutions were purchased from Gibco (Invitrogen; Auckland, New Zealand) with the exception of the fetal calf serum, which was purchased from HyCone (Logan, UT, USA). The studies were performed using established cultures of radiation-induced fibrosarcoma (RIF) murine cells. The media for cell line maintenance was MEM alpha, supplemented with penicillin (100 units ml-1), streptomycin (100 μg ml-1) and l-glutamine (2 mM). The studies involved incubating the cells for 18 h with either Photofrin or G2 in the media with 5% Fetal Calf Serum (FCS). Photofrin and G2 were present in the media at 17 μM and 0.4 μM concentrations, respectively, as measured by the absorption at 630 nm. Cells to be illuminated in the absence of photosensitizer were removed from the photosensitizer-containing media after the incubation period (18 h), briefly washed in Hanks Balanced Salt Solution (HBSS), and incubated in photosensitizer-free media with 5% FCS for an additional 1 h. Cells to be illuminated in the presence of photosensitizer were left in the photosensitizer-containing media. Trypsin-EDTA (0.05%) was used to collect cells, which were counted and re-suspended at ~2 × 106 cells ml-1 in the media containing 5% FCS. Cells to be illuminated in the presence of the photosensitizer had Photofrin or G2 added to the media to match their concentrations during the incubation. Aliquots of ~4 × 105 cells were placed in tissue culture dishes (60 mm in diameter) and exposed to light of total doses of 0, 0.5 (100 s), or 2 (400 s) J cm-2. The light source was a dye laser (Laserscope, San Jose, CA, USA ) operating at 630 nm (Rhodamine 640). The pump was a frequency-doubled Nd:YAG laser (Laserscope, San Jose, CA, USA) with KTP frequency-doubling crystal. An irradiance of 5 mW cm-2, as measured by a power-meter (Coherent, Auburn, CA, USA), was delivered via microlens-tipped fibers (CardioFocus, Norton, MA, USA) over the area of the tissue culture dishes. Following the exposure, cell aliquots were spun (as needed) to remove photosensitizer-containing media. Cells were plated at concentrations of 102–106 cells per dish in tissue culture dishes (100 mm in diameter) that contained media with 10% FCS. After 7–10 days of incubation (37 °C, 5% CO2), colonies of cells were fixed/stained (2.5 mg ml-1 methylene blue in 30% alcohol), counted, and the plating efficiency was calculated. In an additional study, the same incubation (in the presence of G2 and using illumination dose of 2 J cm-2) conditions were employed to measure G2-induced phototoxicity over higher probe concentrations, ranging from 2.5 to 20 μM.
Results and discussion
Synthesis
The structures of Pd meso-tetra(3,5-dicarboxyphenyl)tetrabenzoporphyrin (PdTBP or G0) and dendrimers PdTBP-(AGn-PEG)8 (n = 1–3, G1–G3) used in this study are shown in Fig. 1. In the abbreviations: AG designates the arylglycine dendrimer, n is the dendrimer generation, PEG is polyethyleneglycol monomethyl ether (av. MW 350). Selected molecular data for the compounds studied are summarized in Table 1.
Fig. 1.

Structures of phosphorescent PdTBP-based dendrimers. PEG designates residues of polyethyleneglycol monomethyl ether (av. MW 350).
Table 1.
Selected molecular and data for phosphorescent PdTBP-dendrimers
| Gen # | Abbreviation | MW/Daa | Number of peripheral PEG groups | Av. mol. diameter/Åb |
|---|---|---|---|---|
| G0 | PdTBP | 1270 | 0 | – |
| G1 | PdTBP-(AG1-PEG)8 | 8358 | 16 | 28 |
| G2 | PdTBP-(AG2-PEG)8 | 17214 | 32 | 38 |
| G3 | PdTBP-(AG3-PEG)8 | 34918 | 64 | 55 |
Calculated for ideal dendrimer modified with PEG residues of MW 350.
Estimated from molecular mechanics simulations for folded states of ideal dendrimers (ESI†).
The probes were synthesized as described previously13 and characterized by standard methods. The synthesis of porphyrin-dendrimers proceeds via the convergent route, in which ester-terminated AG-dendrons, bearing the amino-groups at their focal points, are attached to the octacarboxyporphyrin core using an optimized peptide coupling reaction, based on the HBTU/DIPEA chemistry. This procedure is very effective for Gen 1 and Gen 2 dendrimers; however, for larger dendrimers complete dendrimerization of porphyrins becomes difficult, since the cores contain eight closely positioned anchor points.13 Following hydrolysis of the ester groups, the peripheral carboxyls on the dendrimers were modified with polyethyleneglycol monomethyl ether (av. MW 350). Esterification of the dendrimers could leave some of the peripheral carboxylic groups non-esterified. MALDI spectra of the resulting PEGylated compounds (Fig. S6, ESI†) show broad peaks, whose maxima correspond to ~90% of the carboxyls converted to PEG350 esters. Thus, compounds used in this study, especially G2 and G3, should not be considered as monodisperse dendrimers; however, the non-monodisperse nature of these materials does not affect conclusions about the relationship between their average size and photophysical/photochemical behavior (see below).
Photophysical properties
The photophysical properties of the PdTBP-dendrimers were examined in buffered aqueous solutions (phosphate buffer, 50 mM) at pH 7.0–7.2, except for compound G0, which is soluble only at higher pH (pH ~ 9). The absorption spectra of compounds G0–G3 are shown in Fig. 2. The spectra are normalized by the values at the Q-band maxima and scaled by the molar absorption coefficient of G0 compound (ε ~ 9.0 × 104 M-1 cm-1) to facilitate comparisons.29 In the visible region, the spectra of all dendrimers are dominated by the absorption of the core PdTBP and in general resemble the spectrum of G0. The Q-band maxima of G1–G3 (637 nm) are shifted by 174 cm-1 to the red relative to the maximum of G0 (630 nm) and coincide with each other. In the Soret band region, the red-shift is larger (304 cm-1) and it progresses from G0 (441 nm) to G1 (445 nm) to G2–G3 (447 nm). These shifts are most likely caused by the gradual change in the porphyrin environment (e.g. increase in hydrophobicity, interactions between the porphyrin π-system and aromatic fragments of the dendritic arms, displacement of solvent, etc.) upon increase in the size of the dendritic jacket and more effective core encapsulation. Similar changes have been observed previously for other dendritically-encapsulated porphyrins.30,31
Fig. 2.

Absorption spectra of compounds G0–G3 in buffered aqueous solutions at 298 K.
The Soret/Q intensity ratios for all G1–G3 compounds are noticeably lower than for G0. This decrease could be to some extent related to the band broadening in the case of the dendrimers, probably a result of partial aggregation. G3 shows the smallest broadening in the series, and the integrated intensity of its Soret band (77.5 relative units) is very close to that of G0 (82.7 relative units). The small difference could be due to the different core substituents, i.e. carboxyls in G0 and carboxamides in G3.
In Pd porphyrins, S1→T1 intersystem crossing occurs with practically unity efficiency;32 and the emission of PdTBP's is entirely due to the T1→S0 phosphorescence.33,34 Compounds G0–G3 exhibit broad phosphorescence with maxima at 810–815 nm (Fig. 3). A very weak fluorescence at ca. 700 nm was also observed for all the dendrimers in the series, which was most likely originated from a free-base TBP impurity.34
Fig. 3.

Phosphorescence spectra in air-equilibrated aqueous solutions at 298 K. The emission intensities are directly comparable since the spectra were recorded for isoabsorbing solutions at the excitation wavelength (λex = 634 nm).
In the absence of oxygen, phosphorescence lifetimes τ0 and quantum yields ϕ0 (Table 2) were found to increase in the series of the dendrimers with an increase in the dendritic generation, resembling the trend observed for Pt porphyrin-based dendrimers.13 This effect may be caused by a combination of several factors. First, it is possible that the non-radiative deactivation of the T1 state occurs in part with involvement of the solvent (water) molecules, located in the immediate vicinity of the porphyrin. As hydrophobic dendritic branches fold and displace the solvent from the porphyrin environment, this relaxation pathway becomes diminished. Secondly, spin–orbit coupling in Pd porphyrins is known to be related to vibrational activity.34,35 Larger dendritic substituents may affect the vibrations of the core by folding and enclosing the porphyrin into a motion-restricting pocket, thus affecting the rate of T1→S0 intersystem crossing and increasing the probability of radiative relaxation (phosphorescence).
Table 2.
Photophysical and oxygen quenching properties of compounds G0–G3 in aqueous and ethanol–methanol solutions at 298 K
| H2O |
C2H5OH–CH3OH 4: 1 (v/v) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent | λmax(B)/nm | λmax(Q)/nm | λemiss/nm | τ0/μsa | ϕ 0 b | kq/mmHg–1 s–1 (k2/107 M–1 s–1)c | ηq (air)d | τ0/μs | kq/mmHg–1 s–1 (k2/109 M–1 s–1) | ηq (air)d |
| G0 | 441 | 630 | 815 | 100 ± 2 | 0.02 ± 0.005 | 2320 ± 5.2 (146.8 ± 0.33) | 0.97 | 240 ± 3 | 2863 ± 11.6(181.2 ± 0.7) | 0.99 |
| G1 | 445 | 637 | 815 | 240 ± 3 | 0.05 ± 0.01 | 315 ± 0.85 (19.9 ± 0.05) | 0.92 | 260 ± 3 | 1415 ± 5.9 (89.6 ± 0.4) | 0.98 |
| G2 | 447 | 637 | 815 | 289 ± 3 | 0.06 ± 0.01 | 125 ± 0.16 (7.9 ± 0.01) | 0.85 | 360 ± 4 | 611 ± 1.3 (38.7 ± 0.08) | 0.97 |
| G3 | 447 | 637 | 812 | 321 ± 3 | 0.07 ± 0.01 | 71 ± 0.10 (4.5 ± 0.01) | 0.78 | 410 ± 4 | 550 ± 1.5 (34.8 ± 0.09) | 0.97 |
Obtained as intensity-weighted averages of lifetimes, determined from double-exponential fits: τav = (I1τ1 + I2τ2)/(I1 + I2), where I1 and I2 are initial intensities of the decays with lifetimes τ1 and τ2, respectively
Phosphorescence quantum yield in the absence of oxygen measured relative to the fluorescence of tetraphenylporphyrin (H2TPP) in benzene (ϕfl = 0.11).38
See ref. 8 for definition of k2.
Calculated based on the average values of τ0 and kq (eqn (2)).
The emission spectra of dendrimers G0–G3 in air-equilibrated aqueous solutions are shown in Fig. 3. As expected, in the presence of oxygen the increase in the phosphorescence intensity and lifetime with the dendrimer generation is much more pronounced than in the case of deoxygenated solutions. This behavior is consistent with the diffusion-controlled quenching of the T1 state of the core PdTBP by dioxygen. Larger dendrons shield the porphyrin and attenuate the rate of oxygen diffusion more effectively. Similar effects have been observed in other luminescent dendrimers, including phosphorescent [Ru(bpy)2]2+ complexes,36 Pt13 and Pd porphyrins.23,24,26
Because of the presence of macromolecules with different numbers of peripheral PEG groups, the phosphorescence decays of compounds G1–G3 were found to be not purely single exponential. However, they could be well approximated by two-exponential functions. The apparent phosphorescence lifetimes τ were calculated as intensity-weighted averages, resulting in almost ideally linear Stern–Volmer oxygen quenching plots (Fig. 4).
Fig. 4.

Stern–Volmer oxygen phosphorescence quenching plots for compounds G0–G3 in aqueous solutions at 295 K. Lines show the fits of the raw data to the linear equations. Phosphorescence decays were analyzed by two-exponential functions; apparent lifetimes τ were obtained by intensity-weighted averaging.
The relative decrease in the plots’ slopes (Fig. 4) resembles that previously seen in the series of Pt porphyrin-dendrimers.13 The bimolecular quenching rate constant for the unprotected porphyrin G0 was calculated to be 1.47 × 109 M-1 s-1. This quite high value supports the hypothesis that the phosphorescence quenching in these systems is controlled by oxygen diffusion. The Stern–Volmer quenching plot was also constructed for compound G2 using the phosphorescence intensity data. When plotted as I0/I vs. p(O2) and τ0/τ vs. p(O2), the intensity and lifetime based plots fully overlapped (Fig. S5, ESI†), ruling out static quenching. This result is not surprising given that there are no obvious oxygen coordinating sites within the porphyrin-dendrimer, and that molecular oxygen is a very fast diffusing quencher under the conditions of our experiments.
The yield of the phosphorescence quenching and sensitization of singlet oxygen (ηq), assuming that every quenching event leads to singlet oxygen generation (see below), is the ratio of the quenching rate constant (kq × p(O2)) to the total rate of the triplet state deactivation (1/τ). Using eqn (1), ηq can be calculated as:
| (2) |
Quenching constants kq (Table 2) decrease in the series of dendrimers from G0 to G3. Nonetheless, because this decrease is partially offset by the increase in the phosphorescence lifetime τ0, the quenching yields ηq change rather weakly throughout the series. For comparison, the quenching constant kq decreases by more than 30-fold from G0 to G3, while ηq decreases only by about 20%, i.e. from 0.97 (G0) to 0.78 (G3). As a result, the unsubstituted porphyrin G0 and all the dendrimers (G1–G3) can be considered efficient singlet oxygen sensitizers in aqueous solutions, comparable in efficiency to other Pd porphyrins, such as e.g. Pd tetrasulfonatophenylporphyrin (ηq = 0.78 in air-equilibrated aqueous solutions).37
Similar trends in the quenching behavior were observed for compounds G0–G3 in C2H5OH–CH3OH 4 : 1 (v/v) solutions at 298 K (Table 2). The kq values in all cases were found to be higher than in aqueous solutions. As in other dendritic systems,23,24 this effect is likely to be caused by the combination of higher solubility of oxygen in organic solvents39 and less restricted oxygen diffusion through the body of the dendrimer to the triplet core. In the diffusion-limited case, the quenching constant kq is proportional to the product of oxygen solubility and oxygen diffusion coefficient. A much less pronounced decrease in kq with an increase in the dendritic generation (Table 2) suggests that in alcohols AG-dendrimers impose lesser barriers for oxygen diffusion than in water and, therefore, they are likely to be less tightly folded and less dynamically constrained.
Singlet oxygen sensitization
Quenching of the triplet states of PdTBPs in the dendrimers by oxygen can be due to the energy transfer, presumably via the triplet–triplet exchange mechanism, or due to the photoinduced electron transfer (PET), followed by either charge recombination or by escape of the radicals from the solvent cage and subsequent chemistry. The PET pathway was ruled out on the basis of spectroscopic and electrochemical measurements. The redox potentials of the dendrimers were measured in acetonitrile solutions at 298 K for G1 dendrimer by the cyclic voltammetry method (see ESI†). Although dendritic substituents in some cases are known to affect redox potentials of encapsulated cofactors,40,41 for the purpose of our analysis measurement performed for G1 provided a sufficient estimate of the PET driving force for all the dendrimers in the series. Based on the first oxidation potential of G1 (1.46 eV vs. SCE) and the reduction potential of dioxygen (–0.8 eV vs. SCE),42 the charge separated state, corresponding to the oxidation of PdTBP, lies much higher in energy (ca. 2.3 eV) than the PdTBP triplet state (ca. 1.5 eV). Thus, PET is highly endergonic in this system, and the sensitization is likely to occur via the energy transfer pathway.
The generation of singlet oxygen was demonstrated by direct measurements of its phosphorescence (λmax = 1270 nm) upon photosensitization using compounds G0–G3 in air-equilibrated C2H5OH–CH3OH mixtures (4 : 1, v/v) or in D2O. In H2O no emission was observed, which is due to the much lower solubility of oxygen in water than in organic solvents42 and very low quantum yield of singlet oxygen emission (ϕ ~ 10-7).43 In water, singlet oxygen emission is efficiently quenched by OH vibrations as well as by the emission re-absorption by the OH vibrational overtone.44,45 As a result, the lifetime of singlet oxygen in water is very short, only 3.5 μs.46 On the other hand, in D2O the lifetime is significantly longer, i.e. 67 μs.47 Subsequently, singlet oxygen emission could be readily observed in air-equilibrated D2O solutions.
The phosphorescence spectra of singlet oxygen in airequilibrated C2H5OH–CH3OH solutions after subtracting long-wavelength tails of the sensitizers’ phosphorescence are shown in Fig. 5 (for uncorrected spectra, see Fig. S1–S2, ESI†). Because the absorbances of the sensitizers at the excitation wavelength (635 nm) were kept the same for all the compounds in the series, the intensities of singlet oxygen phosphorescence in these experiments can be compared directly. The integrated emission intensities are proportional to the quantum yields of singlet oxygen emission (ηem) as sensitized by compounds G0–G3. These values (ηem) are in turn the products of the yields of the sensitization reaction (ηsens) and singlet oxygen phosphorescence (ηphosO2). As discussed above, sensitization in PdTBP-dendrimers occurs by way of triplet–triplet energy transfer and provides virtually the only pathway for oxygen-dependent quenching of the porphyrin triplet state. Therefore, ηsens can be considered equal to the oxygen quenching efficiency ηq (eqn (2)), and:
| (3) |
Fig. 5.

Phosphorescence of singlet oxygen in air-equilibrated C2H5OH–CH3OH 4 : 1 (v/v) solutions (λmax = 1270 nm) as sensitized by compounds G0–G3. The tail phosphorescence of Pd porphyrin-dendrimers was subtracted from the spectra (see Fig. S1, S2 (ESI) for the raw data). The emission intensities are directly comparable, since the absorbances of the sensitizer solutions at the excitation wavelength (635 nm) were kept equal for all the compounds.
The values ηem are practically the same for all the compounds G0–G3 in both C2H5OH–CH3OH (Fig. 5) as well as in D2O (not shown), which is consistent with only a very modest decrease in the quenching efficiency (hq) in going from G0 to G3 (Table 2).
The described above results demonstrate that all the dendritic compounds in the studied series indeed can produce singlet oxygen by way of photosensitization, and that in spite of the dramatic differences in their quenching rate constants (kq) the efficiencies of singlet oxygen generation are very similar.
Cellular phototoxicity
Having determined that phosphorescent compounds G0–G3 are capable of generating singlet oxygen, we set out to examine how cellular phototoxicity induced by these probes compares to the phototoxicity of known PDT photosensitizers under conditions mimicking those of oxygen imaging and/or PDT. The generation-two PdTBP-dendrimer (G2)48 was selected as a representative probe to be evaluated in a clonogenic survival assay. In vitro clonogenic assays are routinely used to measure the survival and proliferative potential of cells exposed to a drug or other cytotoxic treatment.
As a reference photosensitizer we selected Photofrin – a well-characterized clinically approved drug, used in clinical treatments of Barrett's esophagus and early stage lung cancer, as well as in palliation of obstructive esophageal or endobronchial cancer.49 In a typical clinical application, Photofrin is administered intravascularly, and ~48 h later the diseased tissue is irradiated locally at 630 nm.50–52 After 48 h of circulation, Photofrin substantially, albeit not completely, clears from the blood,53 while accumulating in tumor tissues, where it predominantly localizes in the mitochondria.54 Upon activation with light, it generates singlet oxygen, which causes oxidative damage of the mitochondrial membranes.55 The quantum yield of singlet oxygen sensitized by Photofrin in air-equilibrated aqueous solutions varies across reports but has been estimated as 0.54 ± 0.35.56
Comparisons between efficiencies of phototoxic agents as measured by in vitro assays can be extrapolated to tissue applications, provided equal excitation efficiencies of chromophores under comparison are utilized in both types of experiments. In tissue applications, chromophores must have the same optical density and be excited at the same wavelength to insure the same absorption and scattering distribution of the excitation light throughout the tissue. Under these conditions, excitation efficiencies can be considered equal. In in vitro experiments conducted in ideal or weakly scattering solutions, equal excitation efficiencies are achieved simply by using isoabsorbing (equal optical density) solutions.
Clonogenic studies started with incubation of murine radiation-induced fibrosarcoma (RIF) cells with either Photofrin (reference photosensitizer) or G2 for ~18 h. In order to mimic conditions of in vivo oxygen measurements, for which cells could be surrounded by a solution of G2 in the interstitial space, illumination following the incubation was performed on cells directly in a solution of the photosensitizer (G2 or Photofrin). Alternatively, in order to assess phototoxicity induced by the photosensitizer adhered to or internalized by the cells, after incubation the cells were rinsed for 1 h in the photosensitizer-free media prior to illumination. In both types of experiments, the concentrations of G2 and Photofrin in the incubation media were such that the solutions were isoabsorbing at 630 nm with 0.04 OD (see ESI†). This absorbance corresponds to the concentration of Photofrin of about 10 μg ml-1 or 17 μM (MW 600 g mol-1)56 and ~0.4 μM for G2. Clonogenicity, i.e. survival and replicative potential, was assessed ca. one week later by evaluating the ability of the cells to form colonies. The results are expressed as cell surviving fractions, i.e. the ratio of the number of the colony-forming cells in a treated sample vs. that in the control, which received neither photosensitizer nor light.
The surviving fractions of the colony-forming cells for both types of experiments, i.e. photosensitizer left in the medium during irradiation and photosensitizer removed from the medium before irradiation, are plotted in Fig. 6 as a function of the light exposure. The difference between the photodynamic activity of Photofrin and G2 appears immediately clear from the graph. In the case of Photofrin, increase in the light exposure (from 0.0 to 0.5 to 2 J cm-2) is accompanied by a significant increase in the apparent phototoxicity, especially when irradiation was performed in the medium containing the photosensitizer. In fact, when the cells received the light dose of 2 J cm-2, the induced toxicity was so high that the surviving fraction could not be determined because it was below the lower level sensitivity of the performed assay. In contrast, virtually no toxicity was observed in the case of G2 neither when it was present in the medium nor after replacing the medium with sensitizer-free solution. The surviving fractions (average ± SE) of light-treated cells in the case of G2 ranged from 0.7 ± 0.2 to 1.2 ± 0.4, which is not significantly different from the surviving fractions of cells exposed only to light (without incubation with photosensitizer; 0.7 ± 0.1) or exposed only to G2 (without exposure to light; 1.3 ± 0.1). These results clearly demonstrate that in spite of its ability to produce singlet oxygen, G2 does not act as a PDT drug at light doses, which induce complete cell kill in the case of Photofrin.
Fig. 6.

Clonogenic survival data for RIF cells incubated with Photofrin or G2 during 18 h and irradiated with 630 nm light. Hollow symbols – photosensitizer is absent from the medium during irradiation. Filled symbols – photosensitizer is left in the medium during irradiation. Surviving fractions are calculated as fractions of the colony-forming cells in the treated samples compared to controls that received neither photosensitizer nor light. The data are shown as average ± SE from 3 independent studies. * No surviving cells were found for this condition – the point indicates the lower limit of the assay sensitivity.
Additional studies were performed with the purpose to establish at what concentration G2 does become phototoxic. In these experiments, concentrations of G2 varied from 2.5 μM to 20 μM and the illumination dose was 2 J cm-2. The results (Fig. S5, ESI†) unambiguously demonstrate that G2 is non-phototoxic even when used at concentrations highly exceeding (up to 40 times) those required for imaging.
The lack of apparent cellular phototoxicity of G2 is most likely related to its inability to adhere to cellular membranes and/or diffuse or to be transported into cells. Using microscopic imaging we have determined that incubation of different types of cells (e.g. mouse macrophages, HeLa and human epithelial cells) with G2 or other similar PEGylated dendritic porphyrins,13,57 does not lead to internalization of these probes even if they are present in the media at concentrations as high as 150 μM and if incubations last as long as 24 h. In fact, a special internalization method had to be devised in order to deliver one such probe inside cells to perform intracellular oxygen measurements.57
Thus, PEGylation, which was employed originally to prevent interactions of probe molecules with blood plasma proteins, simultaneously appeared to be an effective tool for diminishing phototoxicity.
Singlet oxygen, which is the initial phototoxic species in Type II PDT, has to be formed in the immediate vicinity of its biological target(s) in order to inflict damaging processes, eventually leading to cell death. The mean free-path of O2 (a1Δg) in aqueous solutions is estimated to be ~150 nm,58 which suggests that if the event of triplet quenching occurs at a distance larger than 150 nm from the cell membrane, formation of O2(a1Δg) is unlikely to have an impact on the cell viability. In cell suspensions or in cell cultures immersed in dilute solutions of G2, the overwhelming fraction of quenching events occurs in the solution bulk, and only a negligible number of O2(a1Δg) molecules are generated sufficiently close to cellular membrane. When the medium is replaced with solution containing no G2, cell damage simply cannot occur because no photosensitizer is left in the sample.
In contrast to PEGylated probes, photosensitizers used in PDT bind to cellular substructures. When subjected to illumination, bound photosensitizer molecules produce O2(a1Δg) in the immediate vicinity of vulnerable cellular targets, inflicting photodamage and ultimately compromising cell viability. In our study, PDT with Photofrin was performed under conditions which resulted in nearly quantitative (>99.99%) cell kill, corresponding to the surviving fraction of 0.0001. However, an isoabsorbing dose of G2 exposed to the same illumination resulted in virtually no cell death (<1%) and the surviving fraction being indistinguishable from non-treated control cells.
The probe concentrations employed in our study closely resemble those used in vivo in standard tissue oxygen measurements by diffuse light, i.e. ~0.1–1.0 μM. In such experiments, a phosphorescent probe is introduced directly into the blood plasma or into the interstitial fluid in vivo, and the phosphorescence lifetime measurements are performed with pulsed (time domain) or sinusoidally modulated (frequency domain) light sources. In a common implementation, using a standard LED-based phosphorometer,28 a single oxygen measurement typically requires averaging of 100–200 phosphorescence decays and takes 0.3–0.6 s to complete. During the measurement, the average power on the sample is about 40 μW (as measured by a power meter), which corresponds to 0.67% excitation duty cycle, and the illuminated area of tissue, ca. 0.6–0.7 cm in diameter (~0.4 cm2), is exposed to the excitation flux of ca. 100 μW cm-2. Assuming that individual measurements are performed every two seconds, photon flux averaged over the entire course of the experiment is about 25 μW cm-2, and after 30 min the illuminated area of tissue receives about 4.5 × 10-2 J cm-2 of energy.
In a typical PDT protocol, tissue treated with a photosensitizer is exposed to light such that the flux on the surface is maintained at a level of ca. 75 mW cm-2. The light dose received by a treated tissue after 30 min of PDT at 75 mW cm-2 is 135 J cm-2. Therefore, the light flux used for PDT is ~750 times higher than the flux used for measuring oxygen, and the total light exposure in PDT is ~3000 times higher than that in oxygen measurements. Based on this example, it is reasonable to expect that if under conditions of PDT phosphorescent probes do not induce phototoxic effects, as is the case for G2 vs. Photofrin, they should be completely safe to use under illumination required in routine tissue oxygen measurement applications. Clearly, there is a sufficient safety margin to allow for significant increase in light flux and/or PEGylated probe concentration should stronger phosphorescence signals be needed for oxygen measurement experiments.
Under conditions far below saturation of the triplet state, the rate of generation of O2(a1Δg) at a selected distance from the cellular membrane is proportional to the product of the excitation flux and probe concentration. Therefore, in spite of the fact that G2 and similar probes do not associate with cell membranes, a threshold can in principle be reached by increasing either the concentration or flux where sufficient amounts of O2(a1Δg) would be generated close to the membranes and produce photodamage. These conditions can be encountered, for example, in scanning microscopy applications,57,59 where phosphorescence is generated by focused high intensity light. One way to overcome the phototoxicity problem inherent to such measurements would be to encapsulate probes inside oxygen-permeable carrier vesicles, designed to capture and scavenge O2(a1Δg).
Especially strong caution needs to be exercised when using phosphorescent probes that can bind to cellular components. There are reports in the literature where such probes are being considered for intracellular oxygen measurements,60,61 whereby the probes permeate cellular membranes and may associate directly with cellular organelles. Our results suggest that phosphorescence quenching and production of singlet oxygen in such systems can easily inflict cellular damage even if measurements are performed using low excitation flux conditions.
Conclusions
In this study we tested a series of dendritically-protected PEGylated phosphorescent probes for their ability to generate singlet oxygen and inflict cellular photodamage. In spite of large differences in the oxygen quenching rate constants, all tested PdTBP-dendrimers were found to be able to sensitize singlet oxygen with approximately equal efficiency. Nevertheless, when compared against commonly used photosensitizer Photofrin in a clonogenic cell-survival assay, phosphorescent probe G2 showed virtually no phototoxicity. The lack of phototoxicity is explained by the inability of PEGylated dendrimers to penetrate cellular membranes and localize inside cells, while irradiation of extracellular probes by diffuse light produces very small amount of singlet oxygen in the vicinity of sensitive cellular targets.
The above results suggest that protected phosphorescent probes can be safely used for oxygen measurements in biological systems in vivo. This conclusion is especially important for applications where excitation of phosphorescence is effected by diffuse light, e.g. in 3D phosphorescence lifetime imaging (PLI),62 which combines phosphorescence quenching with diffuse optical tomography. In this imaging method, light exposures are much lower than those encountered in PDT, and therefore phototoxicity is unlikely to present a problem.
Acknowledgements
Support of the grants HL081273, NS031465, CA085831 and EB007279 from the NIH USA is gratefully acknowledged. Grants MAE DGPCC and MIUR (PRIN 20085ZXFEE) are acknowledged for the financial support in Italy. We thank Dr Louise E. Sinks for many useful discussions.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c0pp00356e
Notes and references
- 1.Vanderkooi JM, Maniara G, Green TJ, Wilson DF. An optical method for measurement of dioxygen concentration based on quenching of phosphorescence. J. Biol. Chem. 1987;262:5476–5482. [PubMed] [Google Scholar]
- 2.Wilson DF. Quantifying the role of oxygen pressure in tissue function. Am. J. Physiol.: Heart Circ. Physiol. 2007;294:H11–H13. doi: 10.1152/ajpheart.01293.2007. [DOI] [PubMed] [Google Scholar]
- 3.Mik EG, Johannes T, Ince C. Monitoring of renal venous Po-2 and kidney oxygen consumption in rats by a near-infrared phosphorescence lifetime technique. Am. J. Physiol.: Renal Physiol. 2008;294:F676–F681. doi: 10.1152/ajprenal.00569.2007. [DOI] [PubMed] [Google Scholar]
- 4.Golub AS, Barker MC, Pittman RN. Microvascular oxygen tension in the rat mesentery. Am. J. Physiol.: Heart Circ. Physiol. 2007;294:H21–H28. doi: 10.1152/ajpheart.00861.2007. [DOI] [PubMed] [Google Scholar]
- 5.Hirai DM, Copp SW, Herspring KF, Ferreira LF, Poole DC, Musch TI. Aging impacts microvascular oxygen pressures during recovery from contractions in rat skeletal muscle. Respir. Physiol. Neurobiol. 2009;169:315–322. doi: 10.1016/j.resp.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Bonanno JA, Clark C, Pruitt J, Alvord L. Tear oxygen under hydrogel and silicone hydrogel contact lenses in humans. Optometry and Vision Science. 2009;86:E936–942. doi: 10.1097/OPX.0b013e3181b2f582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakadžić S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, Ruvinskaya S, Devor A, Lo EH, Vinogradov SA, Boas DA. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat. Methods. 2010;7:755–U125. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.We assume that the Henry's law holds throughout the range of physiological oxygen pressures (0–159.6 mmHg). The bimolecular quenching rate constant (k2) is related to the quenching constant kq as: kq = αk2, where α (M mmHg-1) is the oxygen solubility coefficient ([O2] = αp(O2)). At 298 K and the air pressure of 760 mmHg (oxygen fraction in the air is 21% or 159.6 mmHg), air-equilibrated aqueous solutions are 252 μM in O2 (Fogg and Gerrard, ref. 39) and α = 1.58 × 10-6 M mmHg-1.
- 9.Ogilby PR. Singlet oxygen: there is indeed something new under the sun. Chem. Soc. Rev. 2010;39:3181–3209. doi: 10.1039/b926014p. [DOI] [PubMed] [Google Scholar]
- 10.Zhu TC, Finlay JC. The role of photodynamic therapy (PDT) physics. Med. Phys. 2008;35:3127–3136. doi: 10.1118/1.2937440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitra S, Foster TH. Photochemical oxygen consumption sensitized by a porphyrin phosphorescent probe in two model systems. Biophys. J. 2000;78:2597–2605. doi: 10.1016/S0006-3495(00)76804-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stepinac TK, Chamot SK, Rungger-Brandle E, Ferrez P, Munoz JL, Van Den Bergh H, Riva CE, Pournaras CJ, Wagnieres GA. Light-induced retinal vascular damage by Pd-porphyrin luminescent oxygen probes. Invest. Ophthalmol. Visual Sci. 2005;46:956–966. doi: 10.1167/iovs.04-0500. [DOI] [PubMed] [Google Scholar]
- 13.Lebedev AY, Cheprakov AV, Sakadzic S, Boas DA, Wilson DF, Vinogradov SA. Dendritic phosphorescent probes for oxygen imaging in biological systems. ACS Appl. Mater. Interfaces. 2009;1:1292–1304. doi: 10.1021/am9001698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fréchet JMJ, Tomalia DA, editors. Dendrimers and other dendritic polymers. John Wiley & Sons; Chichester: 2001. [Google Scholar]
- 15.New J. Chem. 2007;(7):31. Special Issue on Dendrimers. [Google Scholar]
- 16.Koo YEL, Cao YF, Kopelman R, Koo SM, Brasuel M, Philbert MA. Real-time measurements of dissolved oxygen inside live cells by organically modified silicate fluorescent nanosensors. Anal. Chem. 2004;76:2498–2505. doi: 10.1021/ac035493f. [DOI] [PubMed] [Google Scholar]
- 17.Zhang G, Chen J, Payne SJ, Kooi SE, Demas JN, Fraser CL. Multi-emissive difluoroboron dibenzoylmethane polylactide exhibiting intense fluorescence and oxygen-sensitive room-temperature phosphorescence. J. Am. Chem. Soc. 2007;129:8942. doi: 10.1021/ja0720255. [DOI] [PubMed] [Google Scholar]
- 18.Borisov SM, Klimant I. Luminescent nanobeads for optical sensing and imaging of dissolved oxygen. Microchim. Acta. 2008;164:7–15. [Google Scholar]
- 19.Wu CF, Bull B, Christensen K, McNeill J. Ratiometric single-nanoparticle oxygen sensors for biological imaging. Angew. Chem., Int. Ed. 2009;48:2741–2745. doi: 10.1002/anie.200805894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coogan MP, Court JB, Gray VL, Hayes AJ, Lloyd SH, Millet CO, Pope SJA, Lloyd D. Probing intracellular oxygen by quenched phosphorescence lifetimes of nanoparticles containing polyacrylamide-embedded Ru(dpp(SO3Na)2)3Cl2. Photochem. Photobiol. Sci. 2010;9:103–109. doi: 10.1039/b9pp00071b. [DOI] [PubMed] [Google Scholar]
- 21.Gillanders RN, Arzhakova OV, Hempel A, Dolgova A, Kerry JP, Yarysheva LM, Bakeev NF, Volynskii AL, Papkovsky DB. Phosphorescent oxygen sensors based on nanostructured polyolefin substrates. Anal. Chem. 2010;82:466–468. doi: 10.1021/ac902406w. [DOI] [PubMed] [Google Scholar]
- 22.McLaurin EJ, Greytak AB, Bawendi MG, Nocera DG. Two-photon absorbing nanocrystal sensors for ratiometric detection of oxygen. J. Am. Chem. Soc. 2009;131:12994–13001. doi: 10.1021/ja902712b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinogradov SA, Lo LW, Wilson DF. Dendritic polyglutamic porphyrins: Probing porphyrin protection by oxygen-dependent quenching of phosphorescence. Chem.–Eur. J. 1999;5:1338–1347. [Google Scholar]
- 24.Rozhkov V, Wilson D, Vinogradov S. Phosphorescent Pd porphyrin-dendrimers: Tuning core accessibility by varying the hydrophobicity of the dendritic matrix. Macromolecules. 2002;35:1991–1993. [Google Scholar]
- 25.Dunphy I, Vinogradov SA, Wilson DF. Oxyphor R2 and G2: phosphors for measuring oxygen by oxygen- dependent quenching of phosphorescence. Anal. Biochem. 2002;310:191–198. doi: 10.1016/s0003-2697(02)00384-6. [DOI] [PubMed] [Google Scholar]
- 26.Rietveld IB, Kim E, Vinogradov SA. Dendrimers with tetrabenzoporphyrin cores: near infrared phosphors for in vivo oxygen imaging. Tetrahedron. 2003;59:3821–3831. [Google Scholar]
- 27.Briñas RP, Troxler T, Hochstrasser RM, Vinogradov SA. Phosphorescent oxygen sensor with dendritic protection and two-photon absorbing antenna. J. Am. Chem. Soc. 2005;127:11851–11862. doi: 10.1021/ja052947c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinogradov SA, Fernandez-Seara MA, Dugan BW, Wilson DF. Frequency domain instrument for measuring phosphorescence lifetime distributions in heterogeneous samples. Rev. Sci. Instrum. 2001;72:3396–3406. [Google Scholar]
- 29.Independent measurements of extinction coefficients of porphyrindendrimers would be inaccurate because of the uncertainty in the determination of their molecular weights.
- 30.Jiang DL, Aida T. Morphology-dependent photochemical events in aryl ether dendrimer porphyrins: Cooperation of dendron subunits for singlet energy transduction. J. Am. Chem. Soc. 1998;120:10895–10901. [Google Scholar]
- 31.Rajesh CS, Capitosti GJ, Cramer SJ, Modarelli DA. Photoinduced electron-transfer within free base and zinc porphyrin containing poly(amide) dendrimers. J. Phys. Chem. B. 2001;105:10175–10188. doi: 10.1021/ol015837t. [DOI] [PubMed] [Google Scholar]
- 32.Eastwood D, Gouterman M. Porphyrins. XVIII. Luminescence of Co, Ni, Pd, Pt complexes. J. Mol. Spectrosc. 1970;35:359–375. [Google Scholar]
- 33.Vogler A, Kunkely H, Rethwisch B. Tetrabenzoporphyrin complexes of Iron, Palladium and Platinum. Inorg. Chim. Acta. 1980;46:101–105. [Google Scholar]
- 34.Lebedev AY, Filatov MA, Cheprakov AV, Vinogradov SA. Effects of structural deformations on optical properties of tetrabenzoporphyrins: Free-bases and Pd complexes. J. Phys. Chem. A. 2008;112:7723–7733. doi: 10.1021/jp8043626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knyukshto VN, Shul'ga AM, Sagun EI, Zen'kevich EI. Spectral manifestations of nonplanarity effects in Pd complexes of porphyrins. Opt. Spectrosc. 2002;92:53–62. [Google Scholar]
- 36.Vögtle F, Plevoets M, Nieger M, Azzellini GC, Credi A, De Cola L, De Marchis V, Venturi M, Balzani V. Dendrimers with a photoactive and redox-active [Ru(bpy)3]2+-type core: Photophysical properties, electrochemical behaviour, and excited-state electron-transfer reactions. J. Am. Chem. Soc. 1999;121:6290–6298. [Google Scholar]
- 37.Mosinger J, Micka Z. Quantum yields of singlet oxygen of metal complexes of meso-tetrakis(sulfonatophenyl)porphine. J. Photochem. Photobiol., A. 1997;107:77–82. [Google Scholar]
- 38.Seybold PG, Gouterman M. Pporphyrins.13. Fluorescence spectra and quantum yields. J. Mol. Spectrosc. 1969;31:1. [Google Scholar]
- 39.Fogg PGT, Gerrard W. Solubility of gases in liquids. Wiley; New York: 1991. [Google Scholar]
- 40.Cameron CS, Gorman CB. Effects of site encapsulation on electrochemical behavior of redox-active core dendrimers. Adv. Funct. Mater. 2002;12:17–20. [Google Scholar]
- 41.Sharma AK, Kim N, Cameron CS, Lyndon M, Gorman CB. Dendritically encapsulated, water-soluble Fe4S4: synthesis and electrochemical properties. Inorg. Chem. 2010;49:5072–5078. doi: 10.1021/ic1002447. [DOI] [PubMed] [Google Scholar]
- 42.Montalti M, Credi A, Prodi L, Gandolfi MT. Handbook of Photochemistry. Taylor & Francis Group; Boca Raton: 2006. [Google Scholar]
- 43.Losev AP, Byteva IM, Gurinovich GP. Singlet oxygen luminescence quantum yields in organic solvents and water. Chem. Phys. Lett. 1988;143:127–129. [Google Scholar]
- 44.Ogilby PR, Foote CS. Chemistry of singlet oxygen. 42. Effect of solvent, solvent isotopic substitution and temperature on the lifetime of singlet molecular oxygen O2(a1Δg) J. Am. Chem. Soc. 1983;105:3423–3430. [Google Scholar]
- 45.Schweitzer C, Schmidt R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem. Rev. 2003;103:1685–1757. doi: 10.1021/cr010371d. [DOI] [PubMed] [Google Scholar]
- 46.Egorov SY, Kamalov VF, Koroteev NI, Krasnovsky AA, Toleutaev BN, Zinukov SV. Rise and decay kinetics of photosensitized singet oxygen luminescence in water - measurements with nanosecond time-correlated single photon-counting technique. Chem. Phys. Lett. 1989;163:421–424. [Google Scholar]
- 47.Ogilby PR, Foote CS. Chemistry of singlet oxygen. 36. Singlet molecular oxygen (1Δg) luminescence in solution following pulsed laser excitation. Solvent deuterium isotope effects on the lifetime of singlet oxygen. J. Am. Chem. Soc. 1982;104:2069–2070. [Google Scholar]
- 48.In applications papers, compound G2 is usually referred to as “Oxyphor G3”, where “G3” is not related to the dendritic generation number.
- 49.Busch TM, Hahn SM. Photodynamic therapy. In: Abeloff MD, Armitage JO, Neiderhuber JE, Kastan MB, McKenna WG, editors. Clinical Oncology. P. M. Gorden Associates; Philadelphia: 2004. [Google Scholar]
- 50.Biel MA. Photodynamic therapy of head and neck cancers. Methods Mol. Biol. 1995;635:281–293. doi: 10.1007/978-1-60761-697-9_18. [DOI] [PubMed] [Google Scholar]
- 51.Overholt BF, Wang KK, Burdick JS, Lightdale CJ, Kimmey M, Nava HR, Sivak MV, Jr., Nishioka N, Barr H, Marcon N, Pedrosa M, Bronner MP, Grace M, Depot M. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett's high-grade dysplasia, Gastrointest. Endosc. 2007;66:460–468. doi: 10.1016/j.gie.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 52.Hahn SM, Putt ME, Metz J, Shin DB, Rickter E, Menon C, Smith D, Glatstein E, Fraker DL, Busch TM. Photofrin uptake in the tumor and normal tissues of patients receiving intraperitoneal photodynamic therapy. Clin. Cancer Res. 2006;12:5464–5470. doi: 10.1158/1078-0432.CCR-06-0953. [DOI] [PubMed] [Google Scholar]
- 53.Bellnier DA, Greco WR, Loewen GM, Nava H, Oseroff AR, Dougherty TJ. Clinical pharmacokinetics of the PDT photosensitizers porfimer sodium (Photofrin), 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (Photochlor) and 5-ALA-induced protoporphyrin IX. Lasers Surg. Med. 2006;38:439–444. doi: 10.1002/lsm.20340. [DOI] [PubMed] [Google Scholar]
- 54.Wilson BC, Olivo M, Singh G. Subcellular localization of Photofrin and aminolevulinic acid and photodynamic cross-resistance in vitro in radiation-induced fibrosarcoma cells sensitive or resistant to photofrin-mediated photodynamic therapy. Photochem. Photobiol. 1997;65:166–176. doi: 10.1111/j.1751-1097.1997.tb01894.x. [DOI] [PubMed] [Google Scholar]
- 55.Buettner GR, Kelley EE, Burns CP. Membrane lipid free radicals produced from L1210 murine leukemia cells by photofrin photosensitization: an electron paramagnetic resonance spin trapping study. Cancer Res. 1993;53:3670–3673. [PubMed] [Google Scholar]
- 56.Dysart JS, Patterson MS. Characterization of Photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro. Phys. Med. Biol. 2005;50:2597–2616. doi: 10.1088/0031-9155/50/11/011. [DOI] [PubMed] [Google Scholar]
- 57.Finikova OS, Lebedev AY, Aprelev A, Troxler T, Gao F, Garnacho C, Muro S, Hochstrasser RM, Vinogradov SA. Oxygen microscopy by two-photon-excited phosphorescence. ChemPhysChem. 2008;9:1673–1679. doi: 10.1002/cphc.200800296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hatz S, Poulsen L, Ogilby PR. Time-resolved singlet oxygen phosphorescence measurements from photosensitized experiments in single cells: Effects of oxygen diffusion and oxygen concentration. Photochem. Photobiol. 2008;84:1284–1290. doi: 10.1111/j.1751-1097.2008.00359.x. [DOI] [PubMed] [Google Scholar]
- 59.Golub AS, Pittman RN. Po-2 measurements in the microcirculation using phosphorescence quenching microscopy at high magnification. Am. J. Physiol.: Heart Circ. Physiol. 2008;294:H2905–H2916. doi: 10.1152/ajpheart.01347.2007. [DOI] [PubMed] [Google Scholar]
- 60.O'Riordan TC, Fitzgerald K, Ponomarev GV, Mackrill J, Hynes J, Taylor C, Papkovsky DB. Sensing intracellular oxygen using near-infrared phosphorescent probes and live-cell fluorescence imaging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1613–R1620. doi: 10.1152/ajpregu.00707.2006. [DOI] [PubMed] [Google Scholar]
- 61.Dmitriev RI, Zhdanov AV, Ponomarev GV, Yashunski DV, Papkovsky DB. Intracellular oxygen-sensitive phosphorescent probes based on cell-penetrating peptides. Anal. Biochem. 2010;398:24–33. doi: 10.1016/j.ab.2009.10.048. [DOI] [PubMed] [Google Scholar]
- 62.Apreleva SV, Wilson DF, Vinogradov SA. Tomographic imaging of oxygen by phosphorescence lifetime. Appl. Opt. 2006;45:8547–8559. doi: 10.1364/ao.45.008547. [DOI] [PMC free article] [PubMed] [Google Scholar]